
Abstract
The activity of the pituitary–adrenal axis can profoundly impact on body composition. This is dramatically seen in Cushing’s syndrome (CS) but changes in body composition are also implicated in depression and alcoholic pseudocushing’s. The pathophysiological mechanisms underlying these changes remain poorly understood. Changes to body composition in CS include increased fat mass, decreased bone mass, thinning of the skin and reduced lean mass. Why these tissues are affected so dramatically is unclear. Additionally, the change in body composition between individuals varies considerably for reasons which are only now becoming evident. This paper reviews the phenotypic changes with altered pituitary–adrenal axis activity and discusses the mechanisms involved. The primary focus is on adipose, bone, muscle and skin since the most dramatic changes are seen in these tissues.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
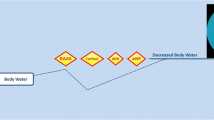
The activity of the pituitary–adrenal axis can have a profound impact on body composition. This is most dramatically seen in patients with Cushing’s syndrome (CS) but changes in body composition are also implicated in other conditions such as depression and alcoholic pseudocushing’s where pituitary–adrenal axis activity is abnormal. The pituitary–adrenal axis might also play a role in determining body composition in otherwise healthy individuals. Despite the changes in CS being well recognized the pathophysiological mechanisms that cause these changes remain poorly understood. Changes to body composition in CS include increased fat mass, decreased bone mass, thinning of the skin and reduced lean mass (Fig. 1) [1, 2].

Effects of glucocorticoids on body composition
Why these particular tissues are affected so dramatically by glucocorticoids remains unclear. Additionally, the relative changes in body composition between individuals can vary considerably for reasons which are only recently becoming evident. This paper will review the phenotypic changes in various tissues that occur with altered pituitary–adrenal axis activity and discuss the pathophysiological mechanisms involved. The primary focus will be on adipose, bone, muscle and skin since the most dramatic changes are seen in these tissues. These are also tissues which originate from a common developmental precursor and for which endogenous glucocorticoids are likely to play a critical role in differentiation.
Effects of excess pituitary–adrenal axis activity on adipose tissue
Most clinicians will be aware of the changes in fat distribution that occur in states of endogenous and exogenous glucocorticoid excess. These changes are predominantly characterized by redistribution of adipose tissue from peripheral to central parts of the body, with a greater increase in visceral fat [1] and loss of subcutaneous fat from the limbs. This redistribution of fat leads to the typical clinical features of a rounded ‘moon’ face, increased fat around the neck with prominence of the dorsal and supraclavicular fat pads ‘buffalo hump’. It has also been reported that there is an overall increase in total body fat mass [2] and a common issue in clinical practice is the discrimination of generalized adiposity due to simple obesity from that of CS. In studies that examined the differences between CS and simple obesity a small reduction in total lean tissue, but not an increase in total body fat, was seen in patients with CS relative to patients with obesity. A reduction in fat and lean tissue mass in the upper limbs were found to be the most discriminatory finding in CS patients compared to people with simple obesity [3]. Increased visceral fat accumulation (measured by waist circumference) has also been reported in patients with adrenal incidentaloma (who may also have ‘subclinical’ Cushing’s with low levels of cortisol excess). Even patients that have minimal increases in serum cortisol levels are susceptible to changes in adipose tissue distribution [4].
Hepatic steatosis (the abnormal deposition of fat within the liver) appears to be a common feature in CS. Using CT scanning in a large series of patients with active CS a prevalence of 20% was observed [5]. The presence of steatosis was correlated with impaired glucose tolerance and increased visceral fat mass. The extent to which the steatosis is related to the patient’s obesity, body fat distribution or altered hormonal profile is unclear. Patients with hepatic steatosis in the context of type II diabetes have been reported to have abnormally increased activity of the pituitary–adrenal axis compared with matched controls suggesting a possible causative role of high cortisol levels [6].
Mechanisms underlying changes in adipose tissue
The mechanisms by which high levels of cortisol alter adipose tissue distribution are unclear. Changes in adipocyte function, e.g. lipoprotein lipase (LPL) activity, preadipocyte differentiation and survival, and altered adipokine secretion have all been implicated as explanations of this phenomenon [7]. In patients with CS, LPL activity seems to be higher in retroperitoneal fat than in subcutaneous adipose tissue and this may be a reason for the tendency to central fat accumulation [2]. This had been previously reported in women with CS, where the enlargement of abdominal fat depots were at least partially due to elevated adipocyte LPL activity and low lipolytic activity in abdominal adipose but not femoral subcutaneous tissue [8]. Lipolysis was found more pronounced in the abdominal fat depots [9]. In humans adipocytes cultured in vitro, glucocorticoids increase LPL activity as well as LPL mRNA expression [10]. Pharmacological doses of glucocorticoids have been shown to stimulate lipolysis in vivo [11, 12].
Studies in vitro have confirmed the stimulatory effects of cortisol upon the differentiation of adipose stromal cells to mature adipocytes [13, 14]. Glucocorticoids can induce exaggerated adipocyte formation and hypertrophy [8, 15]. In the presence of insulin GCs promote preadipocyte differentiation and favour cellular lipid accumulation [16, 17].
The effects of glucocorticoids on fat mass may be indirect in vivo, acting via hormones released from fat or other tissues. For example, circulating levels of leptin and IL-1 receptor antagonist (IL-1ra) has been implicated in the abnormal fat distribution seen in CS. In patients with CS there is an association between total body fat and IL-1ra levels and enhanced levels of IL-1ra could contribute to the leptin resistance in CS [18]. The decrease in fat mass after successful treatment of CS is associated with a decrease in leptin and IL-1ra levels [18]. Leptin gene expression is enhanced by GC and insulin in adipose tissue cultures in vitro [19].
Expression and activity of the 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) enzyme has been implicated in the accumulation of visceral adipose tissue both in simple obesity and in CS [20]. This enzyme interconverts hormonally inactive cortisone with its active counterpart cortisol and in most settings appears to be a net generator of active glucocorticoid. 11β-HSD1 is highly expressed in both adipose stromal cells and mature adipocytes [14]. It was originally proposed that excessive activity of this enzyme in visceral adipose tissue would increase adipose tissue levels of glucocorticoids and thus drive central fat accumulation in some individuals in a similar manner to that seen in CS [21]. Transgenic over-expression of this enzyme in adipose tissue in the mouse appeared to support this hypothesis since this resulted in an increased accumulation of central fat due primarily to an increase in adipocyte size [22]. Studies in humans have been more controversial with some studies showing a positive association between obesity and 11β-HSD1 expression and activity [23–25] whereas others have suggested that obesity is associated with a reduction in 11β-HSD1 expression [26]. The reasons for these apparently contradictory results are currently unclear but might be explained by dynamic changes in 11β-HSD1 activity and expression across differentiation. Although 11β-HSD1 activity is usually considered to be an activator of glucocorticoids preadipocytes have been reported to have a predominantly inactivating capacity when first isolated. This capacity to switch from an inactiving to an activating enzyme appears due to variable expression of a cofactor generating enzyme that is needed for glucocorticoid generation [27].
Another mechanism invoked to explain the preferential expansion of visceral adipose tissue relative to peripheral fat is the differential expression or sensitivity of the glucocorticoid receptor (GR). The GR is expressed in human adipose tissue—initial reports suggested a density higher in visceral than in subcutaneous adipose tissue [28, 29] but more recent studies have refuted this [30]. Although genetic polymorphisms may result in altered HPA axis activity and altered abdominal fat accumulation [31] the relevance of these genetic variants in explaining the phenotypic variation in CS has not been explored.
Role of pituitary adrenal activity in idiopathic obesity
Modest increases in cortisol secretion have been linked to increased body fat and rate of fat accumulation [32, 33]. In children, even after urinary GC excretion was corrected for body surface area (BSA), a significant part of the variability in body fatness could be explained by GC excretion. GC excretion adjusted for BSA has been reported to predict BMI during growth [33]. The association of urinary cortisol output appears stronger with central obesity [34]. The direction of causality however is not clear. In obesity, peripheral cortisol production is suggested to be increased [35], and the HPA-axis is abnormally regulated [36]. Whether increased cortisol production causes obesity or increased obesity affects cortisol production is still unclear.
Despite this association of cortisol production with obesity, there is a weak, negative association of circulating cortisol concentrations and various measures of adiposity including weight, BMI, waist-hip ratio and waist circumference in community-dwelling men [37]. Additionally, changes in cortisol levels and adiposity measures are also negatively associated. In obese patients, there is elevated cortisol secretion, but circulating cortisol levels are typically lower among overweight or obese people [37]. An altered pattern of cortisol secretion with increased amplitude of secretory bursts has been reported in obese subjects despite normal mean circulating levels. An association of obesity with relative insensitivity to glucocorticoid feedback has also been suggested [38].
Cortisol production rates (CPR) and plasma free (non-protein bound) cortisol levels have been examined in individuals of various body weights and ages. Increasing body weight is associated with increasing CPR, which is balanced by enhanced cortisol clearance, resulting in daily plasma free cortisol levels that are invariant to increasing body size. These data suggest that obesity is not characterized by a dysregulation of the HPA axis. CPR/BSA is strongly positively associated with 24 h plasma free cortisol levels suggesting that activation of the HPA axis is a determinant of increased central fat distribution [39].
In summary it appears most likely that idiopathic obesity is associated with increased cortisol production but not the features of CS. This is probably because the increased production of cortisol is matched by an equivalently increased cortisol metabolism and clearance.
Effects of excess pituitary-adrenal axis activity on bone
GC excess has profound effects on the skeleton leading to loss of bone mineral density (BMD) and an increased risk of fracture. The prevalence of osteoporosis (defined as a dual energy absortiometry (DXA) T-score <−2.5) is very high among patients with CS being present in 55% of women with CS [40]. Fractures were reported in 19–50% of CS patients [40, 41] with an overrepresentation of fractures of vertebrae and ribs [42, 43].
Epidemiological studies have examined the relationship between fracture risk and exposure to therapeutic oral glucocorticoids. The risk of developing fractures in patients under treatment with glucocorticoids is increased substantially at all skeleton sites but this was most evident at the spine with at least a fivefold increased risk of fracture [44]. After stopping oral glucocorticoids fracture risk fell rapidly back towards baseline. In this time scale it is unlikely that BMD would return to normal so this suggests that fracture risk with glucocorticoids is in part independent of BMD [45].
On average, BMD is decreased in patients with CS measured by either DXA or quantitative ultrasound [46]. This deficit in BMD is most striking when adjusted for BMI since simple obesity is associated with an increase in BMD [3]. Although BMD is often decreased throughout the entire skeleton [47] bone loss tends to be higher at sites rich in trabecular bone such as the lumbar spine [43, 46, 48–51]. The loss of BMD during exogenous glucocorticoid treatment is often rapid in the first 6–12 months of treatment and then continues at a slower rate [52].
The aetiology of the CS may play a role in the type and extent of bone loss. Bone density was reported to be twofold lower in patients with adrenal CS than in patients with Cushing’s disease even when the differences in age at presentation were taken into account. BMI was higher in women with pituitary disease, which could explain the results [40]. However this difference was not found in other studies [48, 53]. Reasons for these differences may include differences in DHEAS levels between adrenal and pituitary CS and the increased risk of hypogonadism in patients with CS of pituitary origin.
Despite a rapid reduction in fracture risk in patients who cease to use oral glucocorticoids, BMD changes very little in the first 6 months after cure, despite a rapid restoration of bone forming capacity. Thereafter however a remarkable improvement of BMD can be observed in almost all patients [54]. A study on a pair of identical twins, one of whom developed Cushing’s in adolescence demonstrated a dramatic difference in BMD initially that eventually normalized but only many years after successful cure [55]. Despite this improvement in BMD patients cured from CS have an increased prevalence of spine damage, probably as a result of fractures that occurred during the active CS [56]. As with changes in fat mass subtle glucocorticoid excess seen in patients with adrenal incidentalomas have also been linked to bone loss and altered bone turnover markers [46]. An unexpectedly high prevalence of subclinical cortisol excess has been found in patients with idiopathic osteoporosis further suggesting a role for endogenous glucocorticoids in bone loss and fracture [57].
Mechanisms of bone loss
Biochemical markers of bone turnover indicate that CS is associated with a profound decrease in bone formation. Osteocalcin, alkaline phosphatase and PICP (N terminal propeptide of type 1 collagen), are all significantly decreased [49, 58, 59]. Osteocalcin appears to be the most sensitive marker of the effects of endogenous hypercortisolism [60]. The role of bone resorption is controversial but most studies find that resorption is either increased or inappropriately normal for the degree of reduced formation. The urinary cross-linked N-telopeptides of type 1 collagen, a marker of bone resorption was reported to be high in CS, suggesting increased resorption [49]. No differences were found in bone resoprtion parameters between CS and adrenal incidentaloma [60].
Glucocorticoids probably exert their effects on bone through several mechanisms. Glucocorticoid excess reduces sex steroid levels, induces loss of muscle mass and strength and encourages secondary hyperparathyroidism due to reduced calcium absorption and decreased calcium reabsorption in the kidney [43, 51, 61]. Most CS patients have a degree of hypogonadotropic hypogonadism, either directly because of a mass effect in pituitary CS or via inhibition of gonadotropin secretion by glucocorticoids. Women with CS and hypogonadism have significantly decreased BMD in the femoral neck compared to those who are eugonadal. In premenopausal women with a recent diagnosis of CS who continue to menstuate BMD is not reduced compared to healthy controls [62]. Despite these mechanisms the dominant action of glucocorticoid excess on bone appears to be direct effects on bone cells.
Glucocorticoids have complex actions on osteoblasts, the cells responsible for bone formation. They appear to be important in the differentiation of osteoblasts from their uncommitted precursors but also reduce the proliferation of osteoblasts, impair the function of terminally differentiated osteoblasts and induce apoptosis of osteoblasts and osteocytes. The net consequence of glucocorticoid excess is a decreased number of mature osteoblasts [63–65].
Why glucocorticoids appear to be essential for osteoblast differentiation but in excess have detrimental actions is unclear. Recent insights have come from studies examining local generation of glucocorticoids by 11β-HSD1. During differentiation this enzyme is switched on and its activity is able to induce osteoblast differentiation [66]. Importantly the enzyme activity decreases in mature osteoblasts and is only increased again during inflammatory states [67]. It is possible that the presence of high doses of glucocorticoids throughout osteoblast differentiation impairs this coordinated pattern of differentiation.
Clinical experience suggests that some people are relatively resistant to the effects of glucocorticoids on bone whereas some patients with CS present with marked bone loss and fractures without the accompanying features of glucocorticoid excess. The only marker which has been reported to account for such differences is 11β-HSD1. The activity of this enzyme, measured in the urine, predicts the change in bone formation markers in response to prednisolone in healthy males suggesting it is an important mediator of individual susceptibility to glucocorticoid-induced osteoporosis [58].
Effects of excess pituitary–adrenal axis activity on muscle
Proximal muscle weakness is seen in 56–90% of cases of CS [68] and combined with thin skin, easy bruising and osteoporosis is one of the discriminating features to clinically distinguish true CS from pseudo-CS. Harvey Cushing noted muscle weakness in his original patients with Cushing’s disease, but, it was Muller and Kugelberg [69] who performed the first systematic study of the myopathy associated with CS. Several clinical studies have subsequently examined muscle function, histology and metabolism in patients with CS. The effects of GC on muscle are related to dose, type of steroid (9α-fluorinated steroids such as dexamethasone being particularly harmful to muscle) [70], duration of exposure and muscle fibre type [71].
Muscle fibres can be divided into slow twitch oxidative (type I), fast twitch oxidative (type IIa) and fast twitch glycolytic (type IIb) fibres. Type I fibres contain high levels of slow isoform contractile proteins, high volumes of mitochondria, high levels of myoglobin and capillary densities and high oxidative enzyme capacity. Type IIa fibres are characterized by fast contraction with high oxidative capacity and type IIb fibres are characterized by low volumes of mitochondria, high glycolytic enzyme activity, high myosin ATPase activity, increased rate of contraction and low fatigue resistance.
Type II fibre atrophy is the classically described histological abnormality reported in glucocorticoid mediated myopathy [72, 73] however other authors have also described a decrease in type I fibres and an increase in type II fibre number in CS but many of these type II fibres were atrophic [8]. This predominant type II fibre myopathy and atrophy is also seen in hypothyroidism (also conversion of type II fibre to type I fibre) and thyrotoxicosis (also conversion of type I fibre to type II fibre) [74]. In CS a reduced type II mean fibre area and plasma creatinine kinase activity, a myopathic electromyogram and a raised 24 h urinary 3-methylhistidine/creatinine ratio were found [72]. There are a number of ultrastructural changes seen in the muscles of people with myopathy complicating CS [75], these include pronounced mitochondrial damage, with thickening and deep invaginations of the sarcolemmal basement membrane and thickening of the basement membrane of capillaries. Muscle fibres also showed marked disarray and wide interfibrillar spaces containing large vacuoles which represented degenerated mitochondria [75]. Khaleeli et al. [72] reported a sarcolemmal accumulation of glycogen and mitochondria with small deposits of lipofuscin pigment.
Khaleeli et al. [72] also reported that muscle histological abnormalities were more pronounced in the group of patients taking exogenous glucocorticoids for inflammatory conditions such as dermatomyositis. This could be explained by the high cumulative exposure of prednisone (8 mg/day for a mean of 10 years), but an alternative possibility is an induction of 11β-HSD1 which is present and biologically active in human skeletal muscle [76, 77]. 11β-HSD1 enzyme activity (and thus increased tissue conversion of inactive cortisone to active cortisol) might be increased in muscle in inflammatory conditions as shown in fat and bone [67, 78].
Mechanisms underlying glucocorticoid mediated myopathy
Many factors have been described as being important in the development of glucocorticoid-induced myopathy including abnormalities in protein metabolism, myostatin expression, collagen metabolism, mitochondrial function and myosin heavy chain isoform expression.
Glucocorticoid excess increases the rate of whole body proteolysis even during short-term treatment. Glucocorticoids inhibit protein synthesis and amino acid transport into muscle. Leucine concentration in plasma, metabolic clearance rate, turnover and incorporation into protein were all significantly reduced in patients with CS compared with controls. Leucine oxidation rate was similar in both groups suggesting that muscle wasting in CS is primarily due to reduced protein synthesis [79]. Glucocorticoid excess inhibits protein synthesis and stimulates protein degradation in skeletal muscle and is an important factor in the development of muscle atrophy in various catabolic conditions. Glucocorticoid stimulated muscle protein breakdown is primarily caused by ubiquitin-proteasome-dependant proteolysis although calcium-dependent protein degradation may also be involved [80]. Glucocorticoids have also been shown to regulate the concentrations of mRNAs encoding some proteases in muscle cells.
Myostatin (a member of the transforming growth factor-β family) is a potent inhibitor of muscle growth and disruption of the myostatin gene in mice resulted in increased muscle mass due to both muscle hypertrophy and hyperplasia. In rats treated with dexamethasone to induce muscle atrophy, myostatin mRNA expression and protein concentrations were significantly increased [81]. Subsequently myostatin knock out mice were shown to resist glucocorticoid induced muscle atrophy, in association with increased IGF-I and II mRNA expression [82].
Glucocorticoid induced muscle wasting has been found to be greater in aged compared to young rats. They also have more rapid wasting and slower recovery of muscle function following glucocorticoid withdrawal [83]. Protein synthesis was more greatly depressed in old rats and this was associated with activation of the ubiquitin-proteasome proteolytic pathway. This change in protein metabolism might also be explained by leucine resistance on muscle protein synthesis in older rats and the time to recovery of leucine responsiveness following dexamethasone withdrawal was significantly slower in old rats [84]. In rodent models there also appears to be a sexual dimorphism with glucocorticoids leading to increased levels of MyoD1, myogenin and myf-5 in male rats but a decrease in myf-6 in female rats [85].
Glucocorticoids also have a potent effect on myosin heavy chain levels. Glucocorticoid treatment in rats resulted in degradation of muscle contractile proteins and the myosin heavy chain (MyHC) IIB isoform and a decrease in MyHC types II, IIA and IIB synthesis rate [86]. Mitochondrial function is also altered by glucocorticoid therapy. In a micro-array study of 501 human mitochondrial-related genes several were altered by glucocorticoids, the most significantly upregulated gene being monoamine oxidase A. Monoamine oxidase A metabolizes catecholamines and dietary amines and an upregulation in glucocortcoid treated cells leads to an increase in MAO-A mediated hydrogen peroxide production which in turn could lead to muscle cell damage [87].
After correction of hypercortisolism in patients with Cushing’s disease there is an increase in overall muscle and type II mean fibre area [73]. The findings suggest that the increase in muscle mass which may follow surgery is due to an increase in individual cell size. In the early recovery from hypercortisolism the observed decrease of body weight is attributed to a loss of body fat, but the low body cell mass does not normalize within the first 6 months after successful pituitary surgery [88].
Chronic glucocorticoid excess is associated with a number of changes to GH secretion including a decrease in 24 h GH concentrations, decrease peak GH height and decrease peak GH area but normal GH pulse frequency. Despite this patients with CS often have a normal IGF-I concentration (which may be explained by increased tissue sensitivity to GH or by a decrease in GH binding protein) [89]. The deleterious effects on protein metabolism of pharmacologic doses of glucocorticoids administered during a short period, can be prevented by the concomitant administration of GH in normal volunteers (measuring nitrogen balance and isotope dilution techniques) [90]. IGF-I gene transfer into glucocorticoid treated rats also prevented glucocorticoid induced muscle atrophy [91]. IGF-I has been shown to inhibit many proteolytic pathways including lysosomal, protease dependant and calpain dependant proteolysis and regulate the ubiquitin system. These findings provide a potential rationale for the treatment of patients who are recovering from CS with GH and there is some data that this improves the recovery of muscle strength [92].
Effects of excess pituitary–adrenal axis activity on skin
Another discriminating feature of CS is a change in composition and appearance of skin. GC excess results in skin atrophy and fragility leading to striae, easy burning and poor healing [93]. Some skin features depend on the aetiology of CS. In women with pituitary CS, ACTH stimulation of androgen secretion results in hirsutism, male pattern alopecia and acne, and in ectopic ACTH syndrome excessive pigmentation can be seen [93]. In a study involving children and adolescents, the skin manifestations of CS were: purple striae (77%), hirsutism (64%), acne (58%), acanthosis nigricans (28%), ecchymoses (28%), hyperpigmentation (17%) and fungal infections (11%). No correlation between glucocorticoid levels and the severity of skin manifestations were seen. The symptoms and signs decreased dramatically postoperatively and progressively disappeared within a year with the exception of light coloured striae [94].
Mechanisms underlying skin changes in CS
Glucocorticoids increase the catabolism of proteinaceous skin components such as collagen causing skin atrophy [93]. The microscopic findings on skin biopsy are non-specific, with some signs of angiopathy and vasculitis [95]. Abnormalities of dermal collagen are seen with no evidence of elastin destruction [96]. The cell type most likely to be affected is the skin fibroblast. This cell type is important in collagen production and tissue repair. Interestingly prior exposure to elevated glucocorticoid concentrations is not associated with persistent adverse effects on skin fibroblasts and may also have a beneficial outcome in some aspects of cell physiology including increased proliferative capacity in vitro [97].
Mechanisms underlying variability in phenotype in CS
Although the changes in body composition with CS are sometimes obvious then can often be very subtle. Additionally features may be pronounced in some tissues and absent in others even within the same patient. There is also a tendency for children and adolescents with CS to present differently to adults. Some of these differences may relate to variation in levels and dynamics of cortisol secretion, the duration of disease and the modifying effects of excess or deficiency of related pituitary or adrenal hormones. An additional possibility is that there is between and within individual variability in tissue sensitivity to glucocorticoids. Explanations for these differences in sensitivity include differences in GR sensitivity and intracellular metabolism of glucocorticoids.
Mutations in the GR that abolish activity are incompatible with life. Attention has therefore been on subtle genetic polymorphisms with the GR gene or its promoter. Several polymorphisms have been demonstrated that can result in either increased or decreased sensitivity to glucocorticoids [98]. Interestingly the phenotype of patients with these mutations is not marked and is certainly not similar to CS. This is almost certainly because the pituitary–adrenal axis is able to control the level of cortisol in the circulation such that the overall stimulation of GR is similar to that seen in people with normal GR variants. The subtle changes in body composition that are seen tend to support a role of glucocorticoids in body composition with a slight increase in visceral fat, and reductions in BMD and lean mass seen in patients with receptor variants that make the GR more sensitive to glucocorticoids.
Another example of differential sensitivity to glucocorticoids that may affect the expression of the features of CS is the 11β-HSD enzyme system. Expression of 11β-HSD1 is likely to amplify the local tissue of glucocorticoids so it is likely that tissues with high 11β-HSD1 will be preferentially affected in states of glucocorticoids excess. Conversely individuals with low levels of 11β-HSD1 may have partial protection against developing the features of CS. A case has been reported of a woman who developed biochemically proven CS with very few changes in body composition [99]. Biochemical testing after successful cure demonstrated a relatively low total body activity of 11β-HSD1 as assessed by urinary steroid metabolite measurements. A low expression of 11β-HSD1 will have endocrine consequences with the reduced half-life of cortisol potentially being able to offset the increased amount of cortisol produced in CS. 11β-HSD1 expression has now been described in adipose, bone, muscle and skin tissue thus a lack of amplification of glucocorticoid action in these tissues may account for a relative resistance to glucocorticoid induced changes in these tissues.
Developmental explanation of changes in body composition in CS
The changes in body composition seen in CS are primarily due to the abnormal proliferation, differentiation or function of cells that arise from a common precursor. Adipocytes, osteoblasts, myoblasts and fibroblasts all derive from mesenchymal stem cells (as do chondrocytes, themselves dramatically affected by glucocorticoids in CS) (Fig. 2)

Effects of glucocorticoids on cells derived from mesenchymal stem cells
[100]. Glucocorticoids have an important role in the differentiation of mesenchymal stem cells along these different pathways and are important for differentiation in vitro. Glucocorticoids appear to have a permissive role in differentiation with tissue specific factors determining along which pathway cells differentiate [101]. How much these cells rely on circulating glucocorticoids or locally generated glucocorticoids for differentiation in vivo remains unexplored. Additionally the embryological origins of mesenchymal stem cells in the body axis are distinct to those of the limbs and this may have implications for regional disparities in the effects of glucocorticoids on body composition. Interference with the developmental pathway of meschymal stem cells may thus explain why changes in adipose, bone, muscle and skin tissue are the prominent findings in states of systemic glucocorticoid excess.
Conclusions
This review illustrates that the pituitary–adrenal axis has an important role in the determination of body composition in normal individuals and in patients with CS. The reasons why some tissues are more affected by glucocorticoids are becoming clearer and are probably related to a role for glucocorticoids in the normal differentiation of these tissues. An improved understanding of the relationship between pituitary–adrenal axis activity and body composition could lead to better approaches to limit the adverse effects of glucocorticoids and could lead to improved diagnostic approaches in CS.
References
Mayo-Smith W, Hayes CW, Biller BM, Klibanski A, Rosenthal H, Rosenthal DI (1989) Body fat distribution measured with CT: correlations in healthy subjects, patients with anorexia nervosa, and patients with Cushing syndrome. Radiology 170:515–518. Medline
Burt MG, Gibney J, Ho KK (2006) Characterization of the metabolic phenotypes of Cushing’s syndrome and growth hormone deficiency: a study of body composition and energy metabolism. Clin Endocrinol (Oxf) 64:436–443. Medline. doi:10.1111/j.1365–2265.2006.02488.x
Wajchenberg BL, Bosco A, Marone MM et al (1995) Estimation of body fat and lean tissue distribution by dual energy X-ray absorptiometry and abdominal body fat evaluation by computed tomography in Cushing’s disease. J Clin Endocrinol Metab 80:2791–2794. Medline. doi:10.1210/jc.80.9.2791
Garrapa GG, Pantanetti P, Arnaldi G, Mantero F, Faloia E (2001) Body composition and metabolic features in women with adrenal incidentaloma or Cushing’s syndrome. J Clin Endocrinol Metab 86:5301–5306. Medline. doi:10.1210/jc.86.11.5301
Rockall AG, Sohaib SA, Evans D et al (2003) Hepatic steatosis in Cushing’s syndrome: a radiological assessment using computed tomography. Eur J Endocrinol 149:543–548. Medline. doi:10.1530/eje.0.1490543
Zoppini G, Targher G, Venturi C, Zamboni C, Muggeo M (2004) Relationship of nonalcoholic hepatic steatosis to overnight low-dose dexamethasone suppression test in obese individuals. Clin Endocrinol (Oxf) 61:711–715. Medline. doi:10.1111/j.1365–2265.2004.02154.x
Paulsen SK, Pedersen SB, Fisker S, Richelsen B (2007) 11Beta-HSD type 1 expression in human adipose tissue: impact of gender, obesity, and fat localization. Obesity (Silver Spring) 15:1954–1960. Medline
Rebuffe-Scrive M, Krotkiewski M, Elfverson J, Bjorntorp P (1988) Muscle and adipose tissue morphology and metabolism in Cushing’s syndrome. J Clin Endocrinol Metab 67:1122–1128. Medline
Gravholt CH, Dall R, Christiansen JS, Moller N, Schmitz O (2002) Preferential stimulation of abdominal subcutaneous lipolysis after prednisolone exposure in humans. Obes Res 10:774–781. Medline
Fried SK, Russell CD, Grauso NL, Brolin RE (1993) Lipoprotein lipase regulation by insulin and glucocorticoid in subcutaneous and omental adipose tissues of obese women and men. J Clin Invest 92:2191–2198. Medline
Tanaka H, Ichikawa Y, Akama H, Homma M (1989) In vivo responsiveness to glucocorticoid correlated with glucocorticoid receptor content in peripheral blood leukocytes in normal humans. Acta Endocrinol (Copenh) 121:470–476. Medline
Issekutz B Jr, Borkow I (1972) Effect of catecholamines and dibutyryl-cyclic-AMP on glucose turnover, plasma free fatty acids, and insulin in dogs treated with methylprednisolone. Can J Physiol Pharmacol 50:999–1006. Medline
Ahdjoudj S, Lasmoles F, Oyajobi BO, Lomri A, Delannoy P, Marie PJ (2001) Reciprocal control of osteoblast/chondroblast and osteoblast/adipocyte differentiation of multipotential clonal human marrow stromal F/STRO-1(+) cells. J Cell Biochem 81:23–38. Medline. doi:10.1002/1097-4644(20010401)81:1≤23::AID-JCB1021≥3.0.CO;2-H
Bujalska IJ, Kumar S, Hewison M, Stewart PM (1999) Differentiation of adipose stromal cells: the roles of glucocorticoids and 11beta-hydroxysteroid dehydrogenase. Endocrinology 140:3188–3196. Medline. doi:10.1210/en.140.7.3188
MacDougald OA, Lane MD (1995) Transcriptional regulation of gene expression during adipocyte differentiation. Annu Rev Biochem 64:345–373. Medline. doi:10.1146/annurev.bi.64.070195.002021
Ottosson M, Lonnroth P, Bjorntorp P, Eden S (2000) Effects of cortisol and growth hormone on lipolysis in human adipose tissue. J Clin Endocrinol Metab 85:799–803. Medline. doi:10.1210/jc.85.2.799
Mancini T, Doga M, Mazziotti G, Giustina A (2004) Cushing’s syndrome and bone. Pituitary 7:249–252. Medline. doi:10.1007/s11102-005-1051-2
Ueland T, Kristo C, Godang K, Aukrust P, Bollerslev J (2003) Interleukin-1 receptor antagonist is associated with fat distribution in endogenous Cushing’s syndrome: a longitudinal study. J Clin Endocrinol Metab 88:1492–1496. Medline. doi:10.1210/jc.2002-021030
Halleux CM, Servais I, Reul BA, Detry R, Brichard SM (1998) Multihormonal control of ob gene expression and leptin secretion from cultured human visceral adipose tissue: increased responsiveness to glucocorticoids in obesity. J Clin Endocrinol Metab 83:902–910. Medline. doi:10.1210/jc.83.3.902
Stewart PM, Tomlinson JW (2002) Cortisol, 11 beta-hydroxysteroid dehydrogenase type 1 and central obesity. Trends Endocrinol Metab 13:94–96. Medline. doi:10.1016/S1043-2760(02)00566-0
Bujalska IJ, Kumar S, Stewart PM (1997) Does central obesity reflect “Cushing’s disease of the omentum”? Lancet. 349:1210–1213. Medline. doi:10.1016/S0140-6736(96)11222-8
Masuzaki H, Paterson J, Shinyama H et al (2001) A transgenic model of visceral obesity and the metabolic syndrome. Science 294:2166–2170. Medline. doi:10.1126/science.1066285
Rask E, Walker BR, Soderberg S et al (2002) Tissue-specific changes in peripheral cortisol metabolism in obese women: increased adipose 11beta-hydroxysteroid dehydrogenase type 1 activity. J Clin Endocrinol Metab 87:3330–3336. Medline. doi:10.1210/jc.87.7.3330
Engeli S, Bohnke J, Feldpausch M et al (2004) Regulation of 11beta-HSD genes in human adipose tissue: influence of central obesity and weight loss. Obes Res 12:9–17. Medline
Sandeep TC, Andrew R, Homer NZ, Andrews RC, Smith K, Walker BR (2005) Increased in vivo regeneration of cortisol in adipose tissue in human obesity and effects of the 11beta-hydroxysteroid dehydrogenase type 1 inhibitor carbenoxolone. Diabetes 54:872–879. Medline. doi:10.2337/diabetes.54.3.872
Tomlinson JW, Moore JS, Clark PM, Holder G, Shakespeare L, Stewart PM (2004) Weight loss increases 11beta-hydroxysteroid dehydrogenase type 1 expression in human adipose tissue. J Clin Endocrinol Metab 89:2711–2716. Medline. doi:10.1210/jc.2003-031376
Draper N, Walker EA, Bujalska IJ et al (2003) Mutations in the genes encoding 11beta-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase interact to cause cortisone reductase deficiency. Nat Genet 34:434–439. Medline. doi:10.1038/ng1214
Rebuffe-Scrive M, Bronnegard M, Nilsson A, Eldh J, Gustafsson JA, Bjorntorp P (1990) Steroid hormone receptors in human adipose tissues. J Clin Endocrinol Metab 71:1215–1219. Medline
Rebuffe-Scrive M, Lundholm K, Bjorntorp P (1985) Glucocorticoid hormone binding to human adipose tissue. Eur J Clin Invest 15:267–271. Medline
Bujalska IJ, Quinkler M, Tomlinson JW, Montague CT, Smith DM, Stewart PM (2006) Expression profiling of 11beta-hydroxysteroid dehydrogenase type-1 and glucocorticoid-target genes in subcutaneous and omental human preadipocytes. J Mol Endocrinol 37:327–340. Medline. doi:10.1677/jme.1.02048
Dobson MG, Redfern CP, Unwin N, Weaver JU (2001) The N363S polymorphism of the glucocorticoid receptor: potential contribution to central obesity in men and lack of association with other risk factors for coronary heart disease and diabetes mellitus. J Clin Endocrinol Metab 86:2270–2274. Medline. doi:10.1210/jc.86.5.2270
Fraser R, Ingram MC, Anderson NH, Morrison C, Davies E, Connell JM (1999) Cortisol effects on body mass, blood pressure, and cholesterol in the general population. Hypertension 33:1364–1368. Medline
Dimitriou T, Maser-Gluth C, Remer T (2003) Adrenocortical activity in healthy children is associated with fat mass. Am J Clin Nutr 77:731–736. Medline
Marin P, Darin N, Amemiya T, Andersson B, Jern S, Bjorntorp P (1992) Cortisol secretion in relation to body fat distribution in obese premenopausal women. Metabolism 41:882–886. Medline. doi:10.1016/0026–0495(92)90171-6
Stewart PM, Boulton A, Kumar S, Clark PM, Shackleton CH (1999) Cortisol metabolism in human obesity: impaired cortisone–>cortisol conversion in subjects with central adiposity. J Clin Endocrinol Metab 84:1022–1027. Medline. doi:10.1210/jc.84.3.1022
Pasquali R, Vicennati V (2000) The abdominal obesity phenotype and insulin resistance are associated with abnormalities of the hypothalamic-pituitary-adrenal axis in humans. Horm Metab Res 32:521–525. Medline
Travison TG, O’Donnell AB, Araujo AB, Matsumoto AM, McKinlay JB (2007) Cortisol levels and measures of body composition in middle-aged and older men. Clin Endocrinol (Oxf) 67:71–77. Medline. doi:10.1111/j.1365-2265.2007.02837.x
Jessop DS, Dallman MF, Fleming D, Lightman SL (2001) Resistance to glucocorticoid feedback in obesity. J Clin Endocrinol Metab 86:4109–4114. Medline. doi:10.1210/jc.86.9.4109
Purnell JQ, Brandon DD, Isabelle LM, Loriaux DL, Samuels MH (2004) Association of 24-hour cortisol production rates, cortisol-binding globulin, and plasma-free cortisol levels with body composition, leptin levels, and aging in adult men and women. J Clin Endocrinol Metab 89:281–287. Medline. doi:10.1210/jc.2003-030440
Ohmori N, Nomura K, Ohmori K, Kato Y, Itoh T, Takano K (2003) Osteoporosis is more prevalent in adrenal than in pituitary Cushing’s syndrome. Endocr J 50:1–7. Medline. doi:10.1507/endocrj.50.1
Yoshihara A, Okubo Y, Tanabe A et al (2007) A juvenile case of Cushing’s disease incidentally discovered with multiple bone fractures. Intern Med 46:583–587. Medline. doi:10.2169/internalmedicine.46.1824
Kaltsas G, Manetti L, Grossman AB (2002) Osteoporosis in Cushing’s syndrome. Front Horm Res 30:60–72. Medline
Kristo C, Jemtland R, Ueland T, Godang K, Bollerslev J (2006) Restoration of the coupling process and normalization of bone mass following successful treatment of endogenous Cushing’s syndrome: a prospective, long-term study. Eur J Endocrinol 154:109–118. Medline. doi:10.1530/eje.1.02067
van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C (2000) Use of oral corticosteroids and risk of fractures. J Bone Miner Res 15:993–1000. Medline. doi:10.1359/jbmr.2000.15.6.993
Cooper MS (2004) Sensitivity of bone to glucocorticoids. Clin Sci (Lond) 107:111–123. Medline. doi:10.1042/CS20040070
Tauchmanova L, Rossi R, Nuzzo V et al (2001) Bone loss determined by quantitative ultrasonometry correlates inversely with disease activity in patients with endogenous glucocorticoid excess due to adrenal mass. Eur J Endocrinol 145:241–247. Medline. doi:10.1530/eje.0.1450241
Godang K, Ueland T, Bollerslev J (1999) Decreased bone area, bone mineral content, formative markers, and increased bone resorptive markers in endogenous Cushing’s syndrome. Eur J Endocrinol 141:126–131. Medline. doi:10.1530/eje.0.1410126
Minetto M, Reimondo G, Osella G, Ventura M, Angeli A, Terzolo M (2004) Bone loss is more severe in primary adrenal than in pituitary-dependent Cushing’s syndrome. Osteoporos Int 15:855–861. Medline. doi:10.1007/s00198-004-1616-3
Di Somma C, Pivonello R, Loche S et al (2003) Effect of 2 years of cortisol normalization on the impaired bone mass and turnover in adolescent and adult patients with Cushing’s disease: a prospective study. Clin Endocrinol (Oxf) 58:302–308. Medline. doi:10.1046/j.1365-2265.2003.01713.x
Di Somma C, Pivonello R, Loche S et al (2002) Severe impairment of bone mass and turnover in Cushing’s disease: comparison between childhood-onset and adulthood-onset disease. Clin Endocrinol (Oxf) 56:153-158. Medline. doi:10.1046/j.0300-0664.2001.01454.doc.x
Chiodini I, Carnevale V, Torlontano M et al (1998) Alterations of bone turnover and bone mass at different skeletal sites due to pure glucocorticoid excess: study in eumenorrheic patients with Cushing’s syndrome. J Clin Endocrinol Metab 83:1863–1867. Medline. doi:10.1210/jc.83.6.1863
Gennari C, Imbimbo B. (1985) Effects of prednisone and deflazacort on vertebral bone mass. Calcif Tissue Int 37:592–593. Medline. doi:10.1007/BF02554912
van der Eerden AW, den Heijer M, Oyen WJ, Hermus AR (2007) Cushing’s syndrome and bone mineral density: lowest Z scores in young patients. Neth J Med 65:137–141. Medline
Hermus AR, Smals AG, Swinkels LM et al (1995) Bone mineral density and bone turnover before and after surgical cure of Cushing’s syndrome. J Clin Endocrinol Metab 80:2859–2865. Medline. doi:10.1210/jc.80.10.2859
Leong GM, Abad V, Charmandari E et al (2007) Effects of child- and adolescent-onset endogenous Cushing syndrome on bone mass, body composition, and growth: a 7-year prospective study into young adulthood. J Bone Miner Res 22:110–118. Medline. doi:10.1359/jbmr.061010
Faggiano A, Pivonello R, Filippella M et al (2001) Spine abnormalities and damage in patients cured from Cushing’s disease. Pituitary 4:153–161. Medline. doi:10.1023/A:1015362822901
Chiodini I, Mascia ML, Muscarella S et al (2007) Subclinical hypercortisolism among outpatients referred for osteoporosis. Ann Intern Med 147:541–548. Medline
Cooper MS, Blumsohn A, Goddard PE et al (2003) 11beta-hydroxysteroid dehydrogenase type 1 activity predicts the effects of glucocorticoids on bone. J Clin Endocrinol Metab 88:3874–3877. Medline. doi:10.1210/jc.2003-022025
Godschalk MF, Downs RW (1988) Effect of short-term glucocorticoids on serum osteocalcin in healthy young men. J Bone Miner Res 3:113–115. Medline
Francucci CM, Pantanetti P, Garrapa GG, Massi F, Arnaldi G, Mantero F (2002) Bone metabolism and mass in women with Cushing’s syndrome and adrenal incidentaloma. Clin Endocrinol (Oxf) 57:587–593. Medline. doi:10.1046/j.1365-2265.2002.01602.x
Canalis E (1996) Clinical review 83: Mechanisms of glucocorticoid action in bone: implications to glucocorticoid-induced osteoporosis. J Clin Endocrinol Metab 81:3441–3447. Medline. doi:10.1210/jc.81.10.3441
Karavitaki N, Ioannidis G, Giannakopoulos F, Mavrokefalos P, Thalassinos N (2004) Evaluation of bone mineral density of the peripheral skeleton in pre- and postmenopausal women with newly diagnosed endogenous Cushing’s syndrome. Clin Endocrinol (Oxf) 60:264–270. Medline. doi:10.1111/j.1365-2265.2004.01968.x
Canalis E, Bilezikian JP, Angeli A, Giustina A (2004) Perspectives on glucocorticoid-induced osteoporosis. Bone 34:593–598. Medline. doi:10.1016/j.bone.2003.11.026
Cooper MS, Hewison M, Stewart PM (1999) Glucocorticoid activity, inactivity and the osteoblast. J Endocrinol 163:159–164. Medline. doi:10.1677/joe.0.1630159
Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC (1998) Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J Clin Invest 102:274–282. Medline. doi:10.1172/JCI2799
Eijken M, Hewison M, Cooper MS et al (2005) 11beta-Hydroxysteroid dehydrogenase expression and glucocorticoid synthesis are directed by a molecular switch during osteoblast differentiation. Mol Endocrinol 19:621–631. Medline. doi:10.1210/me.2004-0212
Cooper MS, Bujalska I, Rabbitt E et al (2001) Modulation of 11beta-hydroxysteroid dehydrogenase isozymes by proinflammatory cytokines in osteoblasts: an autocrine switch from glucocorticoid inactivation to activation. J Bone Miner Res 16:1037–1044. Medline. doi:10.1359/jbmr.2001.16.6.1037
Newell-Price J, Trainer P, Besser M, Grossman A (1998) The diagnosis and differential diagnosis of Cushing’s syndrome and pseudo-Cushing’s states. Endocr Rev 19:647–672. Medline. doi:10.1210/er.19.5.647
Muller R, Kugelberg E (1959) Myopathy in Cushing’s syndrome. J Neurol Neurosurg Psychiatry 22:314–319. Medline
Lane RJ, Mastaglia FL (1978) Drug-induced myopathies in man. Lancet 2:562–566. Medline. doi:10.1016/S0140-6736(78)92894-5
Mills GH, Kyroussis D, Jenkins P et al (1999) Respiratory muscle strength in Cushing’s syndrome. Am J Respir Crit Care Med 160:1762–1765. Medline
Khaleeli AA, Edwards RH, Gohil K et al (1983) Corticosteroid myopathy: a clinical and pathological study. Clin Endocrinol (Oxf) 18:155–166. Medline
Khaleeli AA, Betteridge DJ, Edwards RH, Round JM, Ross EJ (1983) Effect of treatment of Cushing’s syndrome on skeletal muscle structure and function. Clin Endocrinol (Oxf) 19:547–556. Medline
Kendall-Taylor P, Turnbull DM (1983) Endocrine myopathies. Br Med J (Clin Res Ed) 287:705–708. Medline
Djaldetti M, Gafter U, Fishman P (1977) Ultrastructural observations in myopathy complicating Cushing’s disease. Am J Med Sci 273:273–277. Medline
Jang C, Obeyesekere VR, Dilley RJ, Alford FP, Inder WJ (2006) 11Beta hydroxysteroid dehydrogenase type 1 is expressed and is biologically active in human skeletal muscle. Clin Endocrinol (Oxf) 65:800–805. Medline. doi:10.1111/j.1365-2265.2006.02669.x
Whorwood CB, Donovan SJ, Wood PJ, Phillips DI (2001) Regulation of glucocorticoid receptor alpha and beta isoforms and type I 11beta-hydroxysteroid dehydrogenase expression in human skeletal muscle cells: a key role in the pathogenesis of insulin resistance? J Clin Endocrinol Metab 86:2296–2308. Medline. doi:10.1210/jc.86.5.2296
Tomlinson JW, Moore J, Cooper MS et al (2001) Regulation of expression of 11beta-hydroxysteroid dehydrogenase type 1 in adipose tissue: tissue-specific induction by cytokines. Endocrinology 142:1982–1989. Medline. doi:10.1210/en.142.5.1982
Fitts RH, Romatowski JG, Peters JR, Paddon-Jones D, Wolfe RR, Ferrando AA (2007) The deleterious effects of bed rest on human skeletal muscle fibers are exacerbated by hypercortisolemia and ameliorated by dietary supplementation. Am J Physiol Cell Physiol 293:C313-C320. Medline. doi:10.1152/ajpcell.00573.2006
Louard RJ, Bhushan R, Gelfand RA, Barrett EJ, Sherwin RS (1994) Glucocorticoids antagonize insulin’s antiproteolytic action on skeletal muscle in humans. J Clin Endocrinol Metab 79:278–284. Medline. doi:10.1210/jc.79.1.278
Hong DH, Forsberg NE (1995) Effects of dexamethasone on protein degradation and protease gene expression in rat L8 myotube cultures. Mol Cell Endocrinol 108:199–209. Medline. doi:10.1016/0303-7207(95)03476-N
Joulia-Ekaza D, Cabello G (2007) The myostatin gene: physiology and pharmacological relevance. Curr Opin Pharmacol 7:310-315. Medline. doi:10.1016/j.coph.2006.11.011
Ma K, Mallidis C, Bhasin S et al (2003) Glucocorticoid-induced skeletal muscle atrophy is associated with upregulation of myostatin gene expression. Am J Physiol Endocrinol Metab 285:E363–E371. Medline
Gilson H, Schakman O, Combaret L et al (2007) Myostatin gene deletion prevents glucocorticoid-induced muscle atrophy. Endocrinology 148:452–460. Medline. doi:10.1210/en.2006-0539
Dardevet D, Sornet C, Taillandier D, Savary I, Attaix D, Grizard J (1995) Sensitivity and protein turnover response to glucocorticoids are different in skeletal muscle from adult and old rats. Lack of regulation of the ubiquitin-proteasome proteolytic pathway in aging. J Clin Invest 96:2113–2119. Medline
Rieu I, Sornet C, Grizard J, Dardevet D (2004) Glucocorticoid excess induces a prolonged leucine resistance on muscle protein synthesis in old rats. Exp Gerontol 39:1315–1321. Medline. doi:10.1016/j.exger.2004.06.005
Seene T, Kaasik P, Pehme A, Alev K, Riso EM (2003) The effect of glucocorticoids on the myosin heavy chain isoforms’ turnover in skeletal muscle. J Steroid Biochem Mol Biol 86:201–206. Medline. doi:10.1016/j.jsbmb.2003.08.002
Pirlich M, Biering H, Gerl H et al (2002) Loss of body cell mass in Cushing’s syndrome: effect of treatment. J Clin Endocrinol Metab 87:1078–1084. Medline. doi:10.1210/jc.87.3.1078
Ahtikoski AM, Riso EM, Koskinen SO, Risteli J, Takala TE (2004) Regulation of type IV collagen gene expression and degradation in fast and slow muscles during dexamethasone treatment and exercise. Pflugers Arch 448:123–130. Medline. doi:10.1007/s00424–003–1226–5
Manoli I, Le H, Alesci S et al (2005) Monoamine oxidase-A is a major target gene for glucocorticoids in human skeletal muscle cells. FASEB J 19:1359–1361. Medline
Giustina A, Veldhuis JD (1998) Pathophysiology of the neuroregulation of growth hormone secretion in experimental animals and the human. Endocr Rev 19:717–797. Medline. doi:10.1210/er.19.6.717
Johannsson G, Sunnerhagen KS, Svensson J (2004) Baseline characteristics and the effects of two years of growth hormone replacement therapy in adults with growth hormone deficiency previously treated for Cushing’s disease. Clin Endocrinol (Oxf) 60:550–559. Medline. doi:10.1111/j.1365-2265.2004.02018.x
Phillips PJ, Weightman W (2007) Skin and Cushing syndrome. Aust Fam Physician 36:545–547. Medline
Stratakis CA, Mastorakos G, Mitsiades NS, Mitsiades CS, Chrousos GP (1998) Skin manifestations of Cushing disease in children and adolescents before and after the resolution of hypercortisolemia. Pediatr Dermatol 15:253–258. Medline. doi:10.1046/j.1525-1470.1998.1998015253.x
Reich A, Bednarek-Tupikowska Z, Czarnecka A, Szepietowski JC (2006) Cutaneous manifestations in a patient with a long-term history of untreated ACTH-dependent Cushing’s syndrome. Acta Dermatovenerol Croat 14:30–34. Medline
Groves RW, MacDonald LM, MacDonald DM (1990) Profound digital collagen atrophy: a new cutaneous presentation of adrenal-dependent Cushing’s syndrome. Br J Dermatol 123:667–671. Medline. doi:10.1111/j.1365-2133.1990.tb01486.x
Kletsas D, Pratsinis H, Gioni V, Pilichos K, Yiacoumettis AM, Tsagarakis S (2007) Prior chronic in vivo glucocorticoid excess leads to an anabolic phenotype and an extension of cellular life span of skin fibroblasts in vitro. Ann N Y Acad Sci 1100:449–454. Medline. doi:10.1196/annals.1395.050
van Rossum EF, Lamberts SW (2004) Polymorphisms in the glucocorticoid receptor gene and their associations with metabolic parameters and body composition. Recent Prog Horm Res 59:333–357. Medline. doi:10.1210/rp.59.1.333
Tomlinson JW, Draper N, Mackie J et al (2002) Absence of Cushingoid phenotype in a patient with Cushing’s disease due to defective cortisone to cortisol conversion. J Clin Endocrinol Metab 87:57–62. Medline. doi:10.1210/jc.87.1.57
Oreffo RO, Cooper C, Mason C, Clements M (2005) Mesenchymal stem cells: lineage, plasticity, and skeletal therapeutic potential. Stem Cell Rev 1:169–178. Medline. doi:10.1385/SCR:1:2:169
Oshina H, Sotome S, Yoshii T et al (2007) Effects of continuous dexamethasone treatment on differentiation capabilities of bone marrow-derived mesenchymal cells. Bone 41:575–583. Medline. doi:10.1016/j.bone.2007.06.022
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Fernandez-Rodriguez, E., Stewart, P.M. & Cooper, M.S. The pituitary–adrenal axis and body composition. Pituitary 12, 105–115 (2009). https://doi.org/10.1007/s11102-008-0098-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11102-008-0098-2