
Abstract
The common bean, an important source of protein and minerals for humans, complements cereals both nutritionally and as a rotation crop, supplying nitrogen and reducing soil pathogens. The aim of this study was to develop an operational SNP-based panel for common bean in order to facilitate SSR employment in genetic diversity and population structure analyses, and its use in breeding programs. A set of 88 diverse and important common bean cultivars/lines (53), landraces (33) and wild accessions (2) were genotyped. Overall, the 58 SSRs performed better at evaluating genetic diversity (Ā = 7.38; He = 58.7 %; PI = 1.20E−45) than the 345 SNPs, of which the SSRs dinucleotides (SSR-di) were more informative (Ā = 9.92; He = 72.5 %; PI = 3.40E−26) and a selected set of 13 SSRs (Ā = 15.31/locus; He = 84.5 %; PI = 1.03E−19) allowed for the discrimination of all individuals. For the 345 high-quality scored SNPs a low combined PI (4.70E−119) and high PE (100 %) was obtained for the assessment of parentage and identity. The SNPs were very useful for linkage mapping in inter- (78.2 %) and intra-gene pool (17.7 %) crosses. Both markers afforded high resolution detection of inter-gene pool structure, with greater differentiation based on SNPs (K = 2, F ST = 0.759). The SSRs-di differentiated cultivars/lines and landraces (K = 3) of Mesoamerican origin. A set of 16 SSRs was selected to establish a routine and operational analysis of Genbank accessions allowing an efficient origin-based discrimination of common bean accessions. Operational genotyping panels based on SSRs and SNPs were derived, contributing to the growing integration of genomics with molecular breeding programs of the common bean.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Phaseolus vulgaris (common bean) is the most widely grown species of the genus Phaseolus and is also the most important grain legume in human diets in all continents (Broughton et al. 2003). The bean is a nutritional and cost-effective food source for more than 300 million people in the tropics, and for small-scale farmers it represents an important source of household income (Wortmann et al. 1998). The American continent consumes 43 % of the total bean production worldwide, followed by Asia (34.5 %), Africa (18.5 %), Europe (3.7 %), and Oceania (0.1 %). Developing countries account for ∼87 % of world consumption (Vieira et al. 2010). Brazil is the largest producer of the common bean, with production oscillating from 3.2 to 3.3 million tons/year in the last decade and consumption estimated to be 18.4 kg/inhabitant in 2013. Net Brazilian imports were approximately 100 thousand tons/year until 2006/2007 and increased to 200 thousand tons/year (2012/2013), despite an enormous potential for expansion of domestic production (Silva and Wander 2013). The use of biotechnological tools may contribute to facilitating the development of superior and more competitive genotypes in terms of nutritional and agronomic quality and production costs.
In the initial stages of the pre-breeding process, the use of molecular tools assists in quantification and qualification of genetic diversity. This approach enables the generation of strategies for efficient conservation and use of these genetic resources with a goal of identifying genetically divergent accessions with good agronomic traits that diminish the effects of a narrowing genetic base (Buso et al. 2006). In breeding programs, molecular marker-assisted backcrossing reduces the number of generations required to recover the recurrent parent, thus accelerating the introgression of genomic regions of interest (Oliveira et al. 2008). This backcrossing also facilitates the pyramiding of disease resistance alleles in elite cultivars (Alzate-Marin et al. 2005). In the final stage of breeding programs, molecular markers are useful for the quantification of genetic variability of value for cultivation and use (VCU) lines, determination of genetic identity for registration purposes and cultivar protection, and certification tests of commercial seed lots (Cardoso et al. 2013, 2014).
The common bean (2n = 22, ∼587 Mb) (Schmutz et al. 2014) has a small genome compared to other crop legumes, close to the size of the cowpea bean (Vigna unguiculata) genome, approximately half the size of the soybean (Glycine max), and ∼21 times smaller than the faba bean (Vicia faba) (Ellwood et al. 2008). Over the last 20 years, significant efforts have been made to establish a robust genetic map of the common bean and to use molecular markers for genetic analysis. The first such effort was the development of a genetic map based on RFLP and RAPD markers (Freyre et al. 1998). Steady progress was made in integrating a growing and diverse number of molecular markers using unique biparental populations (Yu et al. 2000; Grisi et al. 2007; Blair et al. 2013a). More recently, a genetic map integrating more than 1000 markers was generated, providing a broad analysis of the entire genome with potential for synteny studies and QTL mapping (Hougaard et al. 2008; Galeano et al. 2011, 2012). Obtaining robust markers with high powers of discrimination that amplify satisfactorily in a diverse genetic background is a continuous process and is dependent on an adequate selection of markers.
Microsatellite or simple sequence repeats (SSRs) markers are widely available for molecular genotyping of the common bean (Gaitán-Solís et al. 2002; Grisi et al. 2007; Garcia et al. 2011). Various studies have developed SSRs from enriched genomic libraries, with emphasis on regions rich in dinucleotides (SSR-di) (Buso et al. 2006). SSRs-di represents the most abundant class of repetitive DNA, enabling the exploitation of molecular polymorphism. A battery of markers currently exists for desired operational characteristics. Recent advances in whole or partial genome sequencing and expressed sequence tags have enabled the identification of a growing number of SSR regions (Hanai et al. 2007). As shown in the study of Müller et al. (2014), it is possible to identify SSRs based on repetitions of polymorphic tetra-, penta-, and hexanucleotides. The use of SSR markers has advanced with the development of simultaneous amplification systems of various loci allied with a semi-automated detection system (Cardoso et al. 2013), enabling their use in more robust genotyping systems. The PhaseolusGenes database includes a comprehensive collection of SSR markers as well as sequences containing microsatellite motifs [http://phaseolusgenes.bioinformatics.ucdavis.edu/].
Single nucleotide polymorphisms (SNPs) comprise the most frequent form of genetic variation, occurring in abundance along the entire genome (Ching et al. 2002). SNPs are biallelically polymorphic. Tri- or tetrallelic polymorphisms may occur, but at a lesser frequency (Brown 2002). Estimates of SNP density in plants vary according to the reproductive habit of the species and the regions they occupy across the genome (Kaczorowski et al. 2008). In soybean, for instance, an estimated 1 SNP occurs for every 2038 and 191 bp in coding and non-coding regions, respectively (Van et al. 2005). The minimum number of SNPs required to successfully determine the most common haplotype groups in soybean varies from 9600 to 13,600 SNPs in cultivars/lines and landraces, respectively (Hyten et al. 2007). The high frequency of SNPs across the genome allows for the development of dense genetic maps and robust association studies that identify genes of interest and more precise genomic approaches (Galeano et al. 2012). In addition, as a result of the reduced mutation rate compared to the SSR markers, as well as the availability of automated detection systems (Thomson et al. 2012; Yu et al. 2011), SNPs are increasingly used as molecular markers in a number of genetic applications in leguminous plant (Loridon et al. 2012; Saxena et al. 2012; Chang et al. 2013; Song et al. 2013). A growing number of SNPs are being reported in the common bean (Hyten et al. 2010; Cortés et al. 2011; Souza et al. 2012; Blair et al. 2013a), thus expanding the possibilities for immediate use of this tool in breeding programs and advancing the knowledge of the genetics of agronomically important traits.
While diverse genetic approaches of SSR marker-based molecular tools are well established for broad use in breeding of the common bean, routine use of the SNP markers remains incipient for this crop. The aim of this study was to evaluate and develop an operational SNP-based panel to be used in complementation with SSR marker analysis to uncover the diversity, genetic structure and for application in breeding of the common bean. In this study, while SSRs performed better at evaluating genetic diversity, the SNPs were more adequate for inter- and intra-gene pool linkage analysis mapping, and presented a greater resolution in the detection of inter-gene pool structure. For the assessment of parentage and individual identification, SNPs allowed a low combined PI (probability of identity) and high PE (probability of exclusion), compared to SSRs, due to the high number of markers used. Focusing on the use for breeding programs worldwide, genotyping panels based on SSR and SNP markers were developed, which provide the means to characterize diverse sets of common bean germplasm and address a wide range of biological questions.
Materials and Methods
Plant Material and DNA Extraction
A total of 88 genotypes of common bean, including 42 commercial cultivars and 11 breeding lines introduced by research institutions from Brazil and abroad, 33 Brazilian landraces and two wild genotypes were characterized by SSR and SNP markers. The genotypes evaluated represent 14 types of grain (Large White, Small White, Carioca, Cranberry, Dark Red Kidney, Jalo, Mulatinho, Pinto, Black, Rajado, Red, Rosinha, Roxo and others) belonging to the Mesoamerican (67 genotypes) and Andean (21 genotypes) gene pools (Supplementary Material 1). Total genomic leaf DNA was isolated as described by Grattapaglia and Sederoff (1994) and quantified by a Qubit spectrophotometer (Invitrogen, Carlsbad, CA, USA). Concentrations were adjusted to 5 and 50 ng/μL for SSR and SNP analyses, respectively.
SSR Genotyping
Genotyping was performed on 58 SSR markers subdivided into two groups: 24 SSRs-di (Supplementary Material 2) and 34 tri- (3), tetra- (9), penta- (8), and hexanucleotide (7) motifs and compound SSRs (7) (Supplementary Material 3). Thirty-four SSRs were derived from BAC-end sequences (BES) developed by Müller et al. (2014) (BES-SSRs). The SSR primers were fluorescently labeled and analyzed in multiplex co-amplification systems composed of three to six loci based on the Standard Dye Set DS-30 for the 24 SSRs-di, and DS-33 for the 34 BES-SSRs (Applied Biosystems, Foster City, CA, USA). SSR amplification was performed with Qiagen Multiplex PCR 2X (Qiagen, Hilden, NRW, Germany) in a GeneAmp Thermal Cycler 9700 (Applied Biosystems), following the parameters established by Müller et al. (2014). Amplified BES-SSR and SSRs-di products were subjected to electrophoresis in the automated DNA analyzers ABI3500 and ABI3100 (Applied Biosystems), respectively. The data were genotyped using the DataCollection 2.0 and GeneMapper 4.1 programs (Applied Biosystems).
Identification of SNPs
A pool of 3487 SNPs was derived from contrasting sequences of the cultivars BAT 93 and Jalo EEP 558 (Hyten et al. 2010). Initial filtering was performed, eliminating the redundant SNPs. To identify repeats, SNPs were aligned against the reference genomes of G. max (Schmutz et al. 2010) obtained from the site Phytozome [http://www.phytozome.net/soybean.php], and P. vulgaris obtained via CYTED (Ibero-American Programme for Science, Technology and Development) [http://www.cyted.org/]. A second step consisted of selection of SNPs for the GGGT (Golden Gate Genotyping Technology) assay design, as described by Grattapaglia et al. (2011). Next, a series of parameters was defined to design oligonucleotides flanking the SNPs, beginning with identification of at least 60 bp upstream and downstream. This process allowed the design of three oligonucleotides, two of which were allele-specific (ASO) for each variant of the SNP, and one of which was locus-specific (LSO) and linked to the 3′ region of the DNA fragment containing the target SNP. Finally, various steps of in silico filtering were performed (Grattapaglia et al. 2011) with a gradual increase of restrictions, such that the SNP pools selected in each filtering (∼300–600 SNPs) were subjected to the Assay Design Tool (ADT, Illumina, San Diego, CA, USA) to obtain a validation score reflecting the probability of genotyping success. The 384 SNPs with the highest scores were recommended for synthesis using the Oligo Pool Assay (OPA VC0013574) on the BeadXpress platform (Illumina). The sequence list of one set of SNP ASO primers and their positions (BLASTN: E value ≤ 1.0E−10) in the reference genome of the common bean, Andean variety (G19833, Schmutz et al. 2014), is shown in Supplementary Material 4. The SNP nomenclature “BARC-PV-000XXXX” from the study of Hyten et al. (2010) was reduced to “PV-XXXX” in this study, with “X” referring to the SNP number.
Genotyping of SNPs
The 384 SNPs were genotyped through the Illumina BeadXpress platform (Kim and Misra 2007), based on the VeraCodeTM technology (Illumina, Inc., CA, USA), using the manufacturer’s protocol. These procedures were conducted in the Biotechnology Laboratory of Embrapa Rice and Beans (CNPAF, Santo Antônio de Goiás, GO, Brazil). Genotyping was performed using GenomeStudio version 1.8.4 (Illumina, Inc., CA, USA) with Gen Call Threshold of 0.25; Call Rate values ranging from 0.6463 to 1.00 and GenTrain ≥0.2535. Clustering of the representative homozygote (AA/BB) and heterozygote (AB) allelic groups for each SNP was conducted in automated fashion a priori, followed by manual adjustment. BAT 93 and Jalo EEP 558 profiles were used as references.
Analysis of Genetic Diversity and Neighbor-Joining Clustering
Genetic diversity of the 88 individuals was analyzed using estimates of allelic frequency, number of alleles (A), number of alleles per locus (Ā), number of private alleles (Ap), expected heterozygosity or gene diversity (H E), observed heterozygosity (H O) and the fixation index (F IS) using the program GenAlex v6.5 (Peakall and Smouse 2012). Genetic probability of identity (PI) and genetic probability of exclusion (PE) for each locus and multiple loci were obtained using the same program. Detection of identical genotypes using SSR markers was analyzed by the software Identity v4 (Sefc et al. 1997). Neighbor-joining cluster analysis (Saitou and Nei 1987) based on the simple matching dissimilarity matrix, was implemented in the program DARwin v5.0.158 (Perrier and Jacquemoud-Collet 2006), with 1000 bootstrap replicates.
Analysis of Population Structure
The genetic structure of the common bean genotypes was analyzed by principal component analysis (PCA) calculated by the program PAST v2.17c (Hammer et al. 2001) based on a simple matching dissimilarity matrix. Genetic differentiation (F ST) in the whole sample was estimated, with a confidence interval of 95 % for 9999 permutations, using GenAlex v6.5 (Peakall and Smouse 2012). The program STRUCTURE v2.3.4 (Pritchard et al. 2000) was used to infer the genotypic structure using Bayesian-based clustering. The individual structures were classified in “K” clusters according to genetic similarity. The admixture model was applied, with correlated allelic frequencies, using no previous population information. The number of tested clusters (K) ranged from 1 to 10, with 10 interactions each. The burn-in period and the number of Markov chain Monte Carlo (MCMC) replications were 500,000 and 1,000,000, respectively. The number of genetic groups was determined (most probable K) based on the criteria proposed by Evanno et al. (2005) using the program STRUCTURE HARVESTER v0.6.93 (Earl and vonHoldt 2012). The software CLUMPP v1.1.2 (Jakobsson and Rosenberg 2007) was used to find consensus among the 10 most probable K interactions, and the output was used directly as input in the display program DISTRUCT v1.1 (Rosenberg 2004). Analysis of molecular variance (AMOVA) was performed to test the structure of genetic diversity based on the different classes of markers, using the program Arlequin v3.5 (Excoffier and Lischer 2010) with 10,000 permutations.
Analysis of Linkage Disequilibrium
Analysis of linkage disequilibrium (LD) was conducted using the program Tassel v.2.1 (Bradbury et al. 2007), with 1000 permutations. The heterozygote genotypes were considered to be missing data, and the molecular marker data were later transformed into haplotype data. To evaluate linkage disequilibrium, the standardized disequilibrium coefficient r 2 was calculated for all the SSR and SNP markers. The loci were considered to be in significant LD if p < 0.001 and r 2 > 0.10. These LD parameters were calculated for the whole sample and, separately, for the Andean and Mesoamerican gene pools. In a second step in LD analysis, the SNP loci with minor allele frequency (MAF) < 0.10 were discarded from the analysis.
Alignment of SSRs and SNPs in the P. vulgaris Genome
To visualize the distribution of SSRs and SNPs, the flanking sequences of SNPs and repetitive sequences of SSRs were aligned against the genome of P. vulgaris, Andean variety (G19833) (Schmutz et al. 2014), [https://www.phytozome.com/commonbean.php] using the BLASTN (Altschul et al. 1990) with E value ≤1.0E−25. The minimum length of alignment required to be included in the result was 70 and 120 bp for SSRs and SNPs, respectively. For each SSR and SNP analyzed, only the hit with the best score was used to develop a map on the Circos program (Krzywinski et al. 2009) to display the genomic position of P. vulgaris that aligned with the flanking regions.
Results
Genetic Diversity
The results showed that, in the common bean, SSRs-di were superior to BES-SSRs at estimating the parameters of mean numbers of alleles (A) and mean gene diversity (H E), as presented in Table 1. Of the 24 SSRs-di, a mean of 9.92 alleles was identified per locus, with H E of 0.725 and H O and F IS of 0.034 and 0.954, respectively, confirming the high homozygosity of the analyzed loci. Of the 34 BES-SSRs, the estimated Ā was 1.8× less when compared to that of the SSRs-di, with a mean of 5.59 alleles and a mean H E of 0.490. However, the indices of H O (0.004) and F IS (0.993) were similar to those of the SSRs-di. Nevertheless, within the two categories of SSRs, some loci yielded high estimates: the SSR-di PV163 had 23 alleles (H E of 0.879) and the BES-SSR PvComp10 had 19 alleles (H E of 0.918). In relation to the number of private alleles (Ap) in 24 SSRs-di, 44 and 118 private alleles (Ap) were identified in 24 SSRs-di exclusively from the Andean and Mesoamerican gene pool, respectively, and 18 and 19 private alleles were detected in the loci PV163 and BM154, respectively. Private alleles were identified in 31 loci of the 34 BES-SSRs, maintaining the same proportion observed for the SSRs-di. Of these, 28 and 97 alleles were exclusively from the Andean and Mesoamerican gene pools, respectively. The PvComp4 and PvComp10 loci were notable, with 15 private alleles each.
Of the 384 SNPs evaluated in this study, 358 (93 %) reached the adequate detection level in BeadXpress, of which 345 (96 %) were polymorphic and 13 (4 %) monomorphic, considering the 88 genotypes of the common bean. Only 26 SNPs were excluded from analyses, seven failed to amplify at least one allele and 19 did not have well-defined clusters. Genotypic analysis grouped by gene pool revealed 124 (36 %) polymorphic SNPs within the Andean group and 332 (96 %) in the Mesoamerican group. With the greatest allele frequency estimated at 0.721, the mean H E calculated for the SNPs (0.390) was approximately 1.9× less in relation to the SSRs-di (0.725) and similar to the BES-SSRs (0.490), ranging from 0.024 for the PV-4825 locus to 0.506 for the PV-5330 locus. The values from the H O and F IS indices were estimated at 0.022 and 0.937, respectively (Table 1). Of the 345 polymorphic SNPs, a set of 234 differentiated the Andean (13 private alleles) from the Mesoamerican (221 private alleles) based on allelic pattern.
The power of exclusion estimated for each marker group was high, reaching 99.9 % for SSRs and 100 % for SNPs (a set of 210 SNPs differentiate all genotypes). The PI based on 24 SSRs-di ranged from 0.013 (BM154) to 0.572 (PV251), and a combined probability of genetic identity was on the order of 3.4E−26. Considering the 34 BES-SSRs, the loci estimates ranged from 0.013 (PvComp10) to 0.755 (PvPenta5), with a combined value of 3.6E−20, slightly higher when compared to the SSRs-di even when using an additional set of 10 SSRs. For the 345 SNPs, the combined PI was estimated at 4.7E−119 (Table 1), with individual values ranging from 0.375 (PV-3045, PV-3353, PV-5330, PV-4473, PV-4220, PV-4881, PV-2954, PV-4496) to 0.954 (PV-4825). As a result of the reduced informative power per individual gene locus, a greater number of SNP markers (biallelic) were required to obtain the same values as SSRs (multiallelic). Thus, the same value of PI estimates based on six SSRs-di markers could be obtained using nine BES-SSRs or 23 SNPs. In general, three times more SNPs than SSRs were needed to obtain the same PI estimate. Considering the 58 SSRs evaluated in this study (24 SSRs-di and 34 BES-SSRs), the combined PI for 13 selected loci with 10 or more alleles and an H E above 0.7 was estimated at 1.03E−19, represented by nine SSRs-di (BM154, PV272, PV87, PV163, PV25, BM143, BM187, BM210, and BM114) and four BES-SSRs (PvComp10, PvComp4, PvTri8, and PvTetra25). These 13 SSRs, with mean allele number of 15.31 per locus and a mean H E of 0.845, enabled differentiation of all 88 genotypes of the common bean.
The potential for practical application of SNPs in genetic mapping was evaluated through the polymorphism test on 11 genotypes that compose eight biparental populations. Of these crosses, five involving parental lines of the Mesoamerican gene pool (intra-gene pool) (BAT 477 × Pérola, Red Mexican × Pérola, CNFP10132 × BRS Cometa, BRS Aurora × PI181996, US Pinto 111 × PI181996) showed, on average, 61 (17.7 %) polymorphic SNPs. The mean number of polymorphic SNPs was 270 (78.2 %) for three inter-gene pool crosses (Andean × Mesoamerican) (AND 277 × Rudá, Ouro Branco × CNFP10132, AND 277 × SEA 5) between genitors. It was observed that of the 345 SNPs, the number of shared inter-gene pool polymorphic markers was 212 (61.4 %), and the intra-gene pool was only one SNP (0.29 %)—the SNP (PV-4505) being polymorphic and common among the eight biparental crosses (intra- and inter-gene pool) (Table 2).
Cluster Analysis
Neighbor-joining trees were separately generated for the three groups of markers (SSRs-di, BES-SSRs and SNPs) as well as for the two types of SSR markers jointly, revealing clustering by germplasm origin (Fig. 1). The upper and lower branches of the clusters are represented by genotypes of Andean and Mesoamerican origins, respectively, in which breeding (cultivar/line) and traditional accessions (landrace) are represented. The accession BRSMG Talismã, a priori classified as of Mesoamerican origin, was placed in this a posteriori analysis together with the Andean group. The SSR and SNP data show that the lines of US Pinto 111 and Red Mexican (derived from the USA) were clustered together and near the Mesoamerican genotypes. The wild Mexican accessions (G23554 and PHAS 8328) clustered together with the PI 181996 line (derived from Guatemala) near the group formed by the North American lines (Fig. 1).

Neighbor-joining dendrograms based on a simple matching dissimilarity matrix representing the grouping of the 88 genotypes of the common bean for the following sets of markers: a 24 SSRs-di, b 34 BES-SSRs, c 58 microsatellites (34 BES-SSRs + 24 SSRs-di), and d 345 SNPs. The Andean genotypes are represented in dark green (cultivar/line) and light green (landrace) and the Mesoamerican in red (cultivar/line), and yellow (landrace). The wild genotypes are indicated in light blue. Lines from the USA are represented in dark blue and the line from Guatemala in purple
Population Structure
Principal component analysis of the distinct groups of markers showed similar results, a clear structuring of the genotypes based on Mesoamerican and Andean origins. The first axis, which explained the largest variation of the data, consistently separated the accessions by origin (Fig. 2), exhibiting extreme values of 65.51 and 91.42 % for the BES-SSRs and SNPs markers, respectively (Figs. 2b, d). The second axis explained from 4.29 to 13.93 % of the molecular variation of SNPs and BES-SSRs, respectively. The separation between cultivars/lines (red) and landraces (yellow) was observed within the accessions of Mesoamerican origin only in the PCA based on SSRs-di (Fig. 2a). High estimates of F ST (P < 0.05) among the Andean and Mesoamerican groups were observed for the classes of markers evaluated in this study (SSRs-di: F ST = 0.341, p value = 0.001; BES-SSRs: F ST = 0.410, p value = 0.001; SNPs: F ST = 0.759, p value = 0.001), supporting the differentiation between gene pools. Considering the differentiation between the cultivars/lines and the landraces (disregarding the two wild genotypes), the F ST calculated from the SSRs-di (F ST = 0.085, p value = 0.001) indicated a moderate genetic differentiation, while the BES-SSRs (F ST = 0.032, p value = 0.001) and SNPs (F ST = 0.021, p value = 0.051) showed only a small differentiation.

Principal component analysis (PCA) of the 88 common bean genotypes based on a simple matching dissimilarity matrix, calculated for a 24 SSRs-di, b 34 BES-SSRs, c 58 microsatellites (34 BES-SSRs + 24 SSRs-di), and d 345 SNPs. The Andean genotypes are represented in dark green (cultivar/line) and light green (landrace) and the Mesoamerican in red (cultivar/line), and yellow (landrace). The wild genotypes are indicated in light blue. Lines from the USA are represented in dark blue and the line from Guatemala in purple
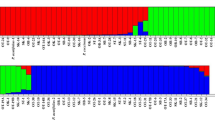
In the STRUCTURE program analysis, the results of ∆K suggested that K = 2 was the most probable clustering type for all groups of markers analyzed individually (SSRs-di, BES-SSRs, total SSRs and SNPs). Figure 3 illustrates the clear division (K = 2) between the genotypes of Andean (green) and Mesoamerican origin (red), with the exception of the accession BRSMG Talismã (ID: 62). This accession was previously identified as of Mesoamerican origin (based on small-seed size) and, a posteriori, placed in the Andean cluster. Through STRUCTURE analysis, the formation of two groups was observed, a small one composed of 21 Andean genotypes, including the BRSMG Talismã of mixed origin, and another consisting of 66 Mesoamerican genotypes. This is consistent with the predominance of Mesoamerican gene pool in the Brazilian landraces and cultivars, as reported in previous studies (Burle et al. 2010; Blair et al. 2013b and Cardoso et al. 2014). Analysis based on SSRs-di (K = 3) revealed a subdivision within the Mesoamerican gene pool represented by cultivars/lines (purple) and landraces (red). Successive subdivisions were observed throughout the analyses. The estimates of K = 4 (pink) and K = 5 (pink) obtained with SNP and SSR markers, respectively, showed a cluster gathering the wild genotypes of Mexican origin (G23554 and PHAS 8328), the North American lines (US Pinto 111 and Red Mexican), and the line from Guatemala (PI 181996). In clusters K = 5 and 6 (yellow), using SNPs, a fragmentation separating the wild and Guatemala genotypes from the accessions of North American origin was observed, corroborating their geographic proximities (Blair et al. 2012). Furthermore, in cluster K = 7 (light blue) and subsequent levels of structure, BES-SSRs showed a separation of the wild germplasm from the USA and Guatemala lines. A greater fragmentation of the cluster (K = 8 to 9) was observed only with the SNP markers. The clusters K = 8 and 9 (brown) represent the group comprising cultivars/lines of Carioca grain type and landraces of Carioca and Mulatinho from the Mesoamerican gene pool, and the K = 4 to 9 (purple bars) represent cultivars/lines and landraces of Roxo/Rosinha grain types. Through ANOVA, the SSRs-di showed 36.5 % of with accession differentiation based on Andean and Mesoamerican origin, followed by the BES-SSRs (49.6 %) and SNPs (87.7 %), with a greater differentiation index (Table 3).

Population structure inferred by the Bayesian approach based on SSRs-di, BES-SSRs, SSRs-di/BES-SSRs, and SNPs markers with K values ranging from 2 to 9. Each solid bar represents single genotypes (samples are ordered as described in Supplementary Material 1). Bars with different colors represent genotypes belonging to different subpopulations, and bars of two colors represent genotypes with admixture. The green and red bars (K = 2) represent Andean and Mesoamerican groups, respectively. Exclusively for the SSRs-di, the purple bars (K = 3) represent cultivars/lines from the Mesoamerican gene pool. The pink bars (as of K = 5 for the SSRs and K = 4 for the SNPs) represent the wild species and lines from USA and Guatemala. For SNPs, the brown bars (K = 8 and 9) represent the group comprising cultivars/lines of Carioca grain type and landraces of Carioca and Mulatinho and the purple bars (K = 4 to 9) represent cultivars/lines and landraces of Roxo/Rosinha grain types
Linkage Disequilibrium
Based on segregation of independent loci the estimates of LD (p < 0.001 and r 2 > 0.10, Table 3) differed between the different groups of germplasm. For the 24 SSRs-di genotyped in the whole sample, 17.75 % were in LD, decreasing to 0.72 % in the Andean group and 1.45 % in the Mesoamerican group. Similar estimates were obtained for the 34 BES-SSRs, of which 21.4 % were in LD in the whole sample, followed by 0.53 % in the Andean and 4.28 % in the Mesoamerican group. In regard to the 345 SNPs genotyped in all samples, 84.92 % were in LD, decreasing to 0.25 % in the Andean and 4.64 % in the Mesoamerican group. In a more stringent analysis of SNPs with MAF > 0.10, a total of 337 markers were analyzed, of which 88.45 % were in LD based on all accessions. Within the Andean group, 290 SNPs were removed and, of the 55 remaining, 3.23 % were in LD. In the Mesoamerican group, 204 SNPs with MAF > 0.10 were removed and, of the 141 analyzed, 15.35 % were in LD. A similar reduction in LD between the entire sample Andean and Mesoamerican subsamples was observed by Kwak and Gepts (2009).
To establish a routine and operational analysis aimed at efficient origin-based discrimination of common bean accessions, a set of 16 SSRs was selected (eight SSRs-di and eight BES-SSRs). These selected SSR markers had one allele with frequency above 85 % in one of the gene pools (highlighted in the Supplementary Material 2 and 3). The STRUCTURE analysis carried out with these SSRs (data not shown) revealed a clear pattern of clustering of the accessions by origin.
Distribution of SSRs and SNPs in the Common Bean Genome
The alignment of 58 SSRs and 345 SNPs against the genome of P. vulgaris (Pv), an Andean variety, is represented in Fig. 4. Among the 24 SSRs-di, 87.5 % (21) were aligned against genome sequences of Pv (except for BM201, BM210, and PV163) and were distributed in 8 of the 11 chromosomes of P. vulgaris. The chromosome Pv2 had the largest number of hits (five SSRs). Of the 34 BES-SSRs, 94.1 % (32) aligned with the genome of Pv (except for PvComp4 and PvPenta8), exhibiting wide genomic distribution with two or more BES-SSRs and at most five markers in Pv10. One hundred percent of SNPs aligned to the Andean genome. Genomic coverage was broad, with 23 (Pv5 and Pv9) to 39 SNPs (Pv1 and Pv3) hits per chromosome. A group of 16 SNPs (highlighted in the Supplementary Material 4) aligned with more than 99 % identity to the genome of Pv (E value = 0) with a length of alignment greater than 350 bp.

Distribution of the flanking regions of SSRs and SNPs through alignment in the genome of Phaseolus vulgaris, Andean variety. The figure includes four concentric circles. The inner circle represents the common bean genome (bars in purple), highlighting the haploid chromosomes (Pv1-11). The histograms represent, from the inside out, the number of BLAST hits encountered by aligning SNPs (red), SSRs-di (green), and BES-SSRs (blue) with the common bean genome. Each bar of the histogram comprises a genomic region of 1 Mb and each thin gray line is equivalent to 1 hit
Discussion
The development of molecular tools for monitoring genetic variability in plant breeding programs greatly increases the efficiency of utilization of available genetic resources. In the face of the diverse classes of currently available markers, knowledge of their genetic informative potential is fundamental to exploit the germplasm diversity. Despite the growing number of SNPs available for P. vulgaris, few studies have examined the potential of this tool for analyzing genetic diversity and structure in this species (Cortés et al. 2011; Blair et al. 2013a). Previously, the most concrete application has been in the development of genetic maps with broad genomic coverage (Galeano et al. 2012). In contrast, the use of SSRs to investigate the genetic diversity and linkage mapping, among other aspects, has been widely described in the literature (Kwak and Gepts 2009; Burle et al. 2010; Yang et al. 2011). Despite the reduced genetic diversity per locus (Cortés et al. 2011; Blair et al. 2013a), the large set of available SNP markers, coupled with high-throughput genotyping systems, makes SNPs attractive for operational application in breeding programs of the common bean (Mammadov et al. 2012). A growing number of studies comparing the performance of SSRs and SNPs are currently being conducted (DeFaveri et al. 2013). For the common bean, the present work is the first to assess the genetic diversity and structure of P. vulgaris using SSR and SNP markers through a comparative study, proposing as a final outcome a set of the most informative markers to be used in an integrative analysis.
SSRs stood out in this study due to their multiallelism and high estimates of H E (SSRs-di Ā = 9.92 and BES-SSRs Ā = 5.59), especially for the SSRs-di (H E = 0.725), which allowed individual discrimination of each of the 88 genotypes studied. For the SNPs, there was reduced H E (Ā = 2, H E = 0.390) and a significantly greater proportion of private alleles among the inter-gene pool genotypes, favoring more precise differentiation by origin. With regard to the potential for sampling allelic diversity within the gene pool, the SSRs-di were more informative and, in two wild accessions alone, 15 private alleles were identified in addition to the 14 identified in the 22 landraces of Mesoamerican origin. Greater estimates of gene diversity in wild, as opposed to domesticated, germplasm (Table 1) have been reported by other authors in autogamous plants (Kwak and Gepts 2009; Li et al. 2010). The greater gene diversity found in the wild accessions is an additional piece of evidence supporting the need for conservation of this biodiversity. Throughout their existence, these accessions developed mechanisms to adapt to biotic and abiotic stress conditions, features which may be amply utilized in research programs.
In this study, the H E of the Mesoamerican gene pool was slightly greater than that of the Andean, in contrast with the results obtained by Blair et al. (2006) using SSRs, and Cortés et al. (2011) and Blair et al. (2013a) using SNPs. It is true that these greater estimates may be a consequence of the greater number (3.2 X) of Mesoamerican accessions characterized, mainly for SNP markers (36 % polymorphic SNP in the Andean group vs. 96 % in the Mesoamerican) due to the biallelic nature of this marker. For the genetic diversity estimates obtained with SSR, considering only the landraces, close values were obtained separately for the gene pools (n = 11; He = 0.318 in the Andean and n = 22; He = 0.384 in the Mesoamerican), in accordance with Burle et al. (2010) that also identified close values between the Mesoamerican (0.33) and the Andean groups (0.30) composed of 221 and 58 accessions of Brazilian landraces, respectively. However, additional genotyping of a large set of samples would provide more realistic information about the usefulness of these SNPs to detect polymorphism in the Andean gene pool. However, regardless of the sample size or the type of marker used, various studies have shown greater H E in the Mesoamerican germplasm (Kwak and Gepts 2009; Mamidi et al. 2011; Bitocchi et al. 2013). According to Mamidi et al. (2013), the two wild gene pools originated from a common ancestor approximately 111,000 years ago, following separate evolutionary paths and subjected to distinct bottleneck effects prior to domestication. Later, during the domestication process, the Andean population was subjected to a bottleneck equivalent to 26 % of the effective size of its ancestral population, while in the Mesoamerican population this bottleneck was 46 %. Together, these factors contributed to a reduction of genetic diversity and an increase in the intra-gene pool population structure. As a consequence of the higher effective size in the Mesoamerican gene pool arising from the greater number of migrants in its initial gene constitution, a greater nucleotide and haplotype diversity was favored (Mamidi et al. 2011; 2013). More recently, a study conducted by Desiderio et al. (2013) using chloroplast SSRs (cpSSRs) also showed greater diversity within the Mesoamerican group, corroborating previous scientific findings and the present study.
The SNP markers, identified between the Andean and Mesoamerican genotypes, provided a greater number of polymorphic markers for purposes of inter- (78.2 %) and intra-gene pool (17.7 %) mapping analysis in P. vulgaris than previous technologies. Similar estimates were obtained by Cortés et al. (2011), who detected a level of inter- and intra-gene pool polymorphism of 71.7 and 15.4 %, respectively, based on analysis of 94 SNP markers. Although the microsatellites are significantly more informative considering individual loci, lower estimates of polymorphism for crosses involving lines belonging to the same gene pool, with indices of approximately 11 %, have been reported using these markers in QTL studies (Garcia et al. 2012; Blair et al. 2011). Considering crosses involving common bean parents from distinct centers of origin, SSRs were less polymorphic when compared to the current SNPs (78.2 %), with polymorphism estimates ranging from 7.7 % (Galeano et al. 2011) to 24 % (Garcia et al. 2011). From a practical perspective, obtaining a large number of SNPs from genotypes that compose the genetic base of interest to the end of maximizing the polymorphism potential of the marker proved an adequate strategy. Thus, it is possible to focus on the development of SNP markers that are genetically informative for populations developed for specific breeding programs that generally belong to the same gene pool (Beebe et al. 2013), but have contrasting phenotypes. Analysis of SNPs through eight biparental combinations in this study identified a significant proportion (61 %) of common polymorphic markers in the inter-gene pool. The availability of these SNPs will facilitate the integration and comparison of genetic maps with greater resolution, which is required for QTL identification.
In this study, SNPs proved the most adequate to differentiate the Andean and Mesoamerican germplasm (F ST = 0.759) compared to SSRs (SSRs-di F ST = 0.341 and BES-SSRs F ST = 0.410). This outcome was a consequence of ascertainment biases existing in the panel of SNPs developed to explore the polymorphism between the universal Andean (Jalo EEP 558) and Mesoamerican (BAT 93) genotypes (Hyten et al. 2010). This results in a tendency of clustering the sampled accessions following the same pattern of differentiation of the target genotypes used to derive the SNPs, as reported for barley (Moragues et al. 2010), rice (Thomson et al. 2012), and the common bean (Blair et al. 2013a). According to Nagasaki et al. (2010), depending on the intent of the study and the origin of sample germplasm, different sets of SNPs should be used. However, the use of common SNP groups is helpful because it allows the standardization of results and consolidation of an integrated database. Thus, the strategy of increasing the genome representation of SNPs through the search for polymorphism in fragments sampled from a great number of races and types of inter- and intra-gene pool grains represents the most effective way of exploiting the genetic variability of P. vulgaris. With regard to the SSRs-di, the greater power of structuring the Mesoamerican germplasm in cultivars/lines and landraces is a consequence of systematic selection of useful markers for accessions primarily from the Mesoamerican gene pool (Grisi et al. 2007; Garcia et al. 2011), which meets the requirements of genetic breeding programs. Similarly, the identification of a larger group of SSRs based on repetitions ≥ trinucleotides, with the same informative/discriminatory potential of the SSRs-di, depends exclusively on an additional effort in selection and characterization of new markers to be incorporated in the genotyping systems.
The structuring of common bean genotypes by origin was observed using SNPs and SSRs, with the greater inter-gene pool variation being demonstrated by SNPs via PCA (91.42 %) and ANOVA (87.7 %). The most likely subdivision was K = 2 according to STRUCTURE, in accordance with previous studies (Kwak and Gepts 2009; Burle et al. 2010; Cardoso et al. 2014; Cortés et al. 2011; Blair et al. 2013a, b). SSRs were more effective at discerning genetic diversity between genetically related germplasm (cultivars/lines and landraces, K = 3 to 7), while SNPs were better at distinguishing the association of subpopulations in Brazilian germplasm through fine-scale structuring (K = 9). The polymorphisms of SSRs and SNPs were generated under different evolutionary mechanisms, exploiting both the fast (∼10−4) and slow (∼10−8−10−9) mutation rates of SSRs and SNPs (Brumfield et al. 2003), respectively, providing unique views of the structure of a given population. In this context, a targeted selection of historically recent SNPs of contrasting allelic frequency among founder lines that contain the diversity encapsulating agronomic traits will be useful for genomic structure determination. In the present study, the BRSMG Talismã cultivar, although considered to be of Mesoamerican origin mainly due to the small-seed size, was predominantly of Andean genetic composition based on our STRUCTURE analyses. The dubious classification of this cultivar reflects its process of development, the recurrent selection, with parental recombination between gene pools. The SNP panel was useful for grouping together cultivars with Carioca grain type and Mulatinho landraces (both Mesoamericans, identified at K = 9), reflecting the process of breeding program crosses. The bicolor seed trait (beige with brown stripes) is typical of the Carioca grain type, which has been commercially cultivated in Brazil since the 1970s. Since then, crosses between Carioca and Mulatinho grain types became frequent, resulting in important cultivars, such as Pérola, which is the most cultivated material in Brazil. In contrast, the genealogies of cultivars with the Mulatinho grain type had little or no overlap with Carioca, which explains their position outside the subgroup described above. The discovery of sub-races in the Brazilian Mesoamerican gene pool by SSRs, denominated Carioca-Jalinho-Mulatinho (M3) and Rosinha-Roxinho (M4) was described by Blair et al. (2013b).
The high level of LD detected for the whole set of samples (p < 0.001 and r 2 > 0.10), ranging from 17.75 to 84.92 %, indicated that the structured gene pool was most likely the main cause of the high LD in the entire sampling. These findings were previously reported by Rossi et al. (2009) and Burle et al. (2010), who estimated an LD of 56 to 80 %, respectively, and also analyzing accessions of both gene pools. Nevertheless, the LDs individually estimated in the Andean and Mesoamerican subgroups were lower and variable as a result of the genetic background of the domesticated accessions. These results indicate that these estimates should not be extrapolated to other studies. A greater LD in the Mesoamerican (23 %, p < 0.01) compared to the Andean group (8 %) was found by Burle et al. (2010) for Brazilian landraces, while Rossi et al. (2009) observed an opposite trend. Breeding efforts in Brazil are predominantly focused on accessions of Mesoamerican origin, favoring greater LD due to a strong founder effect and a domestication process in a secondary center of diversity. This phenomenon is followed by a bottleneck effect derived from the successive selection processes imposed by man, reducing the effective population size. A recent study conducted by Cardoso et al. (2013) found an absence of structuring (F ST = 0.01) among Brazilian cultivars (mainly Mesoamerican) released in the last 26 years (F ST = 0.01) that would be expected to increase LD throughout the entire genome. As the LD is variable over the genome, a general value is not representative and their extensions should be investigated. While genomic regions with high extension of linkage will require a smaller number of SNPs for haplotypic block determination, favoring the association studies and assisted selection, the mapping resolution will tend to be lower, hampering precise QTL and recombinant identifications (Würschum et al. 2011). More detailed information will allow for inferences of LD dynamics in the genome, with implications for the planning of crosses between genitors of common bean breeding programs.
Final Considerations
SSRs have been the markers of choice for genetic studies of the common bean for many years. They are very effective for several applications that require the detection of multiallelic loci and high polymorphism levels. SNP markers genotyped by a high-throughput system have been recently developed but have not yet been routinely incorporated in breeding programs of the common bean. In spite of their lower genetic information content, a large number of SNPs can be simultaneously genotyped, overcoming that limitation. Comparing the ability of individual discrimination of the 88 the common bean genotypes, it was observed that over three times more SNPs were necessary to achieve the same probability of identity obtained with SSRs. Recent efforts in P. vulgaris genome sequencing (Schmutz et al. 2014), in addition to approaches of genotyping-by-sequencing (GbS, Maughan et al. 2010; Elshire et al. 2011) have enabled the development of a large set of SNPs. The SNP detection methods available to date are fast and cost effective with data point per sample around three times less expensive compared to the SSRs. In this study, a SNP panel adequate for inter-gene pool differentiation, linkage and structuring analysis, coupled to accurate calling and high-throughput genotyping was developed and made available to the common bean scientific community. The molecular markers (SSRs and SNPs) made available in this study reached an excellent profile of amplification useful to estimate genetic parameter in common bean germplasm, being encouraged to get tested in other groups of germplasm, especially for programs with predominance of Mesoamerican gene pool. To maintain the integration of genomics as an attractive alternative for the common bean breeding programs, the most appropriate and accessible methods for accurately addressing breeding questions must be utilized.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215(3):403–410
Alzate-Marin AL, Cervigni GDL, Moreira MA, Barros EG (2005) Seleção assistida por marcadores moleculares visando ao desenvolvimento de plantas resistentes a doenças, com ênfase em feijoeiro e soja. Fitopatol Bras 30(4):333–342
Beebe SE, Rao IM, Blair MW, Acosta-Gallegos JA (2013) Phenotyping common beans for adaptation to drought. Front Physiol 4:35. doi:10.3389/fphys.2013.00035
Bitocchi E, Bellucci E, Giardini A, Rau D, Rodriguez M, Biagetti E, Santilocchi R, Spagnoletti Zeuli P, Gioia T, Logozzo G, Attene G, Nanni L, Papa R (2013) Molecular analysis of the parallel domestication of the common bean (Phaseolus vulgaris) in Mesoamerica and the Andes. New Phytol 197(1):300–313. doi:10.1111/j.1469-8137.2012.04377.x
Blair MW, Giraldo MC, Buendía HF, Tovar E, Duque MC, Beebe SE (2006) Microsatellite marker diversity in common bean (Phaseolus vulgaris L.). Theor Appl Genet 113:100–109. doi:10.1007/s00122-006-0276-4
Blair MW, Astudillo C, Rengifo J, Beebe SE, Graham R (2011b) QTL analyses for seed iron and zinc concentrations in an intra-genepool population of Andean common beans (Phaseolus vulgaris L.). Theor Appl Genet 122:511–521. doi:10.1007/s00122-010-1465-8
Blair MW, Soler A, Cortés AJ (2012) Diversification and population structure in common beans (Phaseolus vulgaris L.). PLoS One 7(11):e49488. doi:10.1371/journal.pone.0049488
Blair MW, Cortés AJ, Penmetsa RV, Farmer A, Carrasquilla-Garcia N, Cook DR (2013a) A high-throughput SNP marker system for parental polymorphism screening, and diversity analysis in common bean (Phaseolus vulgaris L.). Theor Appl Genet 126:535–548. doi:10.1007/s00122-012-1999-z
Blair MW, Brondani RPV, Díaz LM, Del Pelosob MJ (2013b) Diversity and population structure of common bean from Brazil. Crop Sci 53:1983–1993. doi:10.2135/cropsci2012.09.0516
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL : software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635. doi:10.1093/bioinformatics/btm308
Broughton WJ, Hernández G, Blair M, Beebe S, Gepts P, Vanderleyden J (2003) Beans (Phaseolus spp.)—model food legumes. Plant Soil 252:55–128
Brown TA (2002) Genomes. Wiley-Liss, Oxford
Brumfield RT, Beerli P, Nickerson DA, Edwards SV (2003) The utility of single nucleotide polymorphisms in inferences of population history. Trends Ecol Evol 18(5):249–256. doi:10.1016/S0169-5347(03)00018-1
Burle ML, Fonseca JR, Kami JA, Gepts P (2010) Microsatellite diversity and genetic structure among common bean (Phaseolus vulgaris L.) landraces in Brazil, a secondary center of diversity. Theor Appl Genet 121:801–813. doi:10.1007/s00122-010-1350-5
Buso GSC, Amaral ZPS, Brondani RPV, Ferreira ME (2006) Microsatellite markers for the common bean Phaseolus vulgaris. Mol Ecol Notes 6:252–254. doi:10.1111/j.1471-8286.2006.01210.x
Cardoso PCB, Veiga MM, Menezes IPP, Valdisser PA, Borba TC, Melo LC, Del Peloso MJ, Brondani C, Vianello RP (2013) Molecular characterization of high performance inbred lines of Brazilian common beans. Genet Mol Res 12(4):5467–5484. doi:10.4238/2013.February.6.4
Cardoso PC, Brondani C, Menezes IP, Valdisser PA, Borba TC, Del Peloso MJ, Vianello RP (2014) Discrimination of common bean cultivars using multiplexed microsatellite markers. Genet Mol Res 13(1):1964–1978. doi:10.4238/2014.March.24.1
Chang S, Hartman GL, Singh RJ, Lambert KN, Hobbs HA, Domier LL (2013) Identification of high-quality single-nucleotide polymorphisms in Glycine latifolia using a heterologous reference genome sequence. Theor Appl Genet 126:1627–1638. doi:10.1007/s00122-013-2079-8
Ching A, Caldwell KS, Jung M, Dolan M, Smith OS, Tingey S, Morgante M, Rafalski AJ (2002) SNP frequency, haplotype structure and linkage disequilibrium in elite maize inbred lines. BMC Genet 3:19. doi:10.1186/1471-2156-3-19
Cortés AJ, Chavarro MC, Blair MW (2011) SNP marker diversity in common bean (Phaseolus vulgaris L.). Theor Appl Genet 123:827–845. doi:10.1007/s00122-011-1630-8
DeFaveri J, Viitaniemi H, Leder E, Merilä J (2013) Characterizing genic and nongenic molecular markers: comparison of microsatellites and SNPs. Mol Ecol Resour 13(3):377–392. doi:10.1111/1755-0998.12071
Desiderio F, Bitocchi E, Bellucci E, Rau D, Rodriguez M, Attene G, Papa R, Nanni L (2013) Chloroplast microsatellite diversity in Phaseolus vulgaris. Front Plant Sci 22(3):312. doi:10.3389/fpls.2012.00312
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4(2):359–361. doi:10.1007/s12686-011-9548-7
Ellwood SR, Phan HT, Jordan M, Hane J, Torres AM, Avila CM, Cruz-Izquierdo S, Oliver RP (2008) Construction of a comparative genetic map in faba bean (Vicia faba L.); conservation of genome structure with Lens culinaris. BMC Genomics 9:380. doi:10.1186/1471-2164-9-380
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6(5):e19379. doi:10.1371/journal.pone.0019379
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. doi:10.1111/j.1365-294X.2005.02553.x
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. doi:10.1111/j.1755-0998.2010.02847.x
Freyre R, Skroch PW, Geffroy V, Adam-Blondon AF et al (1998) Towards an integrated linkage map of common bean. 4. Development of a core linkage map and alignment of RFLP maps. Theor Appl Genet 97:847–856
Gaitán-Solís E, Duque MC, Edwards KJ, Tohme J (2002) Microsatellite repeats in common bean (Phaseolus vulgaris): Isolation, characterization and cross-species amplification in Phaseolus ssp. Crop Sci 42:1228–1236
Galeano CH, Fernandez AC, Franco-Herrera N, Cichy KA, McClean PE, Vanderleyden J, Blair MW (2011) Saturation of an intra-gene pool linkage map: towards a unified consensus linkage map for fine mapping and synteny analysis in common bean. PLoS One 6:e28135. doi:10.1371/journal.pone.0028135
Galeano CH, Cortés AJ, Fernández AC, Soler Á, Franco-Herrera N, Makunde G, Vanderleyden J, Blair MW (2012) Gene-based single nucleotide polymorphism markers for genetic and association mapping in common bean. BMC Genet 13:48. doi:10.1186/1471-2156-13-48
Garcia RAV, Rangel PN, Brondani C, Martins WS, Melo LC, Carneiro MS, Borba TC, Brondani RP (2011) The characterization of a new set of EST-derived simple sequence repeat (SSR) markers as a resource for the genetic analysis of Phaseolus vulgaris. BMC Genet 12:41. doi:10.1186/1471-2156-12-41
Garcia RAV, Rangel PN, Bassinello PZ, Brondani C, Melo LC, Sibov ST, Vianello-Brondani RP (2012) QTL mapping for the cooking time of common beans. Euphytica 186:779–792. doi:10.1007/s10681-011-0587-7
Grattapaglia D, Sederoff R (1994) Genetic linkage maps of Eucalyptus grandis and E. urophylla using a pseudo-testcross strategy and RAPD markers. Genetics 137(4):1121–1137
Grattapaglia D, Silva-Junior OB, Kirst M, de Lima BM, Faria DA, Pappas GJ Jr (2011) High-throughput SNP genotyping in the highly heterozygous genome of Eucalyptus: assay success, polymorphism and transferability across species. BMC Plant Biol 11:65. doi:10.1186/1471-2229-11-65
Grisi MCM, Blair MW, Gepts P, Brondani C (2007) Genetic mapping of a new set of microsatellite markers in a reference common bean (Phaseolus vulgaris) population BAT 93 x Jalo EEP 558. Genet Mol Res 6(3):691–706
Hammer Ø, Harper DAT, Ryan PD (2001) Past: paleontological statistics software package for education and data analysis. Palaeontol Electron 4(1):1–9
Hanai LR, De Campos T, Eduardo L et al (2007) Development, characterization, and comparative analysis of polymorphism at common bean SSR loci isolated from genic and genomic sources. Genome 50:266–277. doi:10.1139/G07-007
Hougaard BK, Madsen LH, Sandal N, Moretzsohn MD, Fredslund J, Schauser L, Nielsen AM, Rohde T, Sato S, Tabata S, Bertioli DJ, Stougaard J (2008) Legume anchor markers link syntenic regions between Phaseolus vulgaris, Lotus japonicus, Medicago truncatula and Arachis. Genetics 179(4):2299–2312. doi:10.1534/genetics.108.090084
Hyten DL, Choi I-Y, Song Q, Shoemaker RC, Nelson RL, Costa JM, Specht JE, Cregan PB (2007) Highly variable patterns of linkage disequilibrium in multiple soybean populations. Genetics 175(4):1937–1944. doi:10.1534/genetics.106.069740
Hyten DL, Song Q, Fickus EW, Quigley CV, Lim JS, Choi IY, Hwang EY, Pastor-Corrales M, Cregan PB (2010) High-throughput SNP discovery and assay development in common bean. BMC Genomics 11:475. doi:10.1186/1471-2164-11-475
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806. doi:10.1093/bioinformatics/btm233
Kaczorowski KA, Kim KS, Diers BW, Hudson ME (2008) Microarray-based genetic mapping using soybean near-isogenic lines and generation of SNP markers in the Rag1 aphid resistance interval. Plant Genome 1:89–98
Kim S, Misra A (2007) SNP genotyping: technologies and biomedical applications. Annu Rev Biomed Eng 9:289–320
Krzywinski M, Schein J, Birol İ, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19:1639–1645. doi:10.1101/gr.092759.109
Kwak M, Gepts P (2009) Structure of genetic diversity in the two major gene pools of common bean (Phaseolus vulgaris L., Fabaceae). Theor Appl Genet 118:979–992. doi:10.1007/s00122-008-0955-4
Li Y-H, Li W, Zhang C, Yang L, Chang RZ, Gaut BS, Qiu LJ (2010) Genetic diversity in domesticated soybean (Glycine max) and its wild progenitor (Glycine soja) for simple sequence repeat and single-nucleotide polymorphism loci. New Phytol 188(1):242–253. doi:10.1111/j.1469-8137.2010.03344.x
Loridon K, Burgarella C, Chantret N, Martins F, Gouzy J, Prospéri JM, Ronfort J (2012) Single-nucleotide polymorphism discovery and diversity in the model legume Medicago truncatula. Mol Ecol Resour 13:84–95. doi:10.1111/1755-0998.12021
Mamidi S, Rossi M, Annam D, Moghaddam S, Lee R, Papa R, McClean P (2011) Investigation of the domestication of common bean (Phaseolus vulgaris) using multilocus sequence data. Funct Plant Biol 38:953–967
Mamidi S, Rossi M, Moghaddam SM, Annam D, Lee R, Papa R, McClean PE (2013) Demographic factors shaped diversity in the two gene pools of wild common bean Phaseolus vulgaris L. Heredity 110(3):267–276. doi:10.1038/hdy.2012.82
Mammadov J, Aggarwal R, Buyyarapu R, Kumpatla S (2012) SNP markers and their impact on plant breeding. Int J Plant Genomics 2012:728398. doi:10.1155/2012/728398
Maughan PJ, Yourstone SM, Byers RL, Smith SM, Udall JA (2010) Single-nucleotide polymorphism genotyping in mapping populations via genomic reduction and next-generation sequencing: proof of concept. Plant Genome J 3:166. doi:10.3835/plantgenome2010.07.0016
Moragues M, Comadran J, Waugh R, Milne I, Flavell AJ, Russell JR (2010) Effects of ascertainment bias and marker number on estimations of barley diversity from high-throughput SNP genotype data. Theor Appl Genet 120(8):1525–1534. doi:10.1007/s00122-010-1273-1
Müller BSF, Sakamoto T, Menezes IPP, Prado GS, Martins WS, Brondani C, Barros EG, Vianello RP (2014) Analysis of BAC-end sequences in common bean (Phaseolus vulgaris L.) towards the development and characterization of long motifs SSRs. Plant Mol Biol 86(4–5):455–470. doi:10.1007/s11103-014-0240-7
Nagasaki H, Ebana K, Shibaya T, Yonemaru J, Yano M (2010) Core single-nucleotide polymorphisms—a tool for genetic analysis of the Japanese rice population. Breeding Sci 60(5):648–655
Oliveira LK, Melo LC, Brondani C, Peloso MJD, Brondani RPV (2008) Backcross assisted by microsatellite markers in common bean. Genet Mol Res 7(4):1000–1010
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research–an update. Bioinformatics 28:2537–2539. doi:10.1093/bioinformatics/bts460
Perrier X, Jacquemoud-Collet JP (2006) DARwin software. Available at. http://darwin.cirad.fr/. Accessed on Feb 23, 2013
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rosenberg NA (2004) Distruct: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138. doi:10.1046/j.1471-8286.2003.00566.x
Rossi M, Bitocchi E, Bellucci E, Nanni L, Rau D, Attene G, Papa R (2009) Linkage disequilibrium and population structure in wild and domesticated populations of Phaseolus vulgaris L. Evol Appl 2:504–522. doi:10.1111/j.1752-4571.2009.00082.x
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Bio Evol 4:406–425
Saxena RK, Penmetsa RV, Upadhyaya HD, Kumar A, Carrasquilla-Garcia N, Schlueter JA, Farmer A, Whaley AM, Sarma BK, May GD, Cook DR, Varshney RK (2012) Large-scale development of cost-effective single-nucleotide polymorphism marker assays for genetic mapping in pigeonpea and comparative mapping in legumes. DNA Res 19:449–461. doi:10.1093/dnares/dss025
Schmutz J, Cannon SB, Schlueter J et al (2010) Genome sequence of the palaeopolyploid soybean. Nature 463(7278):178–183. doi:10.1038/nature08670
Schmutz J, McClean PE, Mamidi S et al (2014) A reference genome for common bean and genome-wide analysis of dual domestications. Nat Genet 46(7):707–713. doi:10.1038/ng.3008
Sefc KM, Steinkellner H, Wagner HW, Glössl J, Regner F (1997) Application of microsatellite markers to parentage studies in grapevine. Vitis 36(4):179–183
Silva OF, Wander AE (2013) O feijão-comum no Brasil passado, presente e futuro. Santo Antônio de Goiás, Embrapa Arroz e Feijão. Documentos 287, ISSN 1678–9644; 287, 63 p
Song Q, Hyten DL, Jia G, Quigley CV, Fickus EW, Nelson RL, Cregan PB (2013) Development and evaluation of SoySNP50K, a high-density genotyping array for soybean. PLoS One 8:e54985. doi:10.1371/journal.pone.0054985
Souza TLPO, Barros EG, Bellato CM, Hwang EY, Cregan PB, Pastor-Corrales MA (2012) Single nucleotide polymorphism discovery in common bean. Mol Breeding 30(1):419–428. doi:10.1007/s11032-011-9632-4
Thomson MJ, Zhao K, Wright M et al (2012) High-throughput single nucleotide polymorphism genotyping for breeding applications in rice using the BeadXpress platform. Mol Breeding 29:875–886. doi:10.1007/s11032-011-9663-x
Van K, Hwang EY, Kim MY, Park HJ, Lee SH, Cregan PB (2005) Discovery of SNPs in soybean genotypes frequently used as the parents of mapping populations in the United States and Korea. J Hered 96:529–535
Vieira RF, Mendes IC, Reis-Junior FB, Hungria M (2010) Symbiotic nitrogen fixation in tropical food grain legumes: Current Status. In: Khan MS, Musarrat J, Zaidi A (eds) Microbes for legume improvement, 1st edn. Springer, Vienna, pp 427–472. doi:10.1007/978-3-211-99753-6_18
Wortmann CS, Kirkby RA, Elude CA, Allen DJ (1998) Atlas of common bean (Phaseolus vulgaris L.) production in Africa. CIAT. http://ciat-library.ciat.cgiar.org/Articulos_CIAT/Atlas_common_bean_Africa.pdf. Accessed 10 August 2014
Würschum T, Maurer HP, Kraft T, Janssen G, Nilsson C, Reif JC (2011) Genome-wide association mapping of agronomic traits in sugar beet. Theor Appl Genet 123:1121–1131. doi:10.1007/s00122-011-1653-1
Yang X, Xu Y, Shah T, Li H, Han Z, Li J, Yan J (2011) Comparison of SSRs and SNPs in assessment of genetic relatedness in maize. Genetica 139:1045–1054. doi:10.1007/s10709-011-9606-9
Yu K, Park SJ, Poysa V, Gepts P (2000) Integration of simple sequence repeat (SSR) markers into a molecular linkage map of common bean (Phaseolus vulgaris L.). J Hered 91(6):429–434
Yu H, Xie W, Wang J, Xing Y, Xu C, Li X, Xiao J, Zhang Q (2011) Gains in QTL detection using an ultra-high density SNP map based on population sequencing relative to traditional RFLP/SSR markers. PLoS One 6:e17595. doi:10.1371/journal.pone.0017595
Acknowledgments
We would like to thank the National Council for Scientific and Technological Development (CNPq) for the grants to CB, EGB and RPV; the Coordination for the Improvement of Higher Education Personnel/Ministry of Education (CAPES/MEC) for the grants to BSFM, the Minas Gerais State Research Foundation (FAPEMIG) for the grant to TS; and the Brazilian Agricultural Research Corporation (EMBRAPA) for financial support for this research.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Müller, B.S.F., Pappas, G.J., Valdisser, P.A.M.R. et al. An Operational SNP Panel Integrated to SSR Marker for the Assessment of Genetic Diversity and Population Structure of the Common Bean. Plant Mol Biol Rep 33, 1697–1711 (2015). https://doi.org/10.1007/s11105-015-0866-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-015-0866-x