
Abstract
The capability of molecular markers to provide information of genetic structure is influenced by their number and the way they are chosen. This study evaluates the effects of single nucleotide polymorphism (SNP) number and selection strategy on estimates of germplasm diversity and population structure for different types of barley germplasm, namely cultivar and landrace. One hundred and sixty-nine barley landraces from Syria and Jordan and 171 European barley cultivars were genotyped with 1536 SNPs. Different subsets of 384 and 96 SNPs were selected from the 1536 set, based on their ability to detect diversity in landraces or cultivated barley in addition to corresponding randomly chosen subsets. All SNP sets except the landrace-optimised subsets underestimated the diversity present in the landrace germplasm, and all subsets of SNP gave similar estimates for cultivar germplasm. All marker subsets gave qualitatively similar estimates of the population structure in both germplasm sets, but the 96 SNP sets showed much lower data resolution values than the larger SNP sets. From these data we deduce that pre-selecting markers for their diversity in a germplasm set is very worthwhile in terms of the quality of data obtained. Second, we suggest that a properly chosen 384 SNP subset gives a good combination of power and economy for germplasm characterization, whereas the rather modest gain from using 1536 SNPs does not justify the increased cost and 96 markers give unacceptably low performance. Lastly, we propose a specific 384 SNP subset as a standard genotyping tool for middle-eastern landrace barley.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The recent advances in genomic technologies and the various sequencing projects have led to a shift from anonymous marker systems to a more direct analysis of sequence-based polymorphism (Rafalski 2002). Single nucleotide polymorphisms have been used extensively in human and animal genetics (Abasht and Lamont 2007; Saxena et al. 2007; Zeggini et al. 2007) and are beginning to be applied in plants (Rostoks et al. 2006). SNPs, like other markers, require an initial screening step to identify a subset of variable nucleotides. Typically, the SNP discovery process involves data mining from expressed sequence tag (EST) collections and/or amplicon sequencing from a limited number of individuals. The utility of the SNP sets obtained is affected by the parameters of this discovery protocol. SNPs are generally identified in a discovery panel, which consists of a small sample of individuals from a population or a particular region. As this panel is only a subset of individuals, only a fraction of polymorphisms will be discovered and when these SNPs are then genotyped on a larger sample of individuals an ‘ascertainment bias’ is introduced (Nielsen 2000). Because the discovery panel is small, the probability that a SNP identified in this panel is a function of the allele frequency, which implies that rare SNPs will go undiscovered compared to more frequent common SNPs. This bias may compromise inferences drawn from diversity studies using these SNPs. As a results any statistical measures that rely on allele frequency including nucleotide diversity, population genetics parameters and linkage disequilibrium, will be affected. For example, because rare SNPs are undiscovered, the average diversity of the sites that are polymorphic is high, and because these same SNPs are missing, the average diversity across all sites is underestimated. Several studies have demonstrated that the SNP discovery process introduces bias into estimates of various population genetic parameters (Nielsen 2000; Schlotterer and Harr 2002; Akey et al. 2003; Rosenblum and Novembre 2007; Storz and Kelly 2008).
A recent international effort in barley has led to the development of a 1536-plex SNP set (Kota et al. 2007; Rostoks et al. 2005, 2006) that uses Illumina GoldenGate BeadArray technology (Fan et al. 2006). In this case, the SNP discovery was based on polymorphisms of eight ‘parental plants’ (Rostoks et al. 2005) and nine cultivars (Rostoks et al. 2006) leading to 3072 SNP in two pilot Oligo Pool Arrays (OPAs), which were distilled into one Production Barley 1536 OPA (BOPA1) after performance checks on 196 genotypes. The germplasm used for the original SNP selection was biased in favour of spring cultivar types from USA and Western Europe, mainly parents of well-characterized doubled haploid mapping populations (Rostoks et al. 2005) and all the genotypes used in the second step were modern cultivars from the US, Europe and Manchurian barleys (Rostoks et al. 2006).
These SNPs have provided extensive genome coverage and have dramatically progressed our understanding of the distribution of genetic diversity within cultivated barley. As noted by Hayes and Szucs 2006, the availability of these 1536 SNPs and the high-throughput genotyping approach are one of the most important advances in barley and will allow the identification of regions harbouring genes that determine important traits in barley. However, this picture and any inferences of diversity will be limited by the quality of the underlying data. Particularly problematic is the use of these SNPs to examine diversity in other populations. In barley we are fortunate to have extensive germplasm collections, namely wild progenitor, locally cultivated landrace collections grown throughout the marginal regions of the Fertile Crescent and extensive cultivated accessions. Understanding the genetic diversity within these germplasm collections, particularly the landrace collections which grow and yield under extreme conditions of temperature and water availability, will be important in the future breeding programmes as drought and extreme temperatures are predicted.
Given the efforts by the barley community to develop this set of SNPs, questions as to the general applicability of the BOPA1 genotyping platform have arisen. For example, how accurate will it be for assessing genetic diversity in material from outside the elite cultivated genepool? The diversity of current elite barley cultivars is both lower and most likely different from that of landraces and wild barleys (Jana and Pietrzak 1988; Matus and Hayes 2002; Russell et al. 2003). Russell et al. (2003) found that a collection of geographically localised Syrian and Jordan landraces had similar levels of SSR diversity to European spring barleys but the spectrum of observed alleles was almost completely different between the two sets of germplasm. Furthermore, many other population studies have also revealed that wild barley is a rich source of diversity (e.g. Brown et al. 1978; Nevo et al. 1986; Jana and Pietrzak 1988). A second question is how many SNPs should one use for a particular application? While it is tempting to apply every available marker to a problem, this could be an expensive mistake if far fewer markers produce the desired outcome in terms of accuracy. In this study, we address both questions and propose a simple method to derive guidelines on optimal choices in the SNP selection used in diversity estimations in different subsets of the barley genepool.
Materials and methods
Plant materials
One hundred and sixty-nine individuals were selected from a collection of 480 Syrian and Jordan landraces that, on the basis of SSR data, represent most of the genetic diversity present in this collection (Russell et al. 2003). 171 H. vulgare cultivars were randomly selected from a set of 500 included in the UK recommended list. Seeds from single plants, ‘bagged’ to prevent cross pollination, were used in this study in order to minimise heterozygosity in the germplasm.
DNA extraction and SNP genotyping
Leaves were collected from single greenhouse grown plants and stored at −80°C prior to DNA extraction using Tepnel Nucleoplex plant DNA extraction kits (Tepnel Life Sciences PLC, Manchester, UK). Two microgram of DNA was used for SNP genotyping with BOPA1 at the Southern California Genotyping Consortium facility at the University of California, Los Angeles, USA. Allele calls were performed with the clustering algorithm GenTrain available in BeadStudio 2.0 (Illumina Inc., http://www.illumina.com). Each SNP was manually checked in BeadStudio because the algorithm was developed for outbreeders and searches for three clusters (two homozygotes and one heterozygote), whereas barley is predominantly an inbreeder with the potential for outbreeding minimised in this study (see above). The 1536 SNPs were checked for quality in both germplasm sets. SNPs with more than 25% missing values or ambiguous map locations on a consensus SNP map (unpublished data) were removed, leaving a final number of 1421 and 1473 SNP loci in the landrace and cultivated germplasm sets, respectively. These SNP sets are referred to as “1536 SNP set” throughout this work.
Design of SNP subsets and data analysis
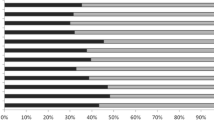
Two subsets of 384 SNPs were selected from the 1536 set. The first was optimised for performance in landrace germplasm (L-SNP-384) and the second for cultivated germplasm (C-SNP-384). Criteria for inclusion in these subsets included: (1) less than 5% missing; (2) minimum allele frequency (MAF) of 0.20 or above and (3) evenly spaced (maximum 10 cM intervals) across the seven chromosomes. The latter criterion could only be implemented for the C-SNP-384 subset because only 386 SNPs were left in for incorporation into L-SNP-384 after filtering by the first two criteria. A third set of 384 SNPs was taken at random (R-SNP-384) from the 1536 set. Finally, 96 SNP subsets (C-SNP-96, L-SNP-96), selected from the retained SNPs after sorting on the basis of high MAF value and a random 96 SNP subset (R-SNP-96), selected at random from the 1536 SNPs were also used in the study. These subsets were checked for their distribution across the chromosomes (Fig. 1) to highlight bias for genomic location.

Frequency of SNP in each barley chromosome included in the Barley OPA1 and 6 subsets of 384 and 96 SNP optimised to reveal the diversity in different barley germplasm types
A genetic diversity index (GD; Weir 1996) was calculated for each SNP as GD = 1 − ∑pi 2, where pi is the frequency of allele i, in each germplasm set. The simple matching similarity [Sij = (a + d)/(a + b + c + d) where a and d are the number of loci where the two genotypes have the same allele (A or B), and b and c are the number of loci where the two genotypes have contrasting alleles] was calculated in each SNP set/germplasm combination and transformed into a distance value (Dij = 1 − Sij) (Weir 1996), which was subsequently used in a principal coordinate analysis (PCO) and Procrustes analysis between pairs of SNP sets within each germplasm group. Correlations between similarity matrices within germplasm sets were also calculated. In order to check the goodness of fit of each set of SNPs describing the diversity present in each germplasm type, the data resolution (DR) coefficient was calculated as described in Van Hintum (2007).
Results
In order to investigate the effects of the ‘origin of SNPs’ and the ‘number of SNPs’ on different germplasm, we first examined the SNP distribution across all accessions. Flapjack (http://bioinf.scri.ac.uk/flapjack) was used to create a visual snapshot (Fig. 2) of the diversity revealed by the 1536 SNP set in cultivated and landrace germplasms. The different patterns between the two germplasm sets are striking with large blocks of red or green present throughout the landraces. This effect was also reflected in the overall diversity values with cultivars having a significantly higher value (0.36) compared to the landraces (0.21) (Table 1). By developing subsets of SNPs we could determine whether acceptable informative data could be obtained with fewer SNPs, and investigate the effects of using SNPs derived from one germplasm set to evaluate diversity in the another set. Therefore, a subset of 384 SNPs (C-SNP-384) with high genetic diversity values (measured as MAF) in the cultivated genepool and even distribution along the barley linkage map was selected. C-SNP-384 performs well in discriminating among cultivar germplasm relative to the 1536, with diversity values of 0.37 and 0.36, respectively, suggesting that fewer markers may be sufficient for estimating the structure of genetic diversity (Table 1). However, both C-SNP-384 and 1536 SNP set reveal lower diversity (0.23 and 0.21, respectively) at the corresponding SNP loci in the landrace germplasm than in the cultivated genepool (Fig. 2; Table 1). These results suggested that the 1536 SNPs in BOPA1 and C-SNP-384 are biased against landrace germplasm with respect to their abilities to reveal genetic diversity. To explore whether the effect was reciprocated, two more subsets of 384 SNPs were designed. L-SNP-384 was optimised to capture landrace diversity and R-SNP-384 was selected at random. L-SNP-384 appeared to reveal more diversity than C-SNP-384 in the landraces with corresponding diversity values of 0.44 compared to 0.23 (Table 1), while there was no obvious difference between them in the cultivated material (0.35 compared to 0.37). Furthermore relatively few SNPs were shared between the germplasm-optimised subsets or between optimised sets and random sets (Table 2). For example, only one-third of the SNPs optimised for landraces 384-SNPs were found within the cultivar optimised 384-SNPs. R-SNP-384 appeared to detect less variability than C-SNP-384 or L-SNP-384 in either germplasm type, but similar values to the 1536 SNPs (Table 1).

Flapjack (http://bioinf.scri.ac.uk/flapjack) graphical genotype view of all SNP loci (full Barley OPA1) on barley chromosome 5H from the complete set of cultivars (171) and landraces (169)
We designed a further set of SNPs (C-SNP-96, L-SNP-96 and R-SNP-96), to explore the possibility of using 96 SNPs for germplasm evaluation via small-scale high-throughput SNP platforms (e.g. Illumina BeadExpress platform). The cultivated and random sets showed similar diversity values as the 384 and 1536 for both the landrace and cultivars (Table 1). Although the L-SNP-96 showed similar diversity value for the cultivars as the L-SNP-384, a maximum value of almost 0.50 was observed among the landrace germplasm. The most likely explanation is that the L-SNP-96 was designed by selecting SNPs from L-SNP-384 that had the highest MAFs and most even distribution across the barley chromosomes.
In order to investigate the effects of SNP number further, we examined diversity on a chromosome by chromosome basis. Genetic diversity values among cultivated and landrace accessions for each of the 384 SNP subsets along with the 1536 SNPs were plotted across each barley chromosome (data not shown). For simplicity three of these are shown in Fig. 3. Within the cultivated germplasm, all sets of SNPs show similar patterns of diversity (Fig. 3, lower plots). In the landrace germplasm, the diversity along the chromosomes was very variable with all SNP subsets, except L-SNP-384 that displayed a consistently higher GD than the others (Fig. 3, upper plots). With the 96-SNP subsets the pattern of diversity along the barley chromosomes was generally comparable with the results observed with the 384 sets, with the exception of the L-SNP-96 appearing biased towards the detection of diversity in landraces, flat lining almost maximum diversity along chromosomes. Its behaviour in the cultivated barley’s appeared similar to that of L-SNP-384.

Genetic diversity index (GD; Weir 1996) (0.0–0.5) along three barley chromosomes (1H, 2H and 3H) using a rolling average of 15 SNPs. Landrace diversity across the 3 chromosomes for different set of 384 SNPs (C-384; L-384; R-384 and all 1536 SNPs) are shown in the upper graphs and the cultivars for the same SNP sets in the lower graphs
To determine whether the different SNP subsets affected the inferred genetic structures within each germplasm type, a simple matching similarity matrix was built for each subset and pair-wise correlations calculated between them (Table 3). Within the landraces (Table 3b) correlations between L-SNP-based similarities and all other SNP sets were generally lower than those among C-SNP, R-SNP and 1536 SNP sets. Relative to the 1536 SNP set, all C- and R-SNP subsets tended to perform well (near or above 0.9 correlation coefficient), whereas the landrace SNP similarity matrices were less well correlated (0.82 and 0.64 for L-SNP-384 and L-SNP-96, respectively). L-SNP-384 similarities were poorly correlated with both R- and C-SNP subsets (averages 0.79 and 0.84, respectively, for 384 and 96 combined). L-SNP-96 showed particularly poor correlation in the landraces with the 1536 set (0.64), both R-SNP subsets (average 0.62) and both C-SNP subsets (average 0.70). Within the cultivated material (Table 3a) the correlations were generally higher, most likely due to the 1536 SNPs originating from cultivated germplasm. For cultivated germplasm all 96 SNP subsets generally showed lower correlation than those observed with their corresponding 384 SNP subsets.
To complement the correlation analysis, we conducted Procrustes analysis of PCOs derived from the genetic distance matrices for the different SNP subsets. Procrustes analysis rotates a test matrix until maximum similarity with a target matrix is obtained by minimising the sum of squared differences between the sample ordinates. It is often used to compare the results of ordination analyses, such as PCO, PCA or multidimensional scaling (Mardia et al. 1979). Figure 4 shows the comparisons between the principal coordinates (PCO) obtained using the 1536 SNP with those based on L-SNP-384, C-SNP-384 and R-SNP-384 in the landrace and cultivated germplasm sets, respectively. In the landraces, there was a significant effect of ‘SNP set’ on the residuals, and therefore the PCO from different sets grouped genotypes somewhat differently (Fig. 4, upper plots). Seven of the 169 genotypes were significantly affected, and these moved further for the L-SNP-384/1536 comparison, compared to comparisons between C-SNP-384 or R-SNP-384 and the 1536 set (Fig. 4, upper plots). These seven accessions are almost identical genetically, differing only in a few SNPs (data not shown). Because very few SNPs discriminate those seven accessions, if different numbers of these are included in each SNP subset, they may greatly impact their location in PCO space. This is reflected in the Procrustes analysis by the length of the arrows (Fig. 4). A procrustes analysis was also performed on the landraces removing those seven lines affected by the SNP set and the genetic structure of the population was consistent when estimated with different SNP sets (data not shown).

Procrustes analysis of the principal coordinate analysis of 169 landrace accessions (upper plots) and 170 barley cultivars (lower plots) rotating the PCO axes from three subsets of 384 SNP [landrace optimised (L-SNP-384), cultivar optimised (C-SNP-384) and randomly selected (R-SNP-384)] onto the full set of 1536 SNP. Dots correspond to the subset based PCO scatter plot and the arrows show where the same dots are located in the 1536 SNP-based PCO scatter
The effect of SNP selection upon the residuals associated with comparisons between the different SNP subsets was not so significant in the cultivated germplasm. The residuals in this analysis were affected mainly by the number of SNPs, with the residuals increasing as the number of SNPs decreases in all subsets (data not shown). We noticed a single anomaly. In the cultivated germplasm, C-SNP-384 classified three lines closer to the rest of the population, while 1536 SNP, R- and L-SNP-384 plotted them as the most negative values on the second principal coordinate (Fig. 4, lower plots). Despite the differences described above, the Procrustes analysis and its accompanying statistics showed a very good agreement between the PCO based on different SNP sets. The values of the correlation-like statistic from the Procrustes analysis were above 0.8 in all pair-wise comparisons (data not shown).
Next, the data resolution statistic DR (van Hintum 2007) was calculated in each subset of SNPs to estimate their reliability in estimating corresponding population structure (Table 4). Very high DR values above 0.97 were observed from the 1536 set in both cultivar and landrace germplasm. Second, the DR statistic for landrace-optimised SNPs in landrace germplasm was also high (0.954) and all 384 SNP subsets performed above 0.9 in both germplasm classes. Lastly, all 96 SNP sets with the possible exception of L-SNP-96 in landraces (0.856) and C-SNP-96 in cultivars (0.790) performed poorly in both germplasm classes.
The 384 and 96 SNP subsets were chosen because these numbers conform to the constraints of most middle-throughput SNP genotyping platforms including the Illumina BeadExpress instrument which is universally employed for scoring Illumina SNP genotyping on this scale. To test whether these are reasonable scales with regard to optimal SNP number for diversity analysis, randomness of chromosome distribution, and correlations between deduced distance matrices we selected 1,000 random SNP sets for 96, 192, 288, 384, 500, 700 and 900 SNPs (7,000 sets in total). First we calculated mean diversity indices and 95% CIs based on 1,000 replicates in cultivars and the landraces. (Supplementary Table 1). Mean D values were highly stable between 384 random SNPs (95% CIs of 0.345/0.366 and 0.193/0.223 for cultivars and landraces, respectively) and SNP number per chromosome was also extremely stable (for example, chromosome 7H had the lowest SNP proportion of 0.116 with a corresponding SD value of 0.0008; data not shown). Next, we explored the correlation (Mantel test) between distance matrices based on each of the 7,000 SNP sets and that based on the 1536 SNP set for both the cultivars and landraces. Supplementary Fig. 1 shows that 384 SNPs yield average r 2 values above 0.98, whereas 96 values are around 0.92. Therefore, 384 SNPs are an excellent compromise between performance and effort with regard to diversity studies.
Discussion
An accurate description of genetic diversity is important for the effective use and management of germplasm resources. We have investigated how different subsets of SNP markers, selected from a collection of 1536 SNPs on BOPA1, an Illumina-based community genotyping platform for barley, reflect measures of genetic diversity in landrace and cultivated germplasm. We explored how SNP selection and the number of SNPs included on the assay platform affect the accuracy of the diversity and population structure estimation for these different germplasm pools. It was immediately clear that BOPA1 reveals lower diversity in the landrace collection used here than a cultivated germplasm set of similar size (Fig. 2). This observation contradicts both previous reports on the genetic diversity found within the same landrace material using SSR markers (Russell et al. 2003) and more general observations that landrace germplasm is more diverse than cultivated accessions in small grain cereal species (Moragues et al. 2007; Brown and Munday 1982). We hypothesised that the reason for this contradiction may be ascertainment bias (Nielsen 2000) caused by the relatively few genotypes used for SNP discovery combined with selection for high performance and MAF in cultivated germplasm. The effects of ascertainment bias on the statistical properties of genotype frequencies has been widely demonstrated (Kuhner et al. 2000; Wakeley et al. 2001; Akey et al. 2003; Nielsen et al. 2004; Clark et al. 2005). To test this hypothesis, we designed subsets of 384 SNPs, one aimed at maximising the diversity detected in landrace germplasm and the other selected at random. A sizeable effect of SNP selection per se was clearly apparent in the diversity estimates (Table 1; Fig. 3). In particular, L-SNP subsets consistently revealed higher diversity levels in landrace material than found for C-SNPs, resolving the apparent contradiction in relative levels of diversity between previous results and our own. Schlotterer and Harr (2002) found that using SNPs discovered only in European Drosophila, European flies exhibited greater variability than African flies; however, when SNPs discovered using both populations the variability in the African population was restored. Similarly, they observed no bias when using the ancestral SNPs on the European populations. This was also the situation when using the landrace-chosen SNPs to examine diversity in the cultivated genepool; any combination of SNP number and origin gave almost identical values ranging from 0.35 to 0.37 in terms of genetic diversity.
Rosenblum and Novembre (2007) found that frequency spectra and summary statistics were highly sensitive to the SNP discovery strategy, necessitating careful selection of the ascertainment panels chosen to jointly sample geographical, phenotypic and genetic diversity. The ascertainment panel of germplasm chosen to develop the 3072 progenitor SNPs from which BOPA1 was derived were not chosen to represent the barley genepool but rather based on the available EST collections that have been deposited into the HarvEST database (http://www.harvest-web.org/). For example, in the HarvEST build 32, there are nine genotypes, (eight elite and one wild barley) with 10,000 or more ESTs, and SNPs were identified from the pair-wise comparisons of these collections. Furthermore, SNP selection for BOPA1 involved screening of barley cultivars exclusively (Rostoks et al. 2006), whose allelic spectrum is poorly matched to that of landrace germplasm. Subsequently, SNPs with MAFs of <8% and that may have been informative in the landrace material were purposely excluded from BOPA1, effectively tailoring the platform for analysis of elite cultivated material. In a recent review, Romero et al. (2009) expressed concern that genetic markers were not selected on a random subset but represent a set that satisfies specific criteria and suggested that despite the wealth of human genotypic data, the accuracy of this data may be problematic. In barley, the criteria for inclusion in BOPA1 was to maximise diversity within the cultivated genepool and so it is likely that we have selected high to intermediate frequency alleles, inflating diversity towards the cultivated genepool. In the human situation, there is a bias towards European diversity and Romero et al. (2009) confirmed the bias affecting SNPs and the underestimation of diversity in the African populations. Several authors have suggested the use of ascertainment bias correction methods. Rosenblum and Novembre (2007) used the methods developed by Nielsen et al. (2004) to correct for bias in studying populations of eastern fence lizards.
Population structure estimates are also an important aspect of germplasm analysis. In this regard, we assessed the effect of SNP selection strategies on the consistency of the population structure. Although, some differences in the classification of genotypes were observed using different subsets of SNPs, the population structures deduced for the two sets of germplasm used in this study was fairly consistent across SNP subsets. While one of the consequences of ascertainment bias is a lowering of the detection rate of rare alleles and an increase in intermediate frequency allele, any inference about population that is reliant on allele frequency will be affected. In particular, tests of natural selection that rely of detecting signatures of selection would be affected. This may be problematic if there are bottlenecks among barley populations. The degree of population differentiation may also be affected, sampling only one population or germplasm type, this would lead to an over representation of SNPs that are intermediate in that population and under represent SNPs which may distinguish between populations (Clark et al. 2005; Rosenblum and Novembre 2007). From previous work we have shown that the allelic composition of cultivated and landrace barleys are very different and if we had only examined alleles which were high to moderate frequency the picture would be very different and most likely underestimate the differences between these two sets (Russell et al. 2003).
Recently, the potential of using association mapping approaches in barley have come to the forefront (Waugh et al. 2009; Hayes and Szucs 2006; Rostoks et al. 2006; Caldwell et al. 2006). Estimating linkage disequilibrium for association approaches to isolating candidate genes is crucial and will be affected by allele frequency. Therefore, the extent of bias will be of concern in relation to measuring linkage disequilibrium. Clark et al. (2005) suggest that the effects of ascertainment bias on LD will be complex and the over sampling of high-frequency SNPs will result in lower LD. However, the effect would likely be small and unlikely to introduce false-positive inferences that are the major problem in such studies.
The other objective of this work was to determine the effects of the number of SNPs used upon the robustness of population structure estimates and to recommend an optimum and cost-efficient number of SNPs to be used in characterization of barley germplasm. The 96 SNP sets gave values for data resolution (DR) in the range of those found using between 75 and 175 AFLP markers and lower than the 10 SSR markers used to assess genetic structure in lettuce (van Hintum 2007). The DR statistic quantifies the consistency of a dataset in describing the genetic structure of the material characterized (van Hintum 2007). It is clear from the data in Table 4 that there is a great improvement in DR when using 384 SNPs compared to 96 SNPs, and any further improvement obtained using 1536 SNPs is marginal at best. We therefore propose that for the majority of questions there will be no significant justification for the additional cost involved in obtaining the extra SNP information.
Conclusions
The results of this study clearly show that there is a bias in the estimation of the diversity present in barley landraces using all of the SNP markers included in the Barley OPA1 chip. This bias can be at least partially nullified by selecting an appropriate subset of SNPs that reveal higher diversity levels in the landrace germplasm. However, this does not alleviate the original bias in the SNP discovery phase and so it may be necessary to design assays that survey specific germplasm sets such as landraces and wild barleys. This may sound like an impossible task as the original SNP discovery was based on individuals who had collections of at least 10,000 EST, but with the next generation sequencing approaches becoming available the possibility of sequencing a panel of landraces and wild barleys may be an achievable task. In terms of the cultivated germplasm, we strongly support the use of a cultivar optimised subset of 384 SNPs instead of all 1536 present on the BOPA1 as a cost-effective tool for genotyping cultivated barley germplasm. Our evidence suggests that there is little advantage to be gained by trying to further optimise the markers in this 384 subset. We caution against the use of any 96-SNP subset, as these deliver much poorer performance as judged by correlations with corresponding data from larger SNP collections.
References
Abasht B, Lamont SJ (2007) Genome-wide association analysis reveals cryptic alleles as an important factor in heterosis for fatness in chicken F2 population. Anim Genet 38:491–498
Akey JM, Zhang K, Xiong M, Jin L (2003) The effect of single nucleotide polymorphism identification strategies on estimates of linkage disequilibrium. Mol Biol Evol 20:232–242
Brown A, Munday J (1982) Population-genetic structure and optimal sampling of land races of barley from Iran. Genetica 58:85–96
Brown A, Nevo E, Zohary D, Dagan O (1978) Genetic variations in natural populations of wild barley. Genetica 49:97–108
Caldwell KS, Russell J, Langridge P, Powell W (2006) Extreme population-dependent linkage disequilibrium detected in an inbreeding plant species, Hordeum vulgare. Genetics 172:557–567
Clark AG, Hubisz MJ, Bustamante CD, Williamson SH, Nielsen R (2005) Ascertainment bias in studies of human genome-wide polymorphism. Genome Res 15:1496–1502
Fan J, Chee MS, Gunderson KL (2006) Highly parallel genomic assays. Nat Rev Genet 7:632–644
Hayes P, Szucs P (2006) Disequilibrium and association in barley: thinking outside the glass. Proc Natl Acad Sci USA 103:18385–18386
Jana S, Pietrzak LN (1988) Comparative assessment of genetic diversity in wild and primitive cultivated barley in a center of diversity. Genetics 119:981–990
Kota R, Varshney RK, Prasad M, Zhang H, Stein N, Graner A (2007) EST-derived single nucleotide polymorphism markers for assembling genetic and physical maps of the barley genome. Funct Integr Genomics 8:223–233
Kuhner MK, Beerli P, Yamato J, Felsenstein J (2000) Usefulness of single nucleotide polymorphism data for estimating population parameters. Genetics 156:439–447
Mardia KV, Kent JT, Bibby JM (1979) Multivariate Analysis. Academic Press, New York
Matus IA, Hayes PM (2002) Genetic diversity in three groups of barley germplasm assessed by simple sequence repeats. Genome 45:1095–1106
Moragues M, Moralejo M, Sorrells M, Royo C (2007) Dispersal of durum wheat [Triticum turgidum ssp. turgidum convar. durum (Desf.) MK] landraces across the Mediterranean basin assessed by AFLPs and microsatellites. Genet Resour Crop Evol 54:1133–1144
Nevo E, Zohary D, Beiles D, Kaplan D, Storch N (1986) Genetic diversity and environmental associations of wild barley, Hordeum spontaneum, in Turkey. Genetica 68:203–213
Nielsen R (2000) Estimation of population parameters and recombination rates from single nucleotide polymorphisms. Genetics 154(2):931–942
Nielsen R, Hubisz MJ, Clark AG (2004) Reconstituting the frequency spectrum of ascertained single-nucleotide polymorphism data. Genetics 168:2373–2382
Rafalski A (2002) Applications of single nucleotide polymorphisms in crop genetics. Curr Opin Plant Biol 5(2):94–100
Romero IG, Manica A, Goudet J, Handley LL, Balloux F (2009) How accurate is the current picture of human genetic variation? Heredity 102:120–126
Rosenblum EB, Novembre J (2007) Ascertainment bias in spatially structured populations: a case study in the eastern fence lizard. J Hered 98:331–336
Rostoks N, Mudie S, Cardle L, Russell J, Ramsay L, Booth A, Svensson J, Wanamaker S, Walia H, Rodriguez E, Hedley P, Liu H, Morris J, Close T, Marshall D, Waugh R (2005) Genome-wide SNP discovery and linkage analysis in barley based on genes responsive to abiotic stress. Mol Genet Gen 274:515–527
Rostoks N, Ramsay L, MacKenzie K, Cardle L, Bhat PR, Roose ML, Svensson JT, Stein N, Varshney RK, Marshall DF, Graner A, Close TJ, Waugh R (2006) Recent history of artificial outcrossing facilitates whole-genome association mapping in elite inbred crop varieties. Proc Natl Acad Sci USA 103:18656–18661
Russell JR, Booth A, Fuller JD, Baum M, Ceccarelli S, Grando S, Powell W (2003) Patterns of polymorphism detected in the chloroplast and nuclear genomes of barley landraces sampled from Syria and Jordan. Theor Appl Genet 107:413–421
Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PIW, Chen H, Roix JJ, Kathiresan S, Hirschhorn JN, Daly MJ, Hughes TE, Groop L, Altshuler D, Almgren P, Florez JC, Meyer J, Ardlie K, Bengtsson Boström K, Isomaa B, Lettre G, Lindblad U, Lyon HN, Melander O, Newton-Cheh C, Nilsson P, Orho-Melander M, Råstam L, Speliotes EK, Taskinen M, Tuomi T, Guiducci C, Berglund A, Carlson J, Gianniny L, Hackett R, Hall L, Holmkvist J, Laurila E, Sjögren M, Sterner M, Surti A, Svensson M, Svensson M, Tewhey R, Blumenstiel B, Parkin M, Defelice M, Barry R, Brodeur W, Camarata J, Chia N, Fava M, Gibbons J, Handsaker B, Healy C, Nguyen K, Gates C, Sougnez C, Gage D, Nizzari M, Gabriel SB, Chirn G, Ma Q, Parikh H, Richardson D, Ricke D, Purcell S (2007) Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 316:1331–1336
Schlotterer C, Harr B (2002) Single nucleotide polymorphisms derived from ancestral populations show no evidence for biased diversity estimates in Drosophila melanogaster. Mol Ecol 11:947–950
Storz JF, Kelly JK (2008) Effects of spatially varying selection on nucleotide diversity and linkage disequilibrium: insights from deer mouse globin genes. Genetics 180:367–379
van Hintum TJL (2007) Data resolution: a jackknife procedure for determining the consistency of molecular marker datasets. Theor Appl Genet 115:343–349
Wakeley J, Nielsen R, Liu-Cordero SN, Ardlie K (2001) The discovery of single nucleotide polymorphisms and inferences about human demographic history. Am J Hum Genet 69:1332–1347
Waugh R, Jannink J-L, Muehlbauer GJ, Ramsay L (2009) The emergence of whole genome association scans in barley. Curr Opin Plant Biol 12(2):218–222
Weir B (1996) Genetic data analysis II: methods for discrete population genetic data. Sinauer Associates, Sunderland, MA
Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, Timpson NJ, Perry JRB, Rayner NW, Freathy RM, Barrett JC, Shields B, Morris AP, Ellard S, Groves CJ, Harries LW, Marchini JL, Owen KR, Knight B, Cardon LR, Walker M, Hitman GA, Morris AD, Doney ASF, McCarthy MI, Hattersley AT (2007) Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 316:1336–1341
Acknowledgments
We would like to acknowledge Drs S. Grando, M. Baum and S. Ceccarelli at International Center for Agricultural Research in the Dry Areas (ICARDA) for providing the Syrian Jordanian landrace collection material. The above work was supported by BBSRC Grant BB/E024726/1 (EXBARDIV) under the ERA-PG Programme ‘Structuring Plant Genomic Research in Europe’. SCRI received Grant-in-Aid from the Scottish Government.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by A. Graner.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Moragues, M., Comadran, J., Waugh, R. et al. Effects of ascertainment bias and marker number on estimations of barley diversity from high-throughput SNP genotype data. Theor Appl Genet 120, 1525–1534 (2010). https://doi.org/10.1007/s00122-010-1273-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-010-1273-1