
Abstract
Microspore production using endogenous developmental programs has not been well studied. The main limitation is the difficulty in identifying genes preferentially expressed in pollen grains at early stages. To overcome this limitation, we collected transcriptome data from anthers and microspore/pollen and performed meta-expression analysis. Subsequently, we identified 410 genes showing preferential expression patterns in early developing pollen samples of both japonica and indica cultivars. The expression patterns of these genes are distinguishable from genes showing pollen mother cell or tapetum-preferred expression patterns. Gene Ontology enrichment and MapMan analyses indicated that microspores in rice are closely linked with protein degradation, nucleotide metabolism, and DNA biosynthesis and regulation, while the pollen mother cell or tapetum are strongly associated with cell wall metabolism, lipid metabolism, secondary metabolism, and RNA biosynthesis and regulation. We also generated transgenic lines under the control of the promoters of eight microspore-preferred genes and confirmed the preferred expression patterns in plants using the GUS reporting system. Furthermore, cis-regulatory element analysis revealed that pollen specific elements such as POLLEN1LELAT52, and 5659BOXLELAT5659 were commonly identified in the promoter regions of eight rice genes with more frequency than estimation. Our study will provide new sights on early pollen development in rice, a model crop plant.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The production of pollen as a complex process requires the coordinated participation of various cell and tissue types. Pollen mother cells produce tetrads of haploid microspores through meiosis, and the microspores (male gametophyte) released from these tetrads become mature pollen via pollen mitosis I (PMI) and pollen mitosis II (PMII) and can then be used for fertilization (Bedinger 1992). The microspore corresponds to a precise stage in the development of the male gamete in higher plants. Microspore development is closely linked with the sporophyte tissue of the male organ, also known as the tapetum, which contributes to the development of the external wall in microspores, which begins after the microspores are released from tetrads. The contribution of the external part has been well studied through genetic studies of many male sterility genes. However, the internal contribution to microspore development is rarely understood because separating the pollen during this stage is very difficult. High-throughput transcriptome data using microarray technology have provided a global understanding of pollen development in rice and Arabidopsis (Becker et al. 2003; Honys and Twell 2003, 2004; Suwabe et al. 2008; Fujita et al. 2010; Wei et al. 2010; Aya et al. 2011). In previous studies, Honys and Twell (2004) and Wei et al. (2010) demonstrated that dynamic changes in the transcriptomes of pollen in Arabidopsis and rice according to developmental progress. Interestingly, the uni-nucleated microspores showed a more similar profile to that of bi-nucleated pollen compared to that of the others, while profiles between mature pollen grains (MPG) and germinating pollen grains (GPG) were more conserved than those of the others (Wei et al. 2010). Furthermore, a microarray technique combined with laser microdissection (44 K LM-microarray) was used to independently identify the transcriptomes of the microspore/pollen and tapetum in rice (Suwabe et al. 2008). Subsequently, 140 male gamete- and tapetum-specific transcripts were identified. Although there was high specificity of identified transcripts, the number of transcripts was quite small because LM microarray technology uses a tiny amount of mRNA as a template and multiple rounds of amplification processes, which can restrict the recovery of endogenous mRNA profiles, as shown in root LM microarray experiments (Suwabe et al. 2008; Takehisa et al. 2012).
A public microarray database, the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/gds), has been collecting microarray data of samples from developing anthers and pollen in rice (Fujita et al. 2010; Deveshwar et al. 2011; Wei et al. 2010). However, it is not yet know how well the transcriptome data for similar anatomical samples from different sources are conserved or diversified. Systematic analysis of similar anatomical transcriptome data available in public databases might provide new insight that has not been provided by individual reports. Especially, the transcriptome data for male gamete development conserved between japonica and indica cultivars are very important for expanding the general understanding of the developmental process.
Combination of meta-expression data and the GUS reporting system is a very effective way to provide novel promoters to manipulate tissue-specific or stress-related traits (Thilmony et al. 2009). Recently, we reported the activity of the promoters of 11 mature pollen-specific genes, OsLPS1 to OsLPS11, in rice. Seven of the these promoters were also operated in a heterologous system using Arabidopsis (Nguyen et al. 2015; Oo et al. 2014). One promoter, OsLPS10 which was showed expression at bi-nucleated microspore stage, was successfully used for the complementation of Arabidopsis gametophytic mutant which was defective in microspore development prior to PMI (Nguyen et al. 2015).
In addition, a number of rice promoters confirmed by the GUS reporting system were evaluated using meta-expression data, indicating that meta-expression profiles based on a large collection of data can be useful sources of novel promoters (Jeong and Jung 2015). Microspore-specific promoters can be an ideal tool in plant breeding, especially in the field of rice hybrid breeding. Male sterile lines can be generated by producing cytotoxin or altering hormone levels using microspore-specific promoters (Oldenhof et al. 1996; Bae et al. 2010). Thus, the discovery of new early microspore-specific promoters that have been well characterized will provide us more options for manipulation of target traits in plants.
Here, we reported the global identification of rice microspore preferential (RMP) genes that are separated from sporophyte anther tissue using meta-expression analysis of anther and pollen samples collected in the public database. In total, 410 RMP genes were identified, but the functional characterization for relating developmental processes was quite limited compared to those showing rice pollen mother cell or tapetum-preferred (RPMCT) genes (263 genes). Among RMP genes, only two have been functionally studied through loss of function approaches, while 28 RPMCT genes have been functionally characterized, nine of which are related to pollen development or sterility. GO analysis revealed that the pyrimidine nucleotide biosynthetic process is very important for microspore development in rice. GUS activity under the control of a RMP gene promoter was used to confirm the tissue-specificity on rice microspore/pollen, suggesting that multiple promoters can be used to manipulate the agronomic traits associated with microspore development.
Results
Meta-expression analysis for genome-wide identification of RMP genes in rice
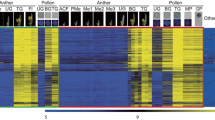
To identify the microspore (early pollen)-preferred genes that are conserved among japonica and indica rice cultivars, we downloaded and analyzed six series of Affymetrix rice microarray data prepared from developing anthers and pollen grains from NCBI GEO (http://www.ncbi.nlm.nih.gov/geo/) (Table S1) (Barrett et al. 2009; Chandran and Jung 2014). The samples were arranged in order according to the process of anther and pollen development. As a control, all of the Affymetrix anatomical meta-expression profiles in the Rice Oligonucleotide Array Database (ROAD; http://www.ricearray.org/expression/meta_analysis.shtml) except for two anther samples were used to analyze the expression patterns in other tissues/organs (Cao et al. 2012). Afterward, we performed K-means clustering (KMC) analysis using the Euclidean distance matrix (EDM) and grouped 57,382 probes into 49 clusters based on expression pattern. As a result, we identified 789 probes from two clusters (42 and 45) (Fig. S1). Further hierarchical clustering analysis using the EDM refined the gene list to 410 genes that showed uni-nucleated (microspore)-preferred expression patterns in both cultivars (Fig. 1; Table S2). In addition, we identified that 263 RPMCT genes include well-known tepetum preferred genes such as tapetum degeneration retardation 1 (TDR1), eternal tapetum 1 (EAT1)/DTD, undeveloped tapetum 1 (UDT1), defective tapetum cell death 1 (DTC1), and persistent tapetal cell 1 (PTC1) and well-known pollen mother cell preferred genes such microspore and tapetum regulator 1 (MTR1), meiosis arrested at leptotene1 (MEL1) and ZIP4 (Table S3) (Jung et al. 2005; Komiya et al. 2014; Niu et al. 2013; Shen et al. 2012; Tan et al. 2012; Yi et al. 2016). Unlike RMP genes, we did not observe significant expression in developing pollen samples, indicating that these genes have major roles in male sporophytic tissues (Fig. 1). Candidate genes are summarized in Supplemental Tables 2 and 3.

Heatmap expression analysis of RMP and RPMCT genes. Expression patterns conserved among indica (brown box) and japonica (green box) types over several stages of development. The number of microarray data is 22 for indica rice (five stages for anthers and three for pollen) and 42 for japonica rice (eight stages for anthers and five for pollen). Anatomical meta-expression data from ROAD were used to analyze the expression patterns in other tissues. ACF archesporial cell-forming stage, BG bi-nucleated gametophyte stage, Fl flowering stage, GP germinating pollen, Me meiotic stage, Me1 meiotic leptotene stage, Me2 meiotic zygotene–pachytene stage, Me3 meiotic diplotene-tetrad stage, MP mature pollen stage, PMe pre-meiosis, TG tri-nucleated pollen stage, UG uni-nucleated gametophyte stage. The yellow color in the heatmap indicates high level of expression; dark-blue low expression
Evaluation of functions of RMP genes in the literature
Recently, the overview of functionally characterized genes in rice online database (OGRO, http://qtaro.abr.affrc.go.jp/ogro) summarized the rice genes that have been genetically characterized (Yamamoto et al. 2012). Of our candidate genes preferentially expressed in early pollen, we found that the functions of two genes were characterized through loss of functions. Of them, cysteine protease 1 (OsCP1) is involved in pollen development (Lee et al. 2004), but OsPHO1;2 regulates the translocation of phosphate from the root to the shoot (Secco et al. 2010). The former has a function which is associated with anatomical expression pattern, but the latter showing differential expression patterns under phosphate starvation is related to a condition-dependent function. In the latter, pollen development under phosphate starvation might be affected and should be addressed by further studies. The analysis for known genes associated with microspore development showed that our candidate genes might have morphological function or stress responses associated with microspore development.
Unlike RMP genes, we found that the functions of 28 RPMCT genes have been elucidated, indicating that pollen mother cell or tapetum development have been actively studied in rice due to the agronomic significance associated with male sterility (Table 1). Of them, 21 genes showed defects in the identity/formation of anthers and floral organs, confirming that tissue-preferred expression patterns are useful guidance for molecular and genetic characterization of the related gene function.
Biological processes specific to RMP genes using GO enrichment analysis and comparison with those of RPMCT genes
We conducted a Gene Ontology (GO) enrichment analysis in ROAD (Cao et al. 2012) in order to query 410 RMP genes. Our objective was to examine GO enrichment within the category of ‘biological process’ with the ROAD GO enrichment tool (http://www.ricearray.org/analysis/go_enrichment.shtml). As a result, we identified 222 GO terms assigned to 132 genes (Table S4). The majority of RMP genes (67.8 %) had limited information on gene functions based on GO. Significant terms in that category were selected with hypergeometric p values ≤0.05 and enrichment values of at least twofold (Table S4; Fig. 2). Of these, 12 GO terms were over-represented in RMP genes. Significantly enriched terms were found for biological processes corresponding to the pyrimidine nucleotide biosynthetic process (50.9 GO fold-enrichment value), ubiquitin-dependent protein catabolic process (10.4), chitin catabolic process (9.6), cell-matrix adhesion (9.1), cellular amino acid metabolic process (8.8), tRNA aminoacylation for protein translation (7.4), lipid transport (5.0), nucleosome assembly (3.8), ion transport (3.6), response to freezing (2.6), carbohydrate metabolic process (2.1), and translation (2.1) (Fig. 2a). The most abundant terms were found for ‘response to freezing,’ representing 24 genes. This was followed by ten genes for translation and nine for ubiquitin-dependent protein catabolic process (Fig. 2a).

GO enrichment analysis of RMP and RPMCT genes. a GO enrichment analysis of RMP genes. b GO enrichment analysis of RPMCT genes. X axis indicates the names of the GO terms, and the Y axis indicates the fold enrichment value. All enriched GO terms were selected under higher than twofold enrichment values and lower than 0.05 hypergeometric p values
To compare the transcriptome expressed in tapetal tissue and pollen mother cell but less expressed in early pollen, we also conducted GO enrichment analysis in biological processes. As a result, we identified 31 GO terms assigned to 137 of 263 genes, finding that the functions of 52.1 % of RPMCT genes can be estimated using GO in the biological process category (Fig. 2b), whereas the others need further investigation. In total, 13 GO terms were over-represented in RPMCT genes. Significantly enriched terms were found for biological processes corresponding to alcohol metabolism (83.5 GO fold-enrichment value), flower development (30.7), cell differentiation (23.4), negative regulation of translation (14.6), gene silencing by RNA (11.7), multicellular organismal development (8.6), ubiquitin-dependent protein catabolic process (6.6), fatty acid biosynthetic process (5.3), cell wall organization (4.4), lipid metabolic process (4.2), lipid transport (4.1), protein ubiquitination (3.9), and regulation of transcription (2.4) (Fig. 2b). The most abundant terms were for ‘regulation of transcription,’ representing 23 genes. This was followed by eight genes for lipid metabolism (Fig. 2b).
Compared to pollen mother cell or tapetum development, microspore development might require pyrimidine nucleotide biosynthesis, nucleosome assembly, cellular amino acid metabolism, protein translation, chitin catabolism, cell-matrix adhesion, ion transport, carbohydrate metabolic process, and response to freezing and translation, while pollen mother cell or tapetum development are more likely to be associated with alcohol metabolism, negative regulation of translation, regulation of transcription, cell differentiation, fatty acid biosynthesis, protein ubiquitination, lipid metabolism, cell wall organization, and multicellular organismal development. Lipid transport, and ubiquitin-dependent protein catabolism are common in both RMP and RPMCT genes, suggesting cooperative features of the tapetum, pollen mother cell, and male-gamete in microspore development.
MapMan analysis of RMP genes compared with RPMCT genes
MapMan allows one to group genes into different functional categories and visualize data through diagrams (Jung and An 2012). To classify RMP (410, green in Fig. 3) and RPMCT (263, red in Fig. 3) genes, we first analyzed the overall overview in the MapMan tool kit. As mentioned in the GO analysis, most of the RMP genes do not have an assigned MapMan term, indicating that most of the candidate genes showing microspore-preferred expression patterns have been less studied than RPMCT genes. Cell wall, metabolism of lipid, secondary metabolite and hormone, miscellaneous function, and RNA MapMan terms are strongly associated with the pollen mother cell or tapetum (Fig. 3; Fig. S2). On the other hand, protein, DNA and nucleotide metabolism MapMan terms are closely related to RMP genes (Fig. 3; Fig. S2). Especially, over-representation of DNA and nucleotide metabolism indicates that microspore development includes active cell division. Over-representation of protein terms in RMP genes indicates its significance in microspore development (Fig. 3). Especially, the ubiquitin and autophagy-dependent degradation overview indicates that three ubiquitin proteins, one E2 ligase, one HECT E3 ligase, seven RING E3 ligases, ten components in SCF E3 ligase complexes, 25 components in BTB E3 ligase complexes, and one component of proteasome are RMP genes (Fig. S3; Table S5), suggesting cooperative function of ubiquitin, E2 ligase, E3 ligases, and proteasome to support microspore development through protein degradation from the tapetum or anther locules. BTB E3 ligase complexes are only identified in RMP genes, suggesting unique roles of BTB E3 ligase complexes in microspore development in rice. In addition, microspore development associated with the protein MapMan term includes the protein translation pathway, as indicated in GO enrichment analysis (Fig. S4). RMP genes for the translation process retain two ribonucleases for RNA processing, two t-RNA ligases, one 60S subunit of ribosomal protein, one for protein synthesis initiation, one for protein elongation, 55 associated with protein degradation, five with protein modification, and one protein for mitochondrial targeting (Table S5). All of these might work together for protein synthesis during microspore development.

MapMan analysis of RMP genes. The overall overview was analyzed with 410 RMP genes and 263 RPMCT genes. Red boxes in these overviews indicate RMP, and green boxes indicate RPMCT genes. Detailed information about the overview is shown in Table S5
Expression patterns of microspore-preferred genes were confirmed through the GUS reporter system
To verify the microspore-preferred expression patterns of our candidate genes, eight candidate genes were selected (Fig. 4b; Table 2). Using PCR reaction with specific primers to amplify the promoter regions of the eight candidate genes, the promoter fragments of eight RMP genes ranging in size from 725 to 2053 bp were generated (Table 2). The promoter fragments were fused to GFP-GUS reporter genes to create the proRMP::GFP-GUS constructs. As shown in Fig. 4a, eight vector constructs were prepared and then transformed into rice callus mediated by Agrobacterium tumefaciens LBA4404 strains (Nguyen et al. 2015). As a result, in total, 316 putative transgenic lines were generated with eight constructs.

Heatmap expression analysis of eight RMP genes for the GUS reporter system and schematic diagrams of promoter-GUS fusion constructs. a Vector maps of the eight promoter-GUS fusion constructs. b Heatmap expression data of eight RMP genes selected for the GUS reporter system
Results of the GUS assay indicated that all eight promoters exhibited very similar GUS expression patterns in pollen grains and anthers, but they were distinct in vegetative organs (Fig. 5a, b). Of them, the promoters of RMP2, RMP7, and RMP8 showed pollen-preferred expression patterns but did not display the GUS signal in leaf, stem, or root tissues (Fig. 5b). On the other hand, promoters of RMP1, RMP4, and RMP6 showed pollen-preferred expression patterns and also exhibited a GUS signal in leaf, stem, and root tissues. In comparison, the promoters of RMP3 and RMP4 showed a GUS signal in leaf and stem tissues but not in the roots, indicating the possible existence of an additional regulatory mechanism different from the microspore-specific genes (Figs. 3, 4). Expression of the other five promoters in other organs/tissues demonstrated unexpected results, possibly caused by wounding and submergence stresses coupled with the GUS assay method. This possibility needs to be tested with meta-expression data under abiotic stress conditions or by analyzing the expression patterns of these genes under wounding or submergence and comparing the results with those in the untreated condition.

Validation of RMP genes using GUS reporter systems and RT-PCR analyses. a Images of flower and pollen from transgenic plants harboring RMP promoter::GUS reporter constructs at sequential stages of pollen development were prepared after GUS staining. b Images of leaf, stem, and leaves from transgenic plants at the vegetative growth stage were also prepared. Promoters from eight RMP genes in Table 2 were used to control GUS expression. UG uni-nucleated gametophyte stage, BG bi-nucleated gametophyte stage, MP mature pollen stage. GUS expression patterns were analyzed from promoter-GUS transgenic lines. Bar in flower image = 0.5 mm; and bar in pollen image = 10 μm. c Real-time RT-PCR analysis of eight genes shown in Fig. 4b. Quantitative real-time PCR analysis was performed for various organs. Transcript levels were normalized to rice UBIQUITIN5 and calculated using the comparative cycle threshold method. Error bars indicate standard deviation (sd). Y axis, relative expression level to rice UBIQUITIN5; X axis, sample names used for analyses. Sh 7-day-old shoots, Ro 7-day-old roots, M/T anthers at meiosis and tetrad, YM anther at young microspore stage, VP anther at vacuolated pollen stage, and MP anther at mature pollen stage
In order to determine GUS signals in the microspores, we first performed DAPI staining for isolated pollen grains at different developmental stages and assigned the pollen to uni-nucleated, bi-nucleated, tri-nucleated, and mature pollen categories. The results indicated that significant levels of GUS signals were first detected at the uni-nucleated microspore stage and were gradually accumulated through the bi-nucleated and tri-nucleated stages, while ultimately reaching the highest level in MPG (Fig. 5a). Due to the accumulating feature of GUS signals, the expression at the tri-nucleated and mature pollen stages might be observed. The patterns of microspore-preferred expression by these genes, as shown in Fig. 2, were confirmed by RT-PCR analysis (Fig. 5c). Of them, RMP1 and RMP8 showed the highest expression in bi-nucleated microspore, while the next highest expression was in young microspores. However, the others showed the highest expression patterns in young microspores, and expression in the seedling roots and shoots was very low, like that in the microarray data. All of our results demonstrated that these meta-expression data for genes preferentially expressed in microspores are highly reliable and are a valuable source of novel promoters for controlling traits associated with pollen maturation.
Analysis of the RMP promoter sequences
The promoter sequences of the RMP1 through RMP8 genes were analyzed using a signal scan search of Plant cis-acting Regulatory DNA Elements (PLACE) program (Higo et al. 1999). The translation start site was counted at +1, and the locations of cis-regulatory elements (CREs) were highlighted for each promoter (Fig. S6). The scanning results revealed numerous CREs in the promoter region of the RMP genes. The functions of identified CREs are grouped and shown in Tables S6 and S7. Among them, the six most abundant CREs detected in RMP promoter regions are listed in Table S6: ACGTATERD1 (ATGC), ARR1AT (NGATT), CAATBOX1 (CAAT), GATABOX (GATA), MYBCORE (CNGTTR), and DOFC OREZM (AAAG). The copy number for each of those CREs is shown in Fig. S7. In detail, we found 16 copies of ACGTATERD1 in RMP4, 12 copies in RMP1, 8 copies in RMP2, RMP3, and RMP6, and 4 copies in RMP7 and RMP8 promoters. For ARR1AT, 26 copies were found in RMP2, whereas the others showed lower copy numbers for this motif as follows: RMP8 (22), RMP4 (21), RMP7 (18), RMP5 (14), RMP1 (12), RMP3 (4). In addition, a high copy number of CAATBOX1 was revealed in RMP2 (34). The other promoters possess fewer: RMP8 (22), RMP1 (18), RMP4 (16), RMP7 (12), RMP5 (11), RMP6 (7), RMP3 (5). We also found 30 copies of GATABOX in RMP1, 20 in RMP4, and 15–17 in RMP5, RMP7, and RMP8. Six copies of MYBCORE were found in the RMP3, RMP5, RMP8 promoters, while four copies were found in RMP1, RMP2, and RMP4. The RMP6 and RMP7 promotors possessed three and five copies, respectively. Futhermore, high copy numbers of DOFCOREZM were detected in the RMP promoters: RMP4 (36), RMP2 (35), RMP5 (33), RMP8 (31), RMP7 (24), RMP1 (15), RMP3 (10), and RMP6 (11). In addition, we analyzed average frequency of these CREs from 211 RMP genes with 2Kb promoter region, and found that ARR1AT, CAATBOX1, and DOFCOREZM have slightly higher portion than the average estimated value of the frequency and the others have slightly lower values (Table S2); ARR1AT has 1.21 (=19.3/16); CAATBOX1, 1.14 (=18.3/16); and DOFCOREZM, 1.27 (=20.3/16). These results indicate that three CREs with increased frequecy might have significant roles in trasncriptinal regulation for early pollen development in rice.
Moreover, 11 CREs related to organ/tissue-preferred expression are listed in Table S7. Of them, anther- or pollen-specific CREs such as GTGANTG10 (GTGA), POLLEN1LELAT52 (AGAAA), SITEIIATCYTC (GNATATNC), 5659BOXLELAT5659 (GAAWTTGTGA) were identified. The results of scanning showed 10–17 copies of GTGANTG10 in the promoter regions of RMP1, RMP2, RMP4, RMP6-8, and 5 and 8 copies in RMP3 and RMP5 were found respectively. Similarly, 13–15 copies of POLLEN1LELAT52 were found in RMP2, RMP4, RMP5, and RMP8. The other promoters had two to eight copies. The SITEIIATCYTC element was found in RMP3 (4), RMP6 (5), and RMP8 (3) but not in the others. One copy of the 5659BOXLELAT5659 element was revealed in the RMP6 promoter. In addition, mesophyll- and meiocyte-preferred CACTFTPPCA1 (YACT) and four root-preferred P1BS (GNATATNC), OSE1-/OSE2-ROOTNODULE (AAAGAT), RHERPATEXPA7 (KCACGW), and ROOTMOTIFTAPOX1 (ATATT) were also identified. The function and frequency of these CREs are listed in Table S7 and Figure S7. Similary to those for highly abundant CREs among selected eight RMP genes, we analyzed the average frequency for ten CREs besides EBOXBNNAPA related to organ/tissue-preferred expression and compared the frequency with the estimated value (Table S2). As a result, we found that four of ten CREs have increased frequency than the estimation; POLLEN1LELAT52 showed 2.23 fold increase (=8.9/4); 5659BOXLELAT5659, 3462.61; OSE1-/OSE2-ROOTNODULE, 1.7 (=1.7/1); and ROOTMOTIFTAPOX1, 2.3 (=9.2/4). These results indicate that four CREs related to pollen- or root-preferred expression with higher fold increase might have more significant roles in transcriptional regulation for early pollen development. In addition, we expect that CREs related to pollen- or root-preferred expression might contribute more than those of highly abundant CREs.
Discussion
Majority of RMP genes are monocot unique genes
For comparative analysis of RMP-preferentially expressed genes between rice and Arabidopsis, we searched for orthologs between rice and Arabidopsis with a database of orthologous groups among rice, Arabidopsis, Brachypodium, maize, Populus, Vitis vinifera, and Sorghum bicolor from the Rice Genome Annotated Project (RGAP; http://rice.plantbiology.msu.edu/annotation_pseudo_ortho.shtml). Subsequently, 25 Arabidopsis orthologue for 410 rice RMP genes identified (Table S9). Majority of RMP genes are monocot unique genes, informing the significance of functional genomic studies for our candidate genes to understand male gamete development at very early stage. Among the 25 Arabidopsis orthologues, there is a report informing that two genes, AT1G31740 and AT5G20710, were preferentially expressed in microspore (Hrubá et al. 2005). However, we did not find any information on the other Arabidopsis orthologues which are active in Arabidopsis young microspores. In addition, we performed rice orthologue search for Arabidopsis DUO1/2/3 and GEX1/2, and analyzed expression patterns of identified the orthologues. There is no probe in Affymatrix array for DUO1 orthologue (LOC_04g46384); DUO3 orthologue (LOC_03g46950) was expressed in most of analyzed tissues/organs; and GEX1 (LOC_09g27040) and GEX2 (LOC_09g25650) were strongly expressed in mature pollen (Fig. S8). Because rice orthologues for DUO3, GEX1 and GEX2 did not exhibit microspore preferential expression in rice and so we did not include them in our candidate gene list. Similar observations were reported in Arabidopsis (Brownfield et al. 2009; Alandete-Saez et al. 2011; Alandete-Saez et al. 2008).
Role of abundant CREs in the promoters of RMP genes
Analyzing upstream promoter regions (725–2053 bp) originated from eight RMP genes using a Signal Scan revealed various CREs. Of them, the ten most abundant were found in all promoters (Table S6; Fig. S6). ACGTATERD1 (5′-ACGT-3′), the ACGT core sequence, has been established as a functional important cis-element that frequently regulates gene expression in synergy with other cis-elements. The ACGT sequence has been found in the ERD1 promoter, required for etiolation-induced increase in LUC activity (Simpson et al. 2003). In addition, ACGT elements are responsible for gene regulation in response to exogenous stress in rice (Qiu et al. 2008; Mehrotra et al. 2013). The binding element ARR1AT (5′-NGATT-3′, where N = G/A/C/T) has been found in both Arabidopsis and rice. ARR1 and ARR2 are cytokinin response regulators that function as transcriptional activators (Sakai et al. 2000). All promoter regions of RMP genes contain a high copy number of ARR1AT (Fig. S6). In addition, a promoter consensus sequence named CAATBOX1 (5′-CAAT-3′) is responsible for the tissue-specific promoter activity that was functionally characterized in the promoter region of the pea legumin gene LegA (Shirsat et al. 1989). The abundance of these cis-acting elements in our RMP promoters suggests that they are basic components of microspore-preferred expression. GATABOX (5′-GATA-3′) is known to be required for high level, light-regulated, and tissue-specific gene expression. GATA transcription factors are a group of DNA-binding proteins distinguished by a zinc finger motif, which have been implicated in light- and nitrate-dependent transcription control (Reyes et al. 2004). The identification of zinc finger transcription factor genes showing the highest expression level in Arabidopsis sperm cells suggests the potential of this regulatory element for pollen-preferred expression (Borges et al. 2008). MYBCORE (5′-CNGTTR-3′) is a binding site for two plant MYB proteins, AtMYB1 and AtMYB2, which were isolated from Arabidopsis. AtMYB2 is involved in the regulation of genes responsive to water stress (Abe et al. 2003). Moreover, MYB-type transcription factors showing high expression in sperm cells suggest the potential involvement of this cis-acting element in pollen development (Borges et al. 2008). In the promoter regions of all RMP genes, MYBCORE was detected from three to six copies (Fig. S6). DOFCOREZM (5′-AAAG-3′) is the target binding site of Dof proteins, which are specific DNA-binding proteins associated with the expression of multiple genes in plants including monocots and dicots (Yanagisawa and Schmidt 1999). In higher plants, many Dof genes have been identified; for example, more than 37 putative genes encoding Dof domain proteins in Arabidopsis and about 30 Dof in rice have been identified (Yanagisawa 2004). The role of the Dof domain protein has been investigated, and it has been shown to be involved in response to stress, light, hormones, seed germination, tissue-specific expression, and in the leaves (Yanagisawa 2000, 2004). Moreover, binding sites for Dof transcription factors have been recorded in the upstream sequences of GEX1 and GEX2, two genes showing sperm-specific expression in Arabidopsis (Engel et al. 2005). High copy number of the DOFCOREZM element were detected in all eight RMP promoters (Fig. S6). Based on the analysis of abundant CREs in RMP promoters, we expect the roles of these CREs to involve basic development, stress response, and homorne response, as well as male-gamete specific function.
Role of pollen-specific CREs identified in promoters of RMP genes
Interestingly, scanning results revealed some CREs involved in anther/pollen-specific expression. For example, GTGANTG10, with sequence 5′-GTGA-3′, was found in all eight promoters with high copy number (5 to 18 copies, Fig. S7). This motif was found in the tobacco promoter conferring late pollen gene g10, which shows homology to pectate lyase and tomato gene Lat56 (Rogers et al. 2001). The presence of a GTGA motif in the rice anther-specific plant lipid transfer protein (OsLTP6) gene promoter might increase GUS expression in rice transgenic plants (Liu et al. 2013). Moreover, the POLLEN1LELAT52 element with sequence 5′-AGAAA-3′ is a known motif required for anther/pollen-specific expression in the tomato Lat52 gene (Bate and Twell 1998). This motif was detected at high copy number in promoter regions of all eight genes (Fig. S7). SITEIIATCYTC (5′-GNATATNC-3′) is a TCP-domain of protein-binding elements in anther- and meristem-specific expression (Welchen and Gonzalez 2005). This element was found only in RMP3, 6 and 8 with 4, 5, and 3 copies, respectively. 5659BOXLELAT5659 (5′-GAAWTTGTGA, W = A/T) is a sequence motif shared between the tomato Lat56 and Lat59 promoters (Twell et al. 1991). This motif is unique for RMP6 with one copy that might be required for male gamete- or tapetum-expressed genes (Hobo et al. 2008) (Fig. S7).
Although many CREs were identified in the promoter regions, only a few CREs have been shown to be involved in the regulation of anther/pollen-specific gene expression. In addition to anther/pollen-preferred CREs, we found that other organ-preferred CREs have the potential for anther/pollen-preferred expression. CACTFTPPCA1 (5′-YACT, Y = T/C) is a tetra-nucleotide motif responsible for mesophyll-specific gene expression of the C4 phosphoenolpyruvate carboxylase gene in C4 plants (Gowik et al. 2004). Recently, this element was also found to be very abundant in the promoter regions of 50 genes preferentially expressed in Arabidopsis male meiocytes (Li et al. 2014). In this study, CACTFTPPCA1 was found in all eight RMP promoters at high copy number compared with other CREs (Table S7; Fig. S7). P1BS (5′-GNATATNC-3′) involved in root-preferred expression was found in RMP4 and RMP7 with 6 and 4 copies, respectively. This element was reported in both monocot and dicot promoters as required for controlling root expression and responsiveness to phosphate deprivation of the phosphate transporter gene (Schunmann et al. 2004; Sobkowiak et al. 2012). Similarly, OSE1-/OSE2-ROOTNODULE (5′-AAAGAT-3′ or 5′-CTCTT-3′) is one of the consensus sequence motifs of organ-specific elements (OSE) characteristic of the promoters activated in the infected cells of root nodules and in the arbuscule-containing cells of mycorrhizal roots (Fehlberg et al. 2005). This CRE was found in all RMP promoters. The RHERPATEXPA7 (5′-KCACGW-3′) detected only in RMP4 and RMP8 is a root hair-specific cis element (Kim et al. 2006). ROOTMOTIFTAPOX1 (5′-ATATT-3′) is a motif found in the rolD promoter of Agrobacterium rhizogenes. The rolD-gus genes were found to have a distinctive expression pattern in roots (Eulgem et al. 1999). It has been reported that genes showing expression in the roots under stress or normal condition also exhibited anther- or pollen-preferred expression patterns, indicating that root-specific CREs identified in the promoters of these eight RMP genes have the potential to regulate microspore-preferred expression.
DNA synthesis is important for microspore development in rice compared to that in the tapetum
In the anther, the tapetum is the innermost of the four sporophytic layers of the anther wall and is in direct contact with the developing male gametophyte. The tapetum contains all of the nutrients for the development and maturation of the microspore (Wang et al. 2015). Single uni-nucleated microspores are then released to produce two daughter cells, a vegetative cell (VC) and a generative cell (GC) at the bi-nucleated stage. The VC exits the cell cycle at the G1 phase, while the GC undergoes further division to produce two sperm cells at PMII (tri-nucleated stage). During the cell cycle, cells are proliferated through four stages, including G1, in which the cell grows and the nucleus has 2C DNA content (where C is a value indicating the amount of DNA contained within a haploid nucleus); S where DNA replicates from 2 to 4C; G2, which is a second growth period; and the M phase that represents mitosis in somatic or germinal cells. Several studies have reported that DNA is synthesized in microspores from G1 until G2 phase of the cell cycle using the immuno-labelling of incorporated Bromo-deoxyuridine in Brassica (Binarova et al. 1993; Pretova et al. 1993). The maximal enrichment of the nucleotide biosynthesis process in RMP genes indicates that rice microspores actively produce DNA during mitotic cell division. Genes to which this GO term can be applied are potential targets to elucidate the mitotic cell division process.
RMP genes are new targets to understand male-gamete development
More than 410 genes preferentially expressed in the uni-nucleated stage and early bi-nucleated stage have been revealed by analyzing meta-expression data, but only two genes were functionally characterized (Lee et al. 2004; Secco et al. 2010). Moreover, GO analysis confirmed that 285 genes (67.9 %) did not have assigned GO terms. On the other hand, 28 of 263 RPMCT genes have been functionally characterized, and 52 % of them have assigned GO terms, indicating that the understanding of RMP genes is quite limited compared to that of RPMCT genes. To date, many genes that are expressed in anther or pollen tissues have been identified in rice. Based on expression patterns, they were grouped as pollen mother cell or tapetum-expressed genes including TDR, Osg6B, RA8, RTS, UDT1, carbon starved anther (CSA), OsLTP6, MTR1, MEL1 and ZIP4 (Yokoi et al. 1997; Jeon et al. 1999; Jung et al. 2005; Luo et al. 2006; Li et al. 2006; Zhang et al. 2010a; Shen et al. 2012; Liu et al. 2013; Niu et al. 2013; Komiya et al. 2014) and late pollen-expressed genes including OsSCP1, OsSCP2, OsSCP3, RIP1, OSIPA, OSIPK, OsLPS1-9, OsLSP10 and OsLPS11 (Park et al. 2006; Han et al. 2006; Gupta et al. 2007; Swapna et al. 2011; Khurana et al. 2013; Oo et al. 2014; Nguyen et al. 2015). However, a few rice microspore-specific or preferred genes such as OsCP1 and OsPH1;2 have been functionally studied (Lee et al. 2004; Secco et al. 2010). Of these, OsCP1 showed the expression patterns in both tapetum and young microspore through previous study using GUS reporter system (Lee et al. 2004) but the GUS activity was majorly detected in microspores and vacuolated pollen grains of OsCP1 promoter trap heterozygous line. Our data also inform that high expression levels in young anthers at microspore stages are mostly contributed by the expression in microspores, but we cannot ignore the contribution by tapetal tissue (Fig. S8). In addition, GUS expression patterns in developing anthers of the heterozygous line were observed later than those of typical tepetum preferred genes such as TDR or UDT1 (Lee et al. 2004). In case of OsPH1;2, the functions were not investigated in term of male gamete development (Secco et al. 2010), and so this gene might be an useful target to study the relationship between phosphate transport and male gamete development.
We compared our list with mature pollen preferred genes and sperm cell preferred genes selected by Russell et al. (2012). Interestingly, we found that 70 genes out of 410 RMP were overlapped with in sperm cell preferred genes identified from recent transcriptome analysis (Russell et al. 2012) (Table S10). Alteration of expression profile was reported during pollen development, expression profile for uni-nucleated microspore was similar to that for bi-nucleate pollen (r = 0.82) but not for mature and GPG (r = 0.13) (Wei et al. 2010). However, some genes might be needed for proper pollen development and continuously expressed in mature pollen as we identified 70 RMP genes in sperm cells of mature pollen, suggesting that functions of some RMP genes continue until mature pollen and germinating pollen stages (Russell et al. 2012). This comparison suggests that most of RMP genes besides a small portion of them might have unique roles in early pollen development.
The development of microspore and tapetum is closely linked, and many rice tapetem preferred gene promoters have been used to study male-sterility and programed cell death. However, knowledge regarding microspore development seems to be limited. The clear understanding of genes expressed in the microspore stage will provide more options to manipulate target genes in microspores or pollen. Our study analyzed eight RMP promoters that have been well characterized in rice. They might be very useful for future studies and practical application in other plants as well as rice.
Materials and methods
Collection of microarray data and identification of RMP genes
We used 64 publicly available rice Affymetrix microarray data prepared from anthers and pollen in NCBI GEO to identify RMP genes. For the comparative transcriptome analyses between rice and Arabidopsis, we downloaded Arabidopsis Affymetrix microarray data series GSE5630, GSE5631, GSE5632, GSE5633, GSE6162, GSE6696, GSE12316, GSE17343, and GSE27281. Intensity values for these six series were first normalized with the Affy package in the R program and then log2-transformed. Affymetrix anatomical meta-expression data with the averaged normalized data were then used for further investigations, e.g., KMC analysis, heatmap construction, and identification of RMP genes. Based on the microarray data, we selected eight candidate genes that were highly expressed in uni-nucleated microspores, designating them as rice microspore-preferred gene 1 (RMP1, LOC_Os01g34920) through RMP8 (LOC_Os07g46950) (Fig. 4a). Whole sequence located in the 5’ upstream region of gene from start codon (ATG) was predicted as its promoter. Full length of promoter regions of RMP1–RMP8 genes were amplified and cloned into pDONR201 donor vector and destination vector pKGWFS7 as described by Oo et al. (2014) to create RMP promoter::GFP-GUS constructs (Fig. 4b). These constructs were transferred into Agrobacterium tumefaciens strain LBA4404 and used to generate transgenic rice plants. The extension primers used are listed in Table S8.
Plant transformation and selection of transgenic plants
Sterilized rice seeds of Dongjin cultivar were cultured on N6D plate medium for 5 days in a growth chamber under conditions of 32 °C and continuous light. Pre-cultured seeds were transformed via Agrobacterium tumefaciens (LBA4404) as described by Nguyen et al. (2015). Pre-cultured seeds were inoculated with Agrobacteria suspension harboring proRMP::GFP-GUS for 2 min with gentle inversion. The seeds were then blotted on sterilized filter papers and co-cultivated on N6D-AS medium for 3 days in the dark at 28 °C. After 3 days of co-cultivation, the seeds were rinsed thoroughly with autoclaved distilled water containing 200 mg/L cefotaxime and placed on N6D-CG medium containing 250 mg/L cefotaxime and 50 mg/L geneticin (Duchefa, Haarlem, The Netherlands) for 4 weeks. Regenerated shoots were selected on REM-CG medium supplement with 250 mg/L cefotaxime and 50 mg/L G-418. Plantlets were transferred to soil and grown in a GMO field.
PCR analysis of transgenic plants
In order to confirm whether transformed plants contained inserted T-DNA, we performed verification by PCR analysis using primers specific to the inserted gene (Table S8). PCR reactions were conducted in conditions of 94 °C/15 s, 55 °C/30 s, and 72 °C/5 m with 35 cycles. PCR products were separated on 1 % agarose gel with a 1 kb ladder marker (Elpis Bio, Daejeon, Korea).
GUS and DAPI staining
GUS activity in vegetative and reproductive organs was performed as described by Han et al. (2006) with minor modifications. Anthers were prefixed in fixative solution (Ethanol 3: Acetic acid 1) overnight at room temperature and subsequently rehydrated through ethanol series, 75, 55 and 35 %. They were then incubated at 60 °C for 1 h in DAPI staining solution supplemented with 20 % methanol and 1.0 µg/mL of DAPI. Pollen grains were briefly centrifuged, transferred to microscope slides, and covered with a cover glass. To determine various stages of development (UC, BC, TC and MP stages), pollen grains were viewed by light and UV epi-illumination using a Nikon ECLIPSE 80i microscope (Nikon, Melville, USA). Pollen images were captured using a ProgRes MFcool camera (Jenoptik, Jena, Germany) at 40× magnification. After GUS staining, images of seedlings and other samples such as leaf, stem, and root were produced using a ProgRes C3 camera (Jenoptik, Jena, Germany) at 0.65× magnification.
MapMan analysis
The MapMan program allows the grouping of genes into different functional categories and visualization of data through various diagrams (Jung and An 2012). To obtain functional classifications, we uploaded RGAP locus IDs for 410 RMP genes and 263 RPMCT genes to the MapMan tool kit. We then investigated the (overall) overview of metabolism, RNA-protein synthesis, and ubiquitin-dependent protein degradation pathway installed in that kit (Fig. 3; Figs. S2–S4). All data are detailed in Table S5.
Promoter sequence analysis
Promoter sequences were analyzed using the PLACE database program (Higo et al. 1999) to detect putative cis-regulatory elements involved in pollen-specific expression. The multiple sequence alignment program (ClustalW2) was also used to compare the promoter sequences.
RNA isolation and RT-PCR analysis
Total RNA was extracted with Tri Reagent (MRC Inc., Cincinnati, OH, USA). For synthesis of cDNA, 1 mg of total RNA was reacted with M-MLV reverse transcriptase (Promega, Madison, USA), 2.5 mM dNTP, and 10 ng of oligo dT. To evaluate the expression patterns of the eight RMP genes that showed GUS activity, we prepared samples from the shoots and roots of 7-day-old seedlings and anthers at four different developmental stages. All primers for RT-PCR are listed in Table S8. The ubiquitin 5 gene (Ubi5) was used as an internal control.
References
Abe H, Urao T, Ito T, Seki M, Shinozaki K, Yamaguchi-Shinozaki K (2003) Arabidopsis AtMYC2 (bHLH) and AtMYB2 (MYB) function as transcriptional activators in abscisic acid signaling. Plant Cell 15:63–78
Alandete-Saez M, Ron M, McCormick S (2008) GEX3, expressed in the male gametophyte and in the egg cell of Arabidopsis thaliana, is essential for micropylar pollen tube guidance and plays a role during early embryogenesis. Mol Plant 1:586–598
Alandete-Saez M, Ron M, Leiboff S, McCormick S (2011) Arabidopsis thaliana GEX1 has dual functions in gametophyte development and early embryogenesis. Plant J 68:620–632
Aya K, Ueguchi-Tanaka M, Kondo M, Hamada K, Yano K, Nishimura M, Matsuoka M (2009) Gibberellin modulates anther development in rice via the transcriptional regulation of GAMYB. Plant Cell 21:1453–1472
Aya K, Suzuki G, Suwabe K, Hobo T, Takahashi H, Shiono K, Yano K, Tsutsumi N, Nakazono M, Nagamura Y, Matsuoka M, Watanabe M (2011) Comprehensive network analysis of anther-expressed genes in rice by the combination of 33 laser microdissection and 143 spatiotemporal microarrays. PLoS One 6:e26162
Bae HK, Kang HG, Kim GJ, Eu HJ, Oh SA, Song JT, Chung IK, Eun MY, Park SK (2010) Transgenic rice plants carrying RNA interference constructs of AOS (allene oxide synthase) genes show severe male sterility. Plant Breed 129:647–651
Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Muertter RN, Edgar R (2009) NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res 37:D885–D890 (database issue)
Bate N, Twell D (1998) Functional architecture of a late pollen promoter: pollen-specific transcription is developmentally regulated by multiple stage-specific and co-dependent activator elements. Plant Mol Biol 37:859–869
Becker JD, Boavida LC, Carneiro J, Haury M, Feijó JA (2003) Transcriptional profiling of Arabidopsis tissues reveals the unique characteristics of the pollen transcriptome. Plant Physiol 133:713–725
Bedinger P (1992) The remarkable biology of pollen. Plant Cell 4:879–887
Binarova P, Straatman K, Hause B, Hause G, van Lammeren AAM (1993) Nuclear DNA synthesis during the induction of embryogenesis in cultured microspores and pollen of Brassica napus L. Theor Appl Genet 87:9–16
Borges F, Gomes G, Gardner R, Moreno N, McCormick S, Feijo JA, Becker JD (2008) Comparative transcriptomics of Arabidopsis sperm cells. Plant Physiol 148:1168–1181
Brownfield L, Hafidh S, Durbarry A, Khatab H, Sidorova A, Doerner P, Twell D (2009) Arabidopsis DUO POLLEN3 is a key regulator of male germline development and embryogenesis. Plant Cell 21:1940–1956
Cao P, Jung KH, Choi D, Hwang D, Zhu J, Ronald PC (2012) The rice oligonucleotide array database: an atlas of rice gene expression. Rice 5:17
Chandran AKN, Jung KH (2014) Resources for systems biology in rice. J Plant Biol 57:80–92
Chen G, Komatsuda T, Ma JF, Nawrath C, Pourkheirandish M, Tagiri A, Hu YG, Sameri M, Li X, Zhao X, Liu Y, Li C, Ma X, Wang A, Nair S, Wang N, Miyao A, Sakuma S, Yamaji N, Zheng X, Nevo E (2011) An ATP-binding cassette subfamily G full transporter is essential for the retention of leaf water in both wild barley and rice. Proc Natl Acad Sci USA 108:12354–12359
Cui R, Han J, Zhao S, Su K, Wu F, Du X, Xu Q, Chong K, Theissen G, Meng Z (2010) Functional conservation and diversification of class E floral homeotic genes in rice (Oryza sativa). Plant J 61:767–781
Deveshwar P, Bovill WD, Sharma R, Able JA, Kapoor S (2011) Analysis of anther transcriptomes to identify genes contributing to meiosis and male gametophyte development in rice. BMC Plant Biol 11:78
Engel ML, Holmes-Davis R, McCormick S (2005) Green sperm. Identification of male gamete promoters in Arabidopsis. Plant Physiol 138:2124–2133
Eulgem T, Rushton PJ, Schmelzer E, Hahlbrock K, Somssich IE (1999) Early nuclear events in plant defence signalling: rapid gene activation by WRKY transcription factors. EMBO J 18:4689–4699. doi:10.1093/emboj/18.17.4689
Fehlberg V, Vieweg MF, Dohmann EM, Hohnjec N, Puhler A, Perlick AM, Kuster H (2005) The promoter of the leghaemoglobin gene VfLb29: functional analysis and identification of modules necessary for its activation in the infected cells of root nodules and in the arbuscule-containing cells of mycorrhizal roots. J Exp Bot 56:799–806
Fujita M, Horiuchi Y, Ueda Y, Mizuta Y, Kubo T, Yano K, Yamaki S, Tsuda K, Nagata T, Niihama M, Kato H, Kikuchi S, Hamada K, Mochizuki T, Ishimizu T, Iwai H, Tsutsumi N, Kurata N (2010) Rice expression atlas in reproductive development. Plant Cell Physiol 51:2060–2081
Gowik U, Burscheidt J, Akyildiz M, Schlue U, Koczor M, Streubel M, Westhoff P (2004) cis-Regulatory elements for mesophyll-specific gene expression in the C4 plant Flaveria trinervia, the promoter of the C4 phosphoenolpyruvate carboxylase gene. Plant Cell 16:1077–1090
Gupta V, Khurana R, Tyagi AK (2007) Promoters of two anther-specific genes confer organ-specific gene expression in a stage-specific manner in transgenic systems. Plant Cell Rep 26:1919–1931
Han MJ, Jung KH, Yi G, Lee DY, An G (2006) Rice Immature Pollen 1 (RIP1) is a regulator of late pollen development. Plant Cell Physiol 47:1457–1472. doi:10.1093/pcp/pcl013
Higo K, Ugawa Y, Iwamoto M, Korenaga T (1999) Plant cis-acting regulatory DNA elements (PLACE) database. Nucl Aacids Res 27:297–300
Hobo T, Suwabe K, Aya K, Suzuki G, Yano K, Ishimizu T, Fujita M, Kikuchi S, Hamada K, Miyano M, Fujioka T, Kaneko F, Kazama T, Mizuta Y, Takahashi H, Shiono K, Nakazono M, Tsutsumi N, Nagamura Y, Kurata N, Watanabe M, Matsuoka M (2008) Various spatiotemporal expression profiles of anther-expressed genes in rice. Plant Cell Physiol 49:1417–1428
Honys D, Twell D (2003) Comparative analysis of the Arabidopsis pollen transcriptome. Plant Physiol 132:640–652
Honys D, Twell D (2004) Transcriptome analysis of haploid male gametophyte development in Arabidopsis. Genome Biol 5:R85
Hrubá P, Honys D, Twell D, Capková V, Tupý J (2005) Expression of beta-galactosidase and beta-xylosidase genes during microspore and pollen development. Planta 220:931–940
Jeon JS, Chung YY, Lee S, Yi GH, Oh BG, An G (1999) Isolation and characterization of an anther-specific gene, RA8, from rice (Oryza sativa L.) Plant Mol Biol 39:35–44
Jeong HJ, Jung KH (2015) Rice tissue-specific promoters and condition-dependent promoters for effective translational application. J Integr Plant Biol 57:913–924
Jung KH, An G (2012) Application of MapMan and RiceNet drives systematic analyses of the early heat stress transcriptome in rice seedlings. J Plant Biol 55:436–449
Jung KH, Han MJ, Lee YS, Kim YW, Hwang I, Kim MJ, Kim YK, Nahm BH, An G (2005) Rice undeveloped tapetum 1 is a major regulator of early tapetum development. Plant Cell 17:2705–2722
Kang HG, Jeon JS, Lee S ,An G (1998) Identification of class B and class C floral organ identity genes from rice plants. Plant Mol Biol 38:1021–1029
Khurana R, Kapoor S, Tyagi AK (2013) Spatial and temporal activity of upstream regulatory regions of rice anther-specific genes in transgenic rice and Arabidopsis. Transgenic Res 22:31–46
Kim DW, Lee SH, Choi SB, Won SK, Heo YK, Cho M, Park YI, Cho HT (2006) Functional conservation of a root hair cell-specific cis-element in angiosperms with different root hair distribution patterns. Plant Cell 18:2958–2970
Ko SS, Li MJ, Sun-Ben Ku M, Ho YC, Lin YJ, Chuang MH, Hsing HX, Lien YC, Yang HT, Chang HC, Chan MT (2014) The bHLH142 transcription factor coordinates with TDR1 to modulate the expression of EAT1 and regulate pollen development in rice. Plant Cell 26:2486–2504
Kobayashi K, Maekawa M, Miyao A, Hirochika H, Kyozuka J (2010) PANICLE PHYTOMER2 (PAP2), encoding a SEPALLATA subfamily MADS-box protein, positively controls spikelet meristem identity in rice. Plant Cell Physiol 51:47–57
Kobayashi K, Yasuno N, Sato Y, Yoda M, Yamazaki R, Kimizu M, Yoshida H, Nagamura Y, Kyozuka (2012) Inflorescence meristem identity in rice is specified by overlapping functions of three AP1/FUL-like MADS box genes and PAP2, a SEPALLATA MADS box gene. Plant Cell 24:1848–1859
Komiya R, Ohyanagi H, Niihama M, Watanabe T, Nakano M, Kurata N, Nonomura K (2014) Rice germline-specific Argonaute MEL1 protein binds to phasiRNAs generated from more than 700 lincRNAs. Plant J 78:385–397
Lee S, Jung KH, An G, Chung YY (2004) Isolation and characterization of a rice cysteine protease gene, OsCP1, using T-DNA gene-trap system. Plant Mol Biol 54:755–765
Li N, Zhang DS, Liu HS, Yin CS, Li XX, Liang WQ, Yuan Z, Xu B, Chu HW, Wang J, Wen TQ, Huang H, Luo D, Ma H, Zhan DB (2006) The rice tapetum degeneration retardation gene is required for tapetum degradation and anther development. Plant Cell 18:2999–3014
Li X, Yang Y, Yao J, Chen G, Li X, Zhang Q, Wu C (2009) FLEXIBLE CULM 1 encoding a cinnamyl-alcohol dehydrogenase controls culm mechanical strength in rice. Plant Mol Biol 69:685–689
Li H, Pinot F, Sauveplane V, Werck-Reichhart D, Diehl P, Schreiber L, Franke R, Zhang P, Chen L, Gao Y, Liang W, Zhang D (2010) Cytochrome P450 family member CYP704B2 catalyzes the Ñ -hydroxylation of fatty acids and is required for anther cutin biosynthesis and pollen exine formation in rice. Plant Cell 22:173–190
Li H, Liang W, Hu Y, Zhu L, Yin C, Xu J, Dreni L, Kater MM, Zhang D (2011a) Rice MADS6 interacts with the floral homeotic genes SUPERWOMAN1, MADS3, MADS58, MADS13, and DROOPING LEAF in specifying floral organ identities and meristem fate. Plant Cell 23:2536–2552
Li H, Yuan Z, Vizcay-Barrena G, Yang C, Liang W, Zong J, Wilson ZA, Zhang D (2011b) PERSISTENT TAPETAL CELL1 encodes a PHD-finger protein that is required for tapetal cell death and pollen development in rice. Plant Physiol 156:615–630
Li J, Yuan J, Li M (2014) Characterization of putative cis-regulatory elements in genes preferentially expressed in Arabidopsis male meiocytes. BioMed Res Int 708364
Liu X, Shangguan Y, Zhu J, Lu Y, Han B (2013) The rice OsLTP6 gene promoter directs anther-specific expression by a combination of positive and negative regulatory elements. Planta 238:845–857
Luo H, Lee JY, Hu Q, Nelson-Vasilchik K, Eitas TK, Lickwar C, Kausch AP, Chandlee JM, Hodges TK (2006) RTS, a rice anther-specific gene is required for male fertility and its promoter sequence directs tissue-specific gene expression in different plant species. Plant Mol Biol 62:397–408
Mehrotra R, Sethi S, Zutshi I, Bhalothia P, Mehrotra S (2013) Patterns and evolution of ACGT repeat cis-element landscape across four plant genomes. BMC Genomics 14:203
Nguyen TD, Oo MM, Moon S, Bae HK, Oh SA, Soh MS, Song JT, Kim JH, Jung KH, Park SK (2015) Expression analysis of two rice pollen-specific promoters using homologous and heterologous systems. Plant Biotechnol Rep 9:297–306
Niu N, Liang W, Yang X, Jin W, Wilson ZA, Hu J, Zhang D (2013) EAT1 promotes tapetal cell death by regulating aspartic proteases during male reproductive development in rice. Nat Commun 4:1445
Oldenhof MT, de Groot PFM, Visser JH, Schrauwen JAM, Wullems GJ (1996) Isolation and characterization of a microspore-specific gene from tobacco. Plant Mol Biol 31:213–225
Oo MM, Bae HK, Nguyen TD, Moon S, Oh SA, Kim JH, Soh MS, Song JT, Jung KH, Park SK (2014) Evaluation of rice promoters conferring pollen-specific expression in a heterologous system, Arabidopsis. Plant Reprod 27:47–58
Park JI, Hakozaki H, Endo M, Takada Y, Ito H, Uchida M, Okabe T, Watanabe M (2006) Molecular characterization of mature pollen-specific genes encoding novel small cysteine-rich proteins in rice (Oryza sativa L.). Plant Cell Rep 25:466–474
Pretova A, De Ruijter NCA, Van Lammeren AAM, Schel JHN (1993) Structural observations during androgenic microspore culture of the 4 cl genotype of Zea mays L. Euphytica 65:61–69
Qiu D, Xiao J, Xie W, Liu H, Li X, Xiong L, Wang S (2008) Rice gene network inferred from expression profiling of plants overexpressing OsWRKY13, a positive regulator of disease resistance. Mol Plant 1:538–551
Reyes JC, Muro-Pastor MI, Florencio FJ (2004) The GATA family of transcription factors in Arabidopsis and rice. Plant Physiol 134:1718–1732
Rogers HJ, Bate N, Combe J, Sullivan J, Sweetman J, Swan C, Lonsdale DM, Twell D (2001) Functional analysis of cis-regulatory elements within the promoter of the tobacco late pollen gene g10. Plant Mol Biol 45:577–585
Russell SD, Gou X, Wong CE, Wang X, Yuan T, Wei X, Bhalla PL, Singh MB (2012) Genomic profiling of rice sperm cell transcripts reveals conserved and distinct elements in the flowering plant male germ lineage. New Phytol 195(3):560–573
Sakai H, Aoyama T, Oka A (2000) Arabidopsis ARR1 and ARR2 response regulators operate as transcriptional activators. Plant J 24:703–711
Schunmann PH, Richardson AE, Vickers CE, Delhaize E (2004) Promoter analysis of the barley Pht1;1 phosphate transporter gene identifies regions controlling root expression and responsiveness to phosphate deprivation. Plant Physiol 136:4205–4214
Secco D, Baumann A, Poirier Y (2010) Characterization of the rice PHO1 gene family reveals a key role for OsPHO1;2 in phosphate homeostasis and the evolution of a distinct clade in dicotyledons. Plant Physiol 152:1693–1704
Shen Y, Tang D, Wang K, Wang M, Huang J, Luo W, Luo Q, Hong L, Li M, Cheng Z (2012). ZIP4 in homologous chromosome synapsis and crossover formation in rice meiosis. J Cell Sci 125:2581–91
Shi J, Tan H, Yu XH, Liu Y, Liang W, Ranathunge K, Franke RB, Schreiber L, Wang Y, Kai G, Shanklin J, Ma H, Zhang D (2011) Defective pollen wall is required for anther and microspore development in rice and encodes a fatty acyl carrier protein reductase. Plant Cell 23:2225–2246
Shirsat A, Wilford N, Croy R, Boulter D (1989) Sequences responsible for the tissue specific promoter activity of a pea legumin gene in tobacco. Mol Gen Genet 215:326–331
Simpson SD, Nakashima K, Narusaka Y, Seki M, Shinozaki K, Yamaguchi-Shinozaki K (2003) Two different novel cis-acting elements of erd1, a clpA homologous Arabidopsis gene function in induction by dehydration stress and dark-induced senescence. Plant J 33:259–270
Sobkowiak L, Bielewicz D, Malecka EM, Jakobsen I, Albrechtsen M, Szweykowska-Kulinska Z, Pacak A (2012) The role of the P1BS element containing promoter-driven genes in Pi transport and homeostasis in plants. Front. Plant Sci 3:58
Song X, Li P, Zhai J, Zhou M, Ma L, Liu B, Jeong DH, Nakano M, Cao S, Liu C, Chu C, Wang XJ, Green PJ, Meyers BC, Cao X (2012) Roles of DCL4 and DCL3b in rice phased small RNA biogenesis. Plant J 69:462–474
Suwabe K, Suzuki G, Takahashi H, Shiono K, Endo M, Yano K, Fujita M, Masuko H, Saito H, Fujioka T, Kaneko F, Kazama T, Mizuta Y, Kawagishi-Kobayashi M, Tsutsumi N, Kurata N, Nakazono M, Watanabe M (2008) Separated transcriptomes of male gametophyte and tapetum in rice: validity of a laser microdissection (LM) microarray. Plant Cell Physiol 49:1407–1416
Swapna L, Khurana R, Kumar SV, Tyagi AK, Rao KV (2011) Pollen-specific expression of Oryza sativa indica pollen allergen gene (OSIPA) promoter in rice and Arabidopsis transgenic systems. Mol Biotechnol 48:49–59
Takehisa H, Sato Y, Igarashi M, Abiko T, Antonio BA, Kamatsuki K, Minami H, Namiki N, Inukai Y, Nakazono M, Nagamura Y (2012) Genome-wide transcriptome dissection of the rice root system: implications for developmental and physiological functions. Plant J 69:126–140
Tan H, Liang W, Hu J, Zhang D (2012) MTR1 encodes a secretory fasciclin glycoprotein required for male reproductive development in rice. Dev Cell 22:1127–1137
Thangasamy S, Chen PW, Lai MH, Chen J, Jauh GY (2012) Rice LGD1 containing RNA binding activity affects growth and development through alternative promoters. Plant J 71:288–302
Thilmony R, Guttman M, Thomson JG, Blechl AE (2009) The LP2 leucine-rich repeat receptor kinase gene promoter directs organ-specific, light-responsive expression in transgenic rice. Plant Biotechnol J 7:867–882
Tseng IC, Hong CY, Yu SM, Ho TH (2013) Abscisic acid- and stress-induced highly proline-rich glycoproteins regulate root growth in rice. Plant Physiol 163:118–134
Tsuchiya T, Toriyama K, Nasrallah ME, Ejiri S (1992) Isolation of genes abundantly expressed in rice anthers at the microspore stage. Plant Mol Biol 20:1189–1193
Twell D, Yamaguchi J, Wing RA, Ushiba J, McCormick S (1991) Promoter analysis of genes that are coordinately expressed during pollen development reveals pollen-specific enhancer sequences and shared regulatory elements. Genes Dev 5:496–507
Wang E, Wang J, Zhu X, Hao W, Wang L, Li Q, Zhang L, He W, Lu B, Lin H, Ma H, Zhang G, He Z (2008) Control of rice grain-filling and yield by a gene with a potential signature of domestication. Nat Genet 40:1370–1374
Wang S, Zhang G, Song Q, Zhang Y, Li Z, Guo J et al (2015) Abnormal development of tapetum and microspores induced by chemical hybridization agent SQ-1 in wheat. PLoS ONE 10:e0119557
Wei LQ, Xu WY, Deng ZY, Su Z, Xue Y, Wang T (2010) Genome-scale analysis and comparison of gene expression profiles in developing and germinated pollen in Oryza sativa. BMC Genomics 11:338
Welchen E, Gonzalez DH (2005) Differential expression of the Arabidopsis cytochrome c genes Cytc-1 and Cytc-2. Evidence for the involvement of TCP-domain protein-binding elements in anther- and meristem-specific expression of the Cytc-1 gene. Plant Physiol 139:88–100
Yamamoto E, Yonemaru J-I, Yamamoto T, Yano M (2012) OGRO: the overview of functionally characterized genes in rice online database. Rice 5:1–10
Yanagisawa S (2000) Dof1 and Dof2 transcription factors are associated with expression of multiple genes involved in carbon metabolism in maize. Plant J 21:281–288
Yanagisawa S (2004) Dof domain proteins: plant-specific transcription factors associated with diverse phenomena unique to plants. Plant Cell Physiol 45:386–391
Yanagisawa S, Schmidt RJ (1999) Diversity and similarity among recognition sequences of Dof transcription factors. Plant J 17:209–214
Yang J, Zhao X, Cheng K, Du H, Ouyang Y, Chen J, Qiu S, Huang J, Jiang Y, Jiang L, Ding J, Wang J, Xu C, Li X, Zhang Q (2012) A killer-protector system regulates both hybrid sterility and segregation distortion in rice. Science 337:1336–1340
Yi J, Moon S, Lee YS, Zhu L, Liang W, Zhang D, Jung KH, An G (2016) Defective tapetum cell death 1 (DTC1) regulates ROS levels by binding to metallothionein during tapetum Degeneration. Plant Physiol 170:1611–1623
Yokoi S, Tsuchiya T, Toriyama K, Hinata K (1997) Tapetum-specific expression of the Osg6B promoter-β-glucuronidase gene in transgenic rice. Plant Cell Rep 16:363–367
Zhang H, Liang W, Yang X, Luo X, Jiang N, Ma H, Zhang D (2010a) Carbon starved anther encodes a MYB domain protein that regulates sugar partitioning required for rice pollen development. Plant Cell 22:672–689
Zhang J, Nallamilli BR, Mujahid H, Peng Z (2010b) OsMADS6 plays an essential role in endosperm nutrient accumulation and is subject to epigenetic regulation in rice (Oryza sativa). Plant J 64:604–617
Zhu L, Shi J, Zhao G, Zhang D, Liang W (2013) Post-meiotic Deficient Anther1 (PDA1) encodes an ABC transporter required for the development of anther cuticle and pollen exine in rice. J Plant Biol 56:59–68
Acknowledgments
We appreciate the support of Dr. Pamela Ronald and Dr. Peijian Cao in developing the meta-expression profiling database for rice. This work was supported by the Next-Generation BioGreen21 Program of the Rural Development Administration in the Republic of Korea (PJ01100401 to KHJ and PJ01194201 to SKP), and by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science and Technology (NRF-2013R1A1A2006040 to KHJ).
Author contributions
T.D.N., S.M., S.K.P., G.A., M.S.S., J.T.S., and K.H.J. designed the research. T.D.N., S.M., A.K.N.C., V.N.T.N., Y.S.G., and S.A.O. performed experiments. T.D.N. and S.M. analyzed data. T.D.N., S.M., S.A.O., S.K.P., and K.H.J. wrote the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Tien Dung Nguyen and Sunok Moon have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Nguyen, T.D., Moon, S., Nguyen, V.N.T. et al. Genome-wide identification and analysis of rice genes preferentially expressed in pollen at an early developmental stage. Plant Mol Biol 92, 71–88 (2016). https://doi.org/10.1007/s11103-016-0496-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-016-0496-1