
Abstract
Idiopathic Pulmonary Fibrosis (IPF) is a chronic, progressive, and often fatal form of interstitial lung disease. It is characterized by injury with loss of lung epithelial cells and abnormal tissue repair, resulting in replacement of normal functional tissue, abnormal accumulation of fibroblasts and myofibroblasts, deposition of extracellular matrix, and distortion of lung architecture which results in respiratory failure. Despite improvements in the diagnostic approach to IPF and active research in recent years, the molecular mechanisms of the disease remain poorly understood. This highly lethal lung disorder continues to pose major clinical challenges since an effective therapeutic regimen has yet to be identified and developed. For example, a treatment modality has been based on the assumption that IPF is a chronic inflammatory disease, yet most available anti-inflammatory drugs are not effective in treating it. Hence researchers are now focusing on understanding alternative underlying mechanisms involved in the pathogenesis of IPF in the hope of discovering potentially new pharmaceutical targets. This paper will focus on lung tissue repair, regeneration, remodeling, and cell types that may be important to consider in therapeutic interventions and includes a more detailed discussion of the potential targets of current therapeutic attack in pulmonary fibrosis. The discovery that adult bone marrow stem cells can contribute to the formation of differentiated cell types in other tissues, especially after injury, implies that they have the potential to participate in tissue remodeling, and perhaps regeneration. The current promise of the use of adult stem cells for tissue regeneration, and the belief that once irreversibly damaged tissue could be restored to a normal functional capacity using stem cell-based therapy, suggests a novel approach for treatment of diverse chronic diseases. However this optimism is tempered by current evidence that the pathogenesis of pulmonary fibrosis may involve the recruitment of bone marrow-derived fibroblasts, which are the key contributors to the pathogenesis of this chronic progressive disorder. Nevertheless, stem cell-related therapies are widely viewed as promising treatment options for patients suffering from various types of pulmonary diseases. Gender mismatched bone marrow or lung transplant recipients serve as natural populations in which to study the role of bone marrow-derived stem cells in recovery from pulmonary diseases. Understanding the mechanism of recruitment of stem cells to sites of injury, and their involvement in tissue repair, regeneration, and remodeling may offer a novel therapeutic target for developing more effective treatments against this fatal disorder. This article reviews the new concepts in the pathogenesis, current and future treatment options of pulmonary fibrosis, and the recent advances regarding the roles of stem cells in lung tissue repair, regeneration, and remodeling.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Idiopathic Pulmonary Fibrosis (IPF) is the most common form of interstitial lung diseases of unknown origin. It is associated with an extremely poor prognosis for survival in most patients. Life expectancy after diagnosis varies, but is on average less than 5 years (1,2). A recent study has estimated its prevalence ranges from 14 to 42.7 cases and incidence ranges from 6.8 to 16.3 cases per 100,000 persons (3). Despite extensive research efforts over the past decades, no currently available therapy has been shown to either reverse or even halt the progression of this disorder (4).
Although during the past decades significant advances have been made in the understanding of the pathogenesis of IPF, the exact mechanisms underlying the development of IPF still remain unknown (5). More importantly, concepts of pathogenesis have shifted within the past decade. Previously, the long held belief was that chronic inflammation plays an essential role in the pathogenesis of IPF (4–16). In this paradigm IPF is characterized by some degree of lung inflammation and abnormal tissue repair, resulting in replacement of normal functional tissue with abnormal accumulation of fibroblasts and myofibroblasts and deposition of collagen and other extracellular matrix (ECM) in the interstitial and alveolar spaces (6–16). This process is known to involve an intricate cytokine network that activates and mediates interactions between multiple cell types resulting in the elevation of collagen gene expression and abnormal deposition of collagen in the lung (4–16).
Another hypothesis is founded on the fact that IPF results from epithelial cell injury and abnormal wound repair in the absence of preceding inflammation (5,17). A more recent hypothesis that is based on the modification of these two hypotheses postulates that inflammation is subsequent to injury and IPF occurs as a result of the body’s immune response to injury to the lung (5).
Finally, recent studies suggest that the pathogenesis of IPF may be much more complex than was previously thought, and that IPF is more likely to be a syndrome rather than specific disease. The pathologic hallmarks of IPF are fibroblastic foci, which are areas rich in mesenchymal cells and ECM. These foci, which consist of fibroblast-like cells and myofibroblasts, are considered to be a key element in the diagnosis of IPF (16). Understanding the origin of these cells and the mechanism of recruitment should shed further insight into the basis for the development of progressive fibrosis, rather than successful healing and regeneration (16).
Previous studies have suggested that fibroblasts and myofibroblasts in injured and fibrotic lung arise from resident fibroblasts present in the adventitia of perivascular and peribronchial tissue. However, recent emerging evidence suggests that circulating fibrocytes and bone marrow-derived progenitor cells (BM) may be recruited in lung injury and play an important role in the pathogenesis of lung disorders (16,18–29). Elucidation of the biology of adult stem or progenitor cells in the context of remodeling/fibrosis on the one hand, and regeneration of lung parenchyma on the other, has the potential to revolutionize the treatment of patients with lung disorders such as IPF, emphysema, and other fibrotic lung diseases. This article briefly reviews the mechanism and current treatment of IPF and speculates on the potential uses of adult stem cells in the research of lung injury and lung disease therapies. We apologize in advance for any studies omitted due to oversight or space limitations.
STEM CELLS DEFINITION AND BACKGROUND
Stem cells are unique cells which have the capacity for limitless self-renewal and differentiation. Another defining characteristic of these cells is their niche (27). A niche is a subset of cells and extracellular components that can accommodate one or more stem cells indefinitely and control their self-renewal and progeny production in vivo. The stem cells undifferentiated character and capacity for unlimited self-renewal are a result of their interaction with the supportive cells and a unique microenvironment in their niche (26). Excellent recent reviews of stem cells in lung disease have outlined the definition and characterization of these cells (23,25–27,29).
Briefly, stem cells may be classified into two categories: embryonic stem cells (ES) cells, and adult BM-derived stem cells. ES cells are pluripotent cells and are derived from the inner cell mass of the blastocyst stage of embryos (30). They are able to form any cell type and tissue in the animal body and therefore have great therapeutic capacity to regenerate any damaged tissue (23). ES cells isolated from the inner cell mass of a blastocyst can be characterized in vivo by the formation of chimeric embryos when injected into an immunocompromised host, and by differentiation in vitro to form embryoid bodies (31). The therapeutic capacity of these cells that proliferate freely in culture has been shown in the animal model of Parkinsonism (32) and other diseases. Therefore, ES cells and resident lung stem cells may provide novel therapeutic approaches for lung disease. Although ES cells have been suggested for use in tissue regeneration of injured lung, their use is currently mired in substantial ethical controversy because of the need to destroy an embryo in order to harvest these cells (33).
Adult stem/progenitor cells are thought to reside within a tissue and are recruited/activated to differentiate during repair after injury. In the lung, type II alveolar epithelial cells are the putative distal progenitor cells that are responsible for repair after injury to type I alveolar epithelial cells. Although the classical concept is that adult stem cells reside as tissue specific cells in various organs and are committed to differentiate and replace existing cells, recent evidence suggests the possibility that additional contribution from BM-derived stem/progenitor cells cannot be fully excluded, at least for certain differentiated lung cell types.
BM is the source of several distinct stem cell populations: a primary category is hematopoietic stem cells (HSCs), which are traditionally thought to give rise solely to cells of the hematopoietic series, such as leukocytes and erythrocytes. The BM also contains marrow stromal or mesenchymal stem cells (MSCs), which are populations that give rise to a variety of connective tissues localized in different germ layers (27). In addition, the BM contains endothelial stem/progenitor cells and multipotent adult progenitor cells (MAPC). Among these HSCs are the best characterized of the adult stem cells (34). HSCs are clearly multipotent and have the ability to maintain or restore mature circulating blood cells. However, whether they have the capacity to also differentiate to non-hematopoietic cells is of considerable recent interest. With respect to MSCs, there is also evidence of their potential to differentiate into diverse mesenchymal tissues and cells, such as bone, cartilage and adipocytes. However, the true potential for either of these adult BM-derived stem cell populations in lung tissue regeneration remains to be determined. There remains some uncertainty as well as to whether they play a harmful role in IPF, such as by promoting fibrogenesis, or a beneficial reparative and/or regenerative role.
EMBRYONIC STEM CELLS GENERATION OF LUNG CELLS
Pluripotent ES cells offer theoretical promise as a potential source of lung endodermal, mesodermal, and ectodermal cell types for lung regeneration. Murine ES cells cultured as embryonic bodies followed by growth on collagen coated surfaces with growth factors, and replating at an air-liquid-interface can develop into an epithelium with clara, ciliated, basal, and intermediate cells (35). In addition, studies have explored conditions that promote mouse ES cell expression of alveolar type II cell differentiation gene markers (36). More recent studies show that fetal lung tissue with high potential for growth and differentiation could be used for lung regeneration. However, using fetal lung tissue to treat adult human lung diseases may be of ethical concern (37), and may result in problems associated with tissue rejection. Nevertheless, ES cells have clear advantages due to their totipotentiality, but many challenges, both technical and ethical, remain.
ENDOGENOUS AND EXOGENOUS LUNG STEM CELLS
Even though many cells with proliferative capacity exist within the lung and other organs, only a subset of these may be considered stem cells. Endogenous tissue stem cells are undifferentiated cells that reside in tissues and participate in regeneration after injury. The adult lung is a vital and complex organ that normally turns over slowly. The epithelial cells that line the airways are constantly exposed to potential toxic agents and pathogens in the environment, and they must, therefore, be able to respond quickly and effectively to both cellular damage and to the local production of a myriad of mediators. Injury models have suggested that different regions of the respiratory system in the trachea, large airways, distal bronchioles and alveoli are derived from diverse stem or progenitor cells, with differing strategies for maintenance and repair.
Recent studies, have suggested potential lung tissue derivation from extrapulmonary BM-derived stem cells. In a recent workshop on stem cells and lung disease sponsored by the National Heart Lung and Blood Institute (NHLBI) and the Cystic Fibrosis Foundation, participants acknowledged the important role of adult BM-derived stem cells in lung biology and their potential therapeutic role in lung diseases. However, there were controversies with respect to the functional role of adult marrow-derived and other adult stem cell populations in lung remodeling (25). There is agreement that stem and progenitor cells are often loosely defined frequently resulting in confusion and misconceptions about research goals and methodology. The report proposed to further refine the field in terms of reparative or regenerative investigations as a central goal of much of the current research in the field of repair and restoration of diseased lung. In this context a useful suggestion was made to define cellular sources for repair as “reparative cells” instead of “stem cells or progenitor cells” to indicate a population(s) of cells that participate in lung remodeling after injury (25). Much needed criteria are also proposed to prove the existence of reparative cells. They should include identification of the origin of reparative cells using specific markers, demonstration of integration into new tissue, and acquisition of tissue-specific function in the new organ.
The three key components of lung tissue, the endothelial, epithelial and mesenchymal elements, contribute to repair and regeneration in response to injury. These cells proliferate from endogenous reparative cells of normal lung resident cells or may be derived from BM-derived progenitors. A key element in repair is vasculogenesis, and the proliferation of surviving endothelial cells at the sites of injury. The presence of BM-derived circulating stem/reparative cells also has been shown to play a role in vasculogenesis in tissue repair and remodeling (38).
Endogenous reparative cells that participate in maintenance and remodeling of injured lung epithelium have been described for tracheobronchial, bronchiolar and alveolar compartments (39–41). Evidence indicating the existence of putative lung tissue reparative cells comes from studies in animal models in which the majority of reparative cells are depleted through exposure to either physical or chemical agents (42–44). The most distinct regions that have been identified to support populations of lung tissue reparative cells include: bronchoalveolar duct junction (42,44), neuroepithelial bodies in bronchioles (43), and the intercartilaginous regions of tracheobronchial airways (41). Each region contains reparative cells that share the common properties of infrequent proliferation and a relatively undifferentiated phenotype. These cells have been characterized at the cellular level. Endogenous lung reparative cells have also been implicated in the development of lung carcinoma.
With respect to mesenchymal cells, fibroblasts/myofibroblasts are considered to be key contributors of pulmonary fibrosis (PF). Previous studies have suggested that myofibroblasts are derived from preexisting intrapulmonary mesenchymal precursors. However, recent discoveries of the significant presence of BM-derived fibroblasts and of the circulating leukocyte subpopulation known as fibrocytes suggest an additional or alternative extrapulmonary source of fibroblasts in injured lung undergoing fibrosis (16,18,20, 22,24).
Further studies are needed to define the mechanisms regulating activity of endogenous lung reparative cells and the molecular signals that are required to initiate these cells to regenerate after injury. There is also a major need for the development of new technical methods and identification of specific molecular markers with which to identify and characterize these cells (25).
Stem Cells and the Alveolar Epithelium
The distal airways terminate in about 300 million alveolar air sacs in the adult human lung. The surface of alveoli is lined by alveolar epithelium, which consists of alveolar type-1 (AT1) and alveolar type-2 (AT2) cells which occupy about 96 and 4%, respectively of the nearly 70 m2 of gas exchange surface. The cytoplasm of AT1 cells is attenuated to the greatest extent to limit the barrier to gas exchange; the diameter of these cells can reach 100 μm. This feature contributes to the vulnerability to injury of AT1 cells and hence the difficulty in isolating them for studies in vitro (29). Functions of AT1 cells are now known to include water and ion transport, host defense, and tumor suppression (45). AT2 cells carry out the metabolic and other functions necessary to maintain the airspace. It is well known that AT2 cells are responsible for the synthesis and secretion of surfactant, surfactant-associated proteins, cytokines, growth factor receptors, enzymes, and other matrix proteins (46–48).
It was previously thought that AT2 cells were the only progenitor cells of the alveolar epithelium, and that AT1 cells were terminally differentiated cells. If either cell type is lost, the nearest AT2 cells will be stimulated to proliferate and, if necessary, differentiate into AT1 cells (49). Although the AT2 cells are often referred to as the stem cell of the alveolar surface (50–52), it is not clear that all AT2 cells in the adult lung meet the criteria of stem cells. Therefore, as a recent NHLBI/Cystic Fibrosis foundation workshop suggested, the term “reparative cells” would be more informative and accurate than “stem cells” for AT2 cells. In addition, studies on lung embryos have shown co-expression of markers specific for AT1 and AT2 cells on progenitor cells, which suggests that the situation is more complex than was originally thought (53,54).
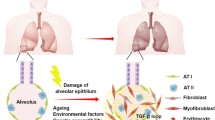
A modified working model of alveolar epithelial kinetics in the adult lung has been proposed (55). Almost all AT2 cells in the normal adult lung are quiescent and can re-enter the cell cycle after stimulation. In this situation, AT2 cells will proliferate and cell division leads to two relatively undifferentiated daughter cells with the potential to revert to either differentiated phenotype. It is also possible for AT1 cells to re-enter the cell cycle process through differentiation to AT2 for use as endogenous stem/reparative cell pools (56–58). Exogenous reparative cells pools, which may come from BM HSCs, could also contribute to the populations of alveolar epithelial cells (AEC) either as AT1 or AT2 cells (Fig. 1).

A schematic drawing stages of alveolar epithelial cells (AEC) apoptosis and resources of stem cells of the type 1 and 2 (AT1 and AT2) alveolar epithelium from endogenous and exogenous pathways and their contribution to lung injury and fibrosis. Activated epithelial cells secrete numerous mediators and pro-apoptotic factors that induced Fas-mediated AEC apoptosis via caspase cascade activation and contribute to fibrotic process. The AT2 cells in normal adult lung are quiescent, but in injured lung AT2 cell regains stem cell function and can re-enter the cell cycle. Cell division leads to two relatively undifferentiated daughter cells with the potential to revert to either differentiated phenotype. It is also possible for AT1 cells to re-enter the cell cycle process through differentiation to AT2 as endogenous stem cell pools. Exogenous stem cells pools, which mostly come from bone marrow HSCs, can also contribute to the populations of AEC either AT1 or AT2 cells.
Both cell types have the potential for apoptosis, which is important for the maintenance of dynamic balance of the epithelial cell population (56). Furthermore, to date is not clear whether there is a subpopulation of AT2 or AT2-like cells which serves as a source of proliferating epithelial cells engaged in epithelial repair after injury, or whether all AT2 cells are capable of proliferating.
Stem Cells in Lung Parenchyma and Fibrotic Lung Disease
The pathogenesis of fibrotic lung disease has recently been considered primarily as an aberrant wound-healing response of the lung parenchyma. Accumulating evidence suggests that parenchymal cells, especially AEC, are likely to be active participants in the process of lung fibrosis. The alveolar and airway epithelial cells are primary targets in lung injury. Abundant histological evidence indicates that there are profound alterations in the alveolar epithelium in chronic fibrosing alveolitis. Changes can be found in both AT1 and AT2 cells. A consistent histological feature of lung tissue in chronic fibrosis is the greatly reduced number of AT1 cells as a result of necrosis or extensive apoptosis, with detachment of cells from the alveolar walls (59,60). AT1 cells appear to be more susceptible to injury than AT2 cells. In severe injury, AT1 cells are lost early after injury, leaving AT2 cells that appear to re-epithelilize the denuded basement membrane by forming an initial layer of cubodial epithelial cells (61,62).
Lung injuries that damage only AT1 cells are generally repairable, whereas injuries that kill both AT1 and AT2 cells are likely to be irreversible and progress to a fatal outcome, presumably as a result of the destruction of the stem/reparative cell population (55,63). Exogenous reparative cell pools, which may come from marrow HSCs, have the potential to also contribute to the population of AEC either as AT1 cells through a straight transdifferentiation (multipotent differentiative cells directly become a different tissue with more complex and specialized structure and function), or as AT2 cells via an oblique transdifferentiation pathway (multipotent differentiative cells indirectly become a different tissue with a more proliferative and immature status) (64,65). In a recent study, Kotton et al. observed that engraftment and transdifferentiation of HSCs were exclusive to AT1 cells in bleomycin-induced pulmonary fibrosis (BIPF) in an animal model (64). They observed that donor marrow-derived cells with the morphological molecular phenotype of AT1 cells had engrafted to the recipient lung parenchyma after 2 days, but there was no engraftment of any AT2 cells found during the experiment (64). This pathway is defined as a straight transdifferentiation.
In contrast, Krause et al. suggest even greater plasticity of BM-derived HSCs by showing that they have the potential not only to repopulate all of the haematopoietic cell lines, but also to engraft to multiple organs (65). This pathway is defined as a straight transdifferentiation. Importantly, in that study over 20% of the epithelial cells in lung parenchyma contain a Y chromosome, indicating their donor BM origin. Functional activity in the engrafted cells is suggested by expression of surfactant protein B, a specific marker of AT2 cells. This degree of plasticity has also been suggested by other studies that have used MSCs or HSCs transplanted into recipient animals with different modalities of lung injury. Recent clinical studies have provided some support for similar plasticity in humans, wherein BM-derived cells are shown to give rise to AT2 cells via an oblique transdifferentiation pathway (66–69). However, despite these findings, other studies have contradicted this claim that HSCs have this level of plasticity, and that BM-derived cell can engraft to lung epithelial cells (70,71). The basis for these contradictory results is unclear, but could at least in part be due to certain differences in study design.
In summary endogenous and exogenous stem/reparative cells may contribute to proliferation of both AT1 and AT2 cells and lung regeneration (64–70). The efficacy of AT2 alveolar repair is inversely related to development of lung fibrosis (55). Figure 1 shows the resource of stem/reparative cells of the alveolar epithelium AT1 and AT2 cells from endogenous and exogenous pathways and their contribution to fibrotic lung.
Regulation of Lung Parenchymal Cells and Fibrosis
Patients with IPF and experimental animal models of lung injury both exhibit evidence of alveolar epithelial injury. Injury to AEC and the cell death accompanying alveolitis have the potential to induce the formation of gaps in the epithelial basement membranes. The migration of fibroblasts through these gaps into the alveolar space induces or contributes to intra-alveolar fibrosis. Interstitial fibrosis and the subsequent intra-alveolar fibrosis result in significant remodeling of the lung. Destruction of alveolar units in IPF is mediated presumably by inflammatory fibrogenic cytokines and immune processes. In severe injury and fibrosis profound alteration in both types of AEC can be found (Fig. 2).

A schematic drawing pathogenetic mechanism of alveolar epithelial cells (AEC) in lung injury alveolitis and fibrosis. An initial injury of known or unknown etiology provokes an unresolved inflammation or AEC damage and alveolitis. Alveolitis is an initial event in the pathogenesis of lung fibrosis. A fibrin clot forms early and serves as a provisional matrix for the migration and proliferation of endogenous AEC stem cells. Bone marrow-derived stem cells HSCs, can also contribute to the populations of AECs. Activated epithelial cells secrete numerous mediators that create a strong profibrotic microenvironment in lung fibrosis inducing AEC injury, alveolitis and apoptosis. Also injured AEC exhibit a defect in the release of some antifibrotic mediators such as prostaglandinE-2 (PGE2). Epithelial cell damage and cell death during alveolitis induce the formation of gaps in the epithelial basement membrane. Progression from the acute phase of lung injury is accompanied by abnormal alveolar reepitalization, tissue repair and remodeling, which may result in progressive fibrosis. AEC produce MMPs that increase basement membrane disruption and allow fibroblast migration, proliferation and differentiation to myofibroblasts. Myofibroblasts secrete extracellular matrix proteins, mainly collagens. An imbalance between MMPs and TIMPs provokes the progressive deposit of the extracellular matrix (ECM) and abnormal collagen accumulation and promotes fibrosis. Alveolar macrophages and AEC secrete TGF-β1, which induces epithelial mesenchymal transition (EMT), and this may contribute to fibroblast/myoflbroblast accumulation in the processes of fibrosis.
The specific molecular signals that are required to initiate AT2 cell division in vivo remain uncertain, although a burgeoning list of possible factors have been suggested by animal models as well as by human studies, including studies of isolated cells in vitro. Several agents, such as fibroblast growth factor 1 (FGF-1), keratinocyte growth factor (KGF), and hepatocyte growth factor (HGF) trigger AT2 mitosis in vivo and in vitro (72,73). The KGF promotes the proliferation and migration of AT2 cells. Local administration of KGF has been shown to significantly reduce the degree of lung injury from hypoxia, bleomycin, and radiation (72,73). Transforming growth factor-β (TGF-β) exerts a negative effect on AT2 cell proliferation and plays a key role in mediating fibrotic tissue remodeling (74,75). Fetal lung expansion induces the differentiation of AT2 into AT1 cells via an intermediate cell type in vitro (76). KGF induces the differentiation of AT2 into AT1 cell phenotype in primary cell culture (77). Retinoic acid stimulates proliferation of AT2 cells and has a positive effect on lung development and maturation, as well as repair and regeneration after injury (78–80).
Alveolar epithelium can be regulated by cytokines, other mediators, and ECM components (81). Alteration in the structure and function of AEC may increase the expression of mediators which regulate the inflammatory and fibrotic response within the lung. AEC of IPF patients can secrete a number of molecules such as cytokines, chemokines, growth factors, their receptors, adhesion molecules, enzymes, other proteins, and ECM components, which may regulate the fibrotic processes in the lung (Table I).
In addition, recent studies have found an association between apoptosis of AEC and lung fibrosis (82), (Fig. 2). It has also been shown that AEC of the fibrotic human lung were composed of both proliferating and dying cells and that apoptotic and necrotic epithelial cells were located in proximity to α-smooth muscle actin (α-SMA) positive interstitial cells (83). Abnormal fibroblast phenotypes isolated from the fibrotic human lung produced factors capable of inducing apoptosis and necrosis of AEC in vitro. This suggests that myofibroblasts may have a role in inducing epithelial cell apoptosis and necrosis.
Furthermore, experimental studies using animal models of BIPF support that initial alveolar epithelial injury may lead to development of fibrosis. Following the initial injury, inflammatory cells accumulate in the alveolar walls. These cells release mediators that stimulate fibroblast activation, migration, and proliferation, as well as ECM accumulation. Inflammatory cells and their products may damage the epithelial cells. In cases of severe or extensive injury, in which abundant AEC apoptosis appears, regeneration of AEC cannot occur, and hence, normal repair and recovery of the lung architecture cannot take place and fibrosis may result (84) (Fig. 2).
Additional support for the direct role of AEC in disease processes also comes from studies in mouse lung explants. Severe epithelial damage induced by prior hypoxic lung injury leads to increased fibroblast proliferation and collagen deposition in culture, whereas less damaged explants did not (85).
In summary, AEC can participate in the initiation of lung inflammation, modulation of the severity of the inflammatory reaction, lead to pulmonary dysfunction when it is damaged, and participate in the abnormal repair of wound processes and fibrosis. Increased understanding of the origin and contributions that AEC make to the pathophysiology of PF may provide directions for the development of new anti-fibrotic agents for treating patients.
ADULT BONE MARROW-DERIVED CELLS IN LUNG DISEASES
While it is well-established that BM HSCs serve as precursors for peripheral blood cells, a number of studies indicate that the pluripotency of BM-derived cells extends further than classic hematopoietic differentiation. Several cell populations derived from BM of adult rodents, particularly HSCs, MSCs, endothelial progenitor cells, skeletal muscle stem cells, myocardial muscle stem cells and circulating fibrocytes, can localize to and acquire phenotypic and functional markers of muscle, bone, liver, skin, and lung (86–93). Therefore, adult stem/reparative cells from BM or peripheral blood can putatively give rise to various circulating, and lineage-restricted progenitor/precursor cells (87–91). Circulating stem/reparative cells that normally express a default pathway of specific differentiation deviate from the normal pathway of differentiation, and transdifferntiate to another lineage (89). Differentiated cells undergo dedifferentiation and regain characteristics of stem cells in order to circulate and redifferentiate in another tissue.
In animal models, relatively small numbers of adult marrow-derived cells transplanted to adult recipient animals have been shown to localize to recipient lungs and to acquire phenotypic characterization of airway or AEC, interstitial cells, and vascular endothelial cells (18–29,86,87,92,94–108). In addition, human lung biopsies taken from BM transplant recipient patients have shown chimerism for both epithelial and endothelial cells (109,110).
With respect to lung injury and fibrosis, studies using animal models have suggested that adult BM-derived cells participate in the generation of lung fibrotic processes (18–22,24). Additionally, evidence has suggested that BM-derived fibroblast precursors and circulating fibrocytes may participate in the process of lung injury, asthma, and fibrosis (18–22,24,111–115). Figure 3 shows the potential of such fibroblast progenitor cells from endogenous and exogenous pathways, and their possible contribution to lung fibrosis.

A schematic drawing of bone marrow derived fibroblast that participates in lung injury, regeneration and fibrosis. The fibroblasts in normal adult lung are quiescent, but in injured lung, fibroblasts regain stem cell function and re-enter the cell cycle. In an injured lung, fibroblast become activated proliferate and differentiate to myofibroblasts. Myofibroblasts secrete extracellular matrix (ECM) and abnormal collagen which accumulates and promotes fibrosis. Alveolar epithelial cells (AEC) may also serve as a source of myofibroblast through epithelial-mesenchymal transition (EMT). It is also possible that exogenous stem cells, like bone marrow-derived fibroblasts, participate in lung injury and these cells are able to engraft into the lung tissue and produce collagen and promote fibrosis. Bone marrow-derived fibroblasts may be recruited to lung tissue by CXC chemokines produced by alveolar macrophages.
Conversely, some evidence suggests a beneficial implication for recruitment of adult marrow-derived cells to the lung with respect to repair and regeneration. In animal models of fibrosis, emphysema, and inflammatory lung injury, BM-derived stem/reparative cells appear to abrogate lung injury (28,99,101–103). Thus, despite numerous studies, the roles of adult marrow-derived cells in lung injury, repair, and remodeling remain unclear and controversial.
Role of Fibroblast/myofibroblast in Pathogenesis of Lung Fibrosis
The fibroblasts of the lung, along with the myofibroblasts, are derived from mesenchymal tissues, and are localized primarily in the interstitial tissue space (116). These cells can be distinguished from each other based on differences in morphology, contractile protein composition, and ECM production. Myofibroblasts express α-SMA and have a phenotype intermediate between fibroblasts and smooth muscle cells (117–122). However, these cells are heterogeneous with respect to their cytokeletal phenotype, especially those found in various fibrotic disease, and fibrotic lesions (117–121).
Fibroblasts are relatively resistant to injury and thus, they are more likely to survive many insults. The primary function of these cells is in repair processes. They regulate the degradation of damaged matrix via secretion of matrix degrading enzymes, and biosynthetically replace it with a new matrix.
During abnormal tissue repair and fibrosis, increased numbers of fibroblasts and myofibroblasts are seen. Fibroblast migration, proliferation, ECM synthesis and degradation, all of which play important roles in chronic inflammation and fibrosis, are regulated by various growth factors and cytokines. Current understanding of the pathogenesis of PF suggests that fibroblasts and myofibroblasts play an important role in the diverse processes that constitute fibrosis. A key common feature found in fibrotic lesions, including PF and wound healing, is the appearance of myofibroblasts at sites of wound healing. Active fibrosis is seen where there is extensive remodeling with active synthesis of ECM components (Fig. 4). It appears that these cells are the primary cellular source of collagen as well as a major source of cytokines in PF. More detailed reviews on the role of fibroblast and myofibroblasts have been presented previously (121,122).

Summary of the key pathogenetic mechanisms of fibroblast/myofibroblast in lung injury and fibrosis. Fibroblast foci, sites of active collagen synthesis, are the pathologic hallmark of IPF. Following an unidentified insult, alveolar epithelial cells (AEC) become injured and abnormal re-epithelialization, tissue repair and remodeling which may result in chronic progressive fibrosis. Increased fibrogenic cytokins, growth factors and other mediators by inflammatory cells and AEC, result in activation, migration and proliferation of fibroblasts and differentiation into myofibroblasts. Myofibroblasts express high levels of cytokines, growth factors, α-SMA and low level of PTEN and telomerase activity. Myofibroblast and AEC produce MMPs that increase basement membrane disruption and allow fibtoblast/myofibroblast migration. Myofibroblast produce extracellular matrix (ECM) proteins and angiotensin II which may promote AEC apoptosis. AEC and alveolar microphages secrete TGF-β1 that induce fibroblast differentiation, increase ECM production and decrease fibtoblast/myofibroblast apoptosis. AECs may also serves as a source of myofibrobalst through epithelial-mesenchymal transition (EMT). Bone marrow-derived fibroblasts may also participate in lung fibroblast accumulation and ECM deposition and enhance fibrosis.
The presence of myofibroblasts in patients with IPF has been documented in both lung tissue biopsies from patients, and in animal models of the disease (121,122). Additionally, a subsequent study in an animal model provided direct evidence that myofibroblasts are the key cellular source of type 1 collagen expression in fibrosis (15).
Moreover, we recently discovered that telomerase is induced and selectively expressed in lung fibroblasts without expression of α-SMA in BIPF in the animal model, suggesting that these telomerase-positive cells may present an intermediate activated phenotype between the quiescent fibroblasts present in normal lungs and the highly activated myofibroblasts induced in lung undergoing active fibrosis (123–125). The differentiation of fibroblast to α-SMA expressing myofibroblasts and their persistence is thought to result in progressive fibrosis, terminating in end-stage lung disease (125–127).
We also have shown that inhibition of induced telomerase activity in fibrotic lung fibroblasts promoted myofibroblast differentiation in these cells (128). Increased fibroblast proliferation and the de novo appearance of myofibroblasts are key features of the fibrotic process in fibrotic lung disease (Fig. 4). Further studies will help clarify the role of telomerase in myofibroblast biology and may provide a novel target for the treatment of IPF.
The Role and Origin of Fibroblasts in Lung Fibrosis
As mentioned above, the hallmark lesions of IPF are the fibroblast foci located in the areas of active fibrogenesis. Fibroblasts are known to be heterogeneous both phenotypically and functionally (129). While kinetic studies suggest their derivation from resident fibroblasts present in the adventitia of perivascular and peribronchial tissue, recent evidence indicates an additional source from circulating blood cells called fibrocytes that possess fibroblast-like properties and that can be chemotactically recruited to sites of tissue injury (18,20,130). Fibrocytes are a unique population of circulating cells of presumed BM-derived stem/reparative cell origin; they represent less than 1% of the circulating pool of leukocytes. They produce ECM proteins including collagen 1 and III (Col-1 and III), vimentin, fibronectin, and myofibroblast-like α-SMA (130). Fibrocytes are characterized by cell markers such as CD markers (CD11a-LFA-1, CD11b-MAC-1, CD13-pan-myeloid-aminopeptidase-N, CD18β2 integrin, CD34-HSC and CD45RO-leukocyte common antigen, CD45-ICAM-1, CD58-LFA-3, CD71-transferrin receptor), MHC class II markers (HLA-DP, HLA-DQ, HLA-DR), chemokine receptors (CCR3, CCR5, CCR7, and CXCR4), and ECM markers (18,20,130–132). After isolation, these cells can be expanded in culture and have been shown to resemble fibroblasts on the basis of their ability to express Col-1. Fibrocytes have been identified in normal tissue as well as in wound and scar tissue, in bronchial walls in asthmatics, in people with various fibrotic diseases, and in basal cell carcinoma (132–136). The major functions of these cells are in tissue repair and regeneration, expression of ECM proteins such as pro Col-1 and III, which are involved in connective tissue formation, abnormal wound healing, and contribution to fibrosis (132–136). These cells promote angiogenesis, and also are a potent antigen presenting cell (APC) for T cell activation. Fibrocytes also secrete a variety of fibrogenic cytokines, growth factors, and other mediators that modulate fibrosis. The major mediators that are expressed by fibrocytes and the potentially relevant roles of these mediators in lung fibrosis are summarized in Table I.
In studies using BM chimera mice created by transplanting BM from enhanced green fluorescent protein (GFP) transgenic mice into lethally irradiated recipient mice, a significant number of lung fibroblasts in bleomycin injured lungs are found to be derived from BM progenitors (18). Histological analysis showed large numbers of GFP-positive cells in fibrotic lesions, and flow cytometry showed that these GFP-positive cells expressed Col-1, telomerase reverse transcriptase, and functional chemokine receptors CXCR4 and CCR7, and can migrate in vitro to their ligands, CCL-12 and CCL-21, respectively. Studies of circulating fibrocytes also indicate their potential as significant sources of lung fibroblasts in response to lung injury (19,20,22,24). Additionally, these fibrocytes appear to play important roles in the development of lung fibrosis (20,22,24). They appear to be present significantly in other fibrotic lung diseases such as asthma, and in other models of lung injury and fibrosis (20,111–115).
In summary, ample and growing evidence indicates that BM progenitors and circulating fibrocytes respond to the signals of lung injury by actively migrating to sites of remodeling. While the phenotype of these cells and their functional roles are not yet entirely (or completely) defined, they have the potential as stem/reparative cells to play a role in the fibroproliferative response leading to lung fibrosis or successful repair and regeneration. Recent literature indicates that both endogenous and exogenous factors can provide the primary stimuli that lead to lung injury and fibrosis. Fibroblasts, myofibroblasts, circulating fibrocytes, chemokines, and growth factors clearly participate in the process of wound healing and functional recovery. Excessive injury to the lung can overcome these reparative processes leading to aberrant wound healing and resulting in PF (Fig. 4). Nonetheless, despite an accumulating number of studies, the roles of adult marrow-derived cells in lung repair and remodeling remain obscure and controversial.
Mechanisms of Recruitment of Bone Marrow-derived Stem Cells to the Lung
Preexisting injury increases recruitment of adult marrow-derived cells to the lung. This suggests that chemotactic signals or other mediators released by injured or remodeling lung serve to attract marrow-derived cells. A number of soluble factors released by injured lung epithelium mediate recruitment of mature circulating leukocytes from BM to the lung (137). Chemokines, their receptors, and angiogenic factors are known to mobilize and recruit both HSCs and endothelial progenitor cells (EPCs) (138–141). It has been noted that increased release of soluble factors as well as increased expression of tissue ligands to which HSCs and EPCs can bind, occurs in a variety of lung injuries. Adult marrow-derived circulating fibrocytes express several chemokine receptors such as CXCR4 and CCR7 and migrate in vitro in response to corresponding ligands (18,20). Adult marrow-derived human MSCs express cell surface adhesion molecules, including vascular cell adhesion molecule-1. In BIPF in an animal model, increased expression of the stromal-derived factor 1 (SDF-1), has been shown, suggesting a role of this signaling pathway in the recruitment of adult marrow-derived cells to the lung (20).
In summary, adult stem/reparative cells circulate to specific tissues in response to injury, and adhere to endothelium in response to expressed adhesion molecules that target these cells to specific tissues. These cells migrate to sites of injury by use of specific chemotactic-gradient-chemokine ligands and receptors, and are integrated into tissue and differentiated for tissue specific function. Major factors involved in stem/reparative cell differentiation include: TGF-β, glucocorticoids, insulin, and endothelin.
Source of Lung Myofibroblasts in Fibrosis
Although the importance of myofibroblasts in the pathogenesis of IPF is clear, myofibroblast origin and their activation process during fibrotic remodeling in vivo remain largely undefined and controversial. Despite ample evidence of myofibroblast derivation from tissue fibroblasts upon appropriate stimulation (e.g. by TGFβ) in vitro, the actual in vivo mechanism or source of myofibroblasts remain uncertain. While there is a potential contribution from circulating fibrocytes; recent studies indicate an additional mechanism may be operative. Epithelial mesenchymal transition (EMT) may be a mechanism for the derivation of the myofibroblast (142,143). EMT is a process in which epithelial cells lose their phenotype and acquire mesenchymal/fibroblast-like properties, with concomitant display of reduced cell adhesion and increased motility. This is a key step during embryonic morphogenesis. A recent study has demonstrated that TGFβ1 induces EMT in AEC in vitro (143). This study also identified cells co-expressing AEC markers and α-SMA in regions of hyperplastic epithelium in IPF lungs, suggesting that EMT may contribute to myofibroblast accumulation during fibrotic processes (143). In addition, more recent findings support the possibility that EMT may be a source of myofibroblasts in IPF lungs (Fig. 2). Lung biopsies from IPF patients exhibited a significant nuclear accumulation of β-catenin in bronchiolar epithelial cells as well as in cuboidal hyperplastic pneumocytes, suggesting involvement of the Wnt/β-catenin signaling pathway (144). β-Catenin signaling plays a role in inducing a mesenchymal phenotype in epithelial cells. A significant expression of N-cadherine by cells overlying fibrotic foci has also been shown (145). A shift of cadherin from the E to the N type is also typical of EMT. Despite these observations, the role of EMT in lung remodeling remains uncertain.
ANIMAL MODELS STUDIES
Recent rapid progress in our understanding of the role of adult marrow-derived cells in lung diseases comes from studies on animal models. The pathologic hallmark of IPF and other fibrotic lung diseases are fibroblast foci, areas rich in mesenchymal cells and ECM. The source of fibroblasts in fibrotic lung has been the focus of intense investigation, but the contribution of intrapulmonary versus extrapulmonary cells in the fibrotic disease process remains unknown. BM chimera mice have been used to investigate the contribution of BM-derived cells in the generation of fibrotic lesion. BM chimera mice have been generated by using BM obtained from GFP transgenic mice and transplanting it into irradiated wild-type recipients (18,146,147). After the demonstration of engraftment using peripheral blood chimerism in excess of 90% donor-derived cells, chimeric mice were exposed to bleomycin lung injury (5–14). Intratracheal instillation of a cancer chemotherapeutic agent, bleomycin sulfate, induces lung injury, inflammation, and fibrosis in mice, with many features similar to those seen in humans with IPF (5–14,148–155). Animal models of BIPF are characterized by AEC injury, which occurs early in pathogenesis, followed by migration of inflammatory cells and fibroblasts into the lung, resulting in extensive accumulation of collagen and ECM, and leading to development of fibrosis.
Histological analysis of fibrotic lesions showed large numbers of GFP-positive cells in fibrotic lesions that expressed Col-1 and chemokine receptors such as CXCR4 and CCR7, and migrated to appropriate ligands. This data also supports studies on human lung injury after bone marrow transplantation (BMT), which has shown that BM-derived cells contribute to pathogenesis, and their requirement is likely regulated by a specific receptor pathway.
Additionally, in recent studies by Ortiz et al., purified BM-derived MSCs from bleomycin-resistant mice were adoptively transferred to susceptible mice after bleomycin injection (156). Donor cells homed to the injured lung and adopted epithelial phenotypes, including that of AT2 cells. In more recent studies in an animal model, mice given bleomycin survived and showed the histological inflammation and fibrosis in the lung that is typical of this model (157). The BM was then suppressed with a single dose of busulfan. When bleomycin was injected 24 h after busulfan, the injury was exaggerated such that two-thirds of the animals did not survive for 14 days, and lung histopathology of animals that did survive to that time tended to be worse than the animals that were not myelosuppressed (28). It was concluded that recovery from bleomycin-induced lung injury requires an intact pool of BM progenitor cells. Delivery of mesenchymal stem/reparative cells to the BM suppressed mice profoundly limited the degree of prolonged injury and fibrosis cause by bleomycin, but persistence of some injury in the BM-suppressed animals suggests that both endogenous and exogenous stem/reparative cells contribute to the repair process (28). Injured lung produces soluble factors that stimulate stem/reparative cell proliferation, which could serve either as a message for expansion of endogenous stem cell pools, or as a local stimulus for expansion of populations of stem cell in the lung (28).
Another impressive piece of evidence is that a single haemopoietic stem cell (lineage depleted and enriched for CD34 and Sca-1 expression by homing to the BM in vivo), when transplanted from an adult male mouse to a lethally irradiated female, not only repopulated all the haemopoietic cell lineages, but also engrafted several organs (158). The engraftment lasted for at least 11 months after BMT. Donor-derived epithelial cells were identified by detection of Y chromosome contributing cells that coexpressed cytokeratins (epithelial markers); they were found in bronchi, gut, and skin, about 4% of the tissue. An interesting finding was that up to 20% of epithelial cells in the lung parenchyma contained the Y chromosome, perhaps as a result of the radiation pneumonitis induced in the process of BMT (159). It was also found that the Y chromosome-containing cell had surfactant B mRNA, which suggests that the donor cells were taking on an AT2 cell phenotype. Despite these findings, studies showing BM to organ engraftment remain controversial. Detection of lineage markers to distinguish resident lung cells and leukocytes is technically difficult, especially in sections of lung. Finally, advanced technical design might be necessary to provide more reliable and reproducible data before the findings are taken into clinical use.
HUMAN STUDIES
Although the characterization and determination of the origin of lung cells (preexisting intrapulmonary versus extrapulmonary stem/reparative cells) involved in the pathogenesis of IPF has largely come from the use of animal disease models in rodents, a few studies in humans have explored the potential source of mesenchymal cell/fibroblast origin in the lung. Clinical tissue specimens taken from patients following sex-mismatched organ transplantations allow a similar model to study in humans. After transplantation of male BM cells to female patients, Y chromosomes have been found in many cell types, including hepatocytes, cardiomyocytes, gut and skin epithelium, and tubular and endothelial cells in the kidney (160–167). However, similar studies have not found cardiomyocytes of BM cell origin (168,169).
Additionally, studies of recipients of sex-mismatched marrow transplants have found donor-derived epithelial and endothelial cells in the lungs (170). In these sex-mismatched lung transplants, cells from bronchial epithelium and AT2 cells have been identified. Donor-derived cells were detected in lung samples taken from as early as 4 days to years after transplantation, which is consistent with animal experiments suggesting that engraftment of the lung is a stable long-term process. However, any previous study has failed to demonstrate chimerism of bronchial and alveolar epithelium (171).
In addition, chimerism or lung engraftment with adult marrow-derived cells has not been found in all human studies, which are contradictory with the results of animal studies. In respect to lung injury and asthma, recent studies have shown that CD34 expressing cells expressed Col-1 and α-SMA in the bronchial mucosa of asthmatic patients, and circulating fibrocytes also expressed CD34 (131,132,135,172–174). These observations suggest that circulating fibrocytes are potential sources for the fibroblasts/myofibroblasts in the remodeling airway lung. Further studies at a single-cell level are needed to further delineate the origin of fibroblasts in fibrotic processes in human lungs.
TREATMENT OF PULMONARY FIBROSIS
IPF is a progressive and irreversible illness. As of yet, there has been no available drug capable of modifying the progressive natural course of IPF, and the current therapy is only minimally effective. IPF continues to pose major clinical challenges, because an effective therapeutic regimen has yet to be determined. The currently accepted approach to treatment of IPF is based on the assumption that it is a chronic, unresolved inflammatory disease. The mainstay of therapy has been the use of corticosteroids with or without immunosuppressive drugs (175). However, it is now clear that therapies with anti-inflammatory drugs are associated with toxicity and do not provide objective benefit (175).
Recent progress in understanding the mechanisms underlying the pathogenesis of fibrosis leads us to expect that inhibitors of pro-fibrogenic cytokines and growth factors may be useful as novel therapeutic agents in controlling undesirable fibrosis. Therefore, potential therapeutic strategies could be developed for use at any of the steps which result in IPF (Fig. 5). These include agents that interfere with the action of inflammatory mediators, agents that prevent epithelial cell damage, agents that prevent fibroblast proliferation and collagen synthesis, agents that down-regulate myofibroblast differentiation, agents that inhibit angiogenesis or stimulate angiostatic CXC chemokines, and agents that intervene with one or more key events in the pathogenesis or signal transduction pathways of IPF. In addition, in lung injury, increased apoptosis of epithelial cells can contribute to fibrosis, while on the other hand, decreased fibroblast or myofibroblast apoptosis promotes fibrosis. Hence, there is little doubt that the development of novel therapeutic strategies for PF may include agents that promote fibroblast apoptosis or prevent epithelial apoptosis.

Possible therapeutic approaches according to target pathways in pulmonary fibrosis. The major mechanistic steps in fibrosis leading to accumulation of collagen and ECM deposition are indicated by the bold arrow. Treatments directed to any one of these steps are listed in the respective adjacent boxes and indicated by crossed arrows. Any treatment directed at any one of these steps may have an impact on additional mechanistic steps. The various therapeutic agents involved are described in the text.
Additionally, current studies suggest that circulating fibrocytes, which are derived from BM, behave like mesenchymal stem cells and migrate into sites of lung injury. These cells expressed Col-1 and α-SMA which are hallmarks of myofibroblasts, and contributed to lung fibrosis (18–22,24). The results of these studies may lead us to development of novel therapeutic agents for treatment of IPF that block fibrocytes from sites of lung injury.
Finally, the challenge of future targets for therapeutic intervention is to reconcile different pathogenic pathways, and we strongly suspect that no single approach will be sufficient for this lethal disease with few therapeutic options. The aim of this section of the review is to identify candidate pathways that might offer novel therapeutic targets, changing the natural course of this disease, and to give more detail on the roles of stem cells as promising novel therapy. The major therapeutic agents in lung fibrosis include anti-inflammatory drugs, anti-fibrotic and anti-cytokine agents, receptor antagonists, signal transduction pathways inhibitors, anti-apoptosis agents, and angiogenic and MMPs inhibitors (Table II). In addition, recent advances in stem cell biology and technology will eventually turn cell transplantation into a useful treatment for patients with lung diseases.
ANTI-INFLAMMATORY AGENTS
Anti-inflammatory therapies continue to be used despite there being little evidence of inflammation in the pathogenesis of IPF (176–179). Although corticosteroid with or without immunosuppressive drugs were the mainstay of therapy for IPF for decades, their efficacy is unproven and toxicities are substantial (175,180,181). In a majority of cases of IPF, corticosteroid therapy is only partially effective, and most patients deteriorate despite therapy.
Additionally, there is no controlled trial using corticosteroids alone for the treatment of IPF (182,183). Therefore, any conclusive evidence supporting the use of corticosteroid therapy for the treatment of IPF is lacking (182,184). However, given the poor prognosis of the disease and the lack of readily available alternatives and efficacious treatment, a therapeutic trial with anti-inflammatory medications is still justified (177). Generally, patients with IPF respond better to therapy if they exhibit more inflammation and less fibrosis. Current treatment approaches have been recently reviewed (2,175,176,180–185).
ANTI-FIBROTIC AND ANTI-CYTOKINE AGENTS
Recently, there was a paradigm shift in the treatment of IPF, with more of a trend toward using anti-fibrotic treatment, based on the concept that the disease is a fibrotic process with a lack of significant inflammation (17). The reason for patients’ failure to respond to anti-inflammatory therapy with corticosteroids may be that the lung is in a stage of fibrosis rather than inflammation at the time of diagnosis. Thus, anti-fibrotic drugs that interfere with or modulate further progression of lung fibrosis may have potential to improve respiratory function (176,182,186). Anti-cytokine therapeutic strategies are directed at abrogating the activities of the targeted cytokines that have diverse regulatory activities in several processes that comprise fibrosis. This has been attempted by targeting one or more key steps in cytokine synthesis and binding to cognate receptors.
The most promising potential anti-fibrotic agents, have been recently reviewed (3,176,182,185,187) and are either recommended for consideration in clinical trials, or will require additional evaluation prior to a trial. The major anti-fibrotic and anti-cytokine agents that have been used in the treatment of IPF include: colchicine, penicillamine, pirfenidone, TGFβ antagonist, anti-tumor necrosis factor α (TNFα) interferon-γ (IFN-γ) and connective tissue growth factor antagonist, (188–197) (Table II).
Pirfenidone
Pirfenidone is an anti-fibrotic agent that inhibits TGF-β induced collagen synthesis in vitro (190), decreases lung fibroblast proliferation, and downregulates pro-fibrotic cytokines (182,190). Pirfenidone can diminish BIPF by downregulating platelet-derived growth factor (PDGF) expression (190,191). In clinical trials with IPF patients, it was found that pirfenidone was able to stabilize both respiratory function and symptomology. A multiple center study is currently underway.
TGF-β Antagonist
There has been some evidence that both anti-TGF-β antibodies and TGF-β soluble receptors could partially inhibit fibrosis in bleomycin model (191). Although the above therapeutic approaches were efficient in animal models, humans could develop immune reactions against the antibodies. However, recent studies have been designed to investigate whether GC1008, an antibody that neutralized TGFβ, is safe in treating patients with IPF (187).
TNF-α Antagonist
TNF-α has been found to be significantly elevated in BIPF. There is ample evidence that both anti-TNF-α antibodies and TNF-α soluble receptors could inhibit fibrosis in animal models (13,192). TNF-α-induced cytokine networks such as TGFβ, interleukin-5 (IL-5), and eosinophil recruitment in BIPF can all be inhibited by anti-TNF-α antibodies (13). A recent clinical phase 1 trial has reported improvement of lung function after treatment with a soluble TNF receptor in patients with IPF. A phase II double-blind, parallel, placebo-controlled randomized study of the efficacy and safety of a compound that blocks TNF-α in IPF patients is underway. This compound works by binding to TNF-α cell surface receptors, thus inhibiting the initiation of intracellular signaling.
IFN-γ-1b
IFN-γ-1b is an attractive therapeutic candidate because it regulates both macrophage and fibroblast function. It down regulates molecules associated with fibrosis, inflammation, and angiogenesis (193,194). In addition, IFN-γ therapy reduced TGFβ expression in lung biopsies from IPF patients. Prolonged survival has now been suggested in three controlled clinical trials of IFN-γ-1b in the treatment of IPF (195). Another phase III randomized, placebo-controlled trial studying survival outcomes in IPF with IFN-γ-1b is currently underway.
Connective Tissue Growth Factor Antagonist (Anti-CTGF)
CTGF has a crucial role in the IPF pathway by triggering the production of collagen and fibronectin, which causes fibrotic lesion development (196). Human monoclonal antibodies that neutralize the activity of CTGF and modulate fibrosis in lung and kidney have been developed. An open-label, phase 1, safety and tolerability dose-escalating study of FG-3019, a therapeutic antibody designed to block the profibrotic activity of CTGF, has been recently completed in patients with mild-to moderate IPF. The drug was found to be safe and well tolerated (197). A phase II trial of FG-3019 in IPF is currently underway.
RECEPTOR ANTAGONIST
Soluble cytokine receptors or cytokine-binding proteins that could compete with cellular receptors for any available secreted cytokine should effectively inhibit the cytokine’s biological activities.
Decorin
Decorin is a small proteoglycan that binds and reduces biological activities of all isoforms of TGFβ (198), and has been shown to inhibit the fibrotic response using adenovirus mediated delivery of decorin cDNA (199). The advantages of decorin as a potential therapeutic agent in IPF patients are its aerosol administration and the minimal risk of immunological reactions.
Imatinib Mesylate (PDGF Receptor Antagonist)
Imatinib mesylate inhibits activation of the PDGF receptor significantly, and reduces BM fibrosis in humans (200). A phase II randomized, double blind, placebo-controlled study of the clinical effects of imatinib mesylate given orally to patients with IPF has been completed. The efficacy of the biological agent has been encouraging (182), but susceptibility to infections has been a major concern.
Endothelin Receptor 1 Antagonist
The endothelial cell-derived endothelin-1 (ET-1) is a potent mitogen for endothelial cells and vascular smooth muscle cells. ET-1 is strongly upregulated in IPF lungs and is expressed by epithelial cells. Some studies have suggested that inhibition of this mediator could have anti-fibrotic effect (201). Recently, studies have shown that ET-1 and its receptors act as angiogenic regulators, representing a new target for anti-angiogenic therapy (202). Recently bosentan, a non-selective ET A and B receptor antagonist, is being studied in a multi-center phase II/III study in IPF patients.
INHIBITION OF SIGNAL TRANSDUCTION
The importance of the TGFβ receptor-activated family of latent transcription factors called Smads in mediating signal transduction of TGFβ has been discussed elsewhere (154,203–206). In addition, it has been found that the expression of Smad 3 and 7 is down regulated during the process of lung fibrosis (207,208). Because TGFβ is a key regulator of tissue remodeling, altered expression of its intracellular signaling molecule Smad 7 (a major inhibitory regulator in the Smad family) is likely to play an important role in the pathogenesis of PF (208). In addition, recent studies have found that over expression of both Smad 7 and IL-7 antagonize TGFβ signaling and attenuate fibrosis in BIPF in mice (207,208).
Antagonist of TGF-β Mediated Signals
It is well known that TGF-β signals from the membrane to the nucleus by both Smad-dependent and-independent pathways (209–211). Systemic administration of a kinase inhibitor attenuated fibrotic response by modulating the pro-survival signaling phenotype of fibroblasts and myofibroblast in vivo (212). In addition, IFN-γ signaling via the Jak1/STAT1 pathway rapidly increases expression of Smad 7, subsequently inhibiting Smad 3 phosphorylation, a critical step in TGF-β signaling to the nucleus. Thus, in addition to strategies to block the action of TGF-β extracellularly, it may be possible to modulate the intracellular signaling pathway of TGF-β.
ANTI-APOPTOSIS
Current evidence suggests that increased and continuous epithelial cell apoptosis, and decreased fibroblast/myofibroblast apoptosis occurred in the process of PF (213). Additionally, recent studies have shown that myofibroblasts from patients with fibrotic lung disease secrete soluble factors (angiotensin peptides) that induce apoptosis of human AEC (214). In addition, recent findings have suggested that myofibroblast-induced epithelial cell apoptosis via an oxidant-mediated mechanism may promote epithelial injury, aberrant repair responses, and progressive fibrosis (215). Pharmacological studies have shown inhibition of both bleomycin-induced apoptosis and fibrosis using broad-spectrum caspase inhibitors (216–218) and blockers of the antigotensin pathways (218). Animal studies have shown that administration of amiodarone (a benzofuran derivative) induces both apoptosis of AEC and lung fibrosis and this effect may be inhibited by captopril (angiotensin converting enzyme inhibitor) (218). Although the mechanisms of AEC apoptosis in PF are not completely understood, the roles of several molecules have been suggested, including Fas activation, angiotensin pathways, activation of T cell-derived perforin and IL-13 stimulation, and activation of TGFβ (219). Fas has been shown to be expressed on AEC from the lungs of IPF patients and in BIPF in animal models (220,221). A recent study has also shown that overexpression of heme oxygenase 1 suppresses BIPF in mice and inhibits AEC apoptosis (222).
Induce Apoptosis in Fibroblasts
HMG-CoA reductase inhibitors (statins) induce apoptosis in normal and fibrotic lung fibroblasts. Another putative anti-fibrotic effect of statins is related to their ability to inhibit the expression of CTGF (223).
ANTIANGIOGENESIS
The existence of neovascularization in IPF has been identified in the recent decades. It is still unclear whether the primary vascular abnormality in IPF is a lack of or an excess of neovascularization. Ample evidence suggests that the heterogeneity in vascularization in IPF may, on the one hand support fibroproliferation, and on the other hand, inhibit normal repair mechanisms. If neovascularization plays a key role in abnormal matrix remodeling, therapy directed at either inhibition of angiogenic or augmentation of angiostatic CXC chemokines could be helpful in IPF (224–229). Indeed, recently an imbalance in the levels of angiogenic chemokines as compared with angiostatic chemokines favoring net angiogenesis has been demonstrated with IPF (224). More recent data suggests an increase in an angiostatic chemokine (IFN-inducible T-cell-α chemoattractant/CXCL11) in bronchial biopsies after IFN-γ1b treatment (226,230). Furthermore, increased levels of the angiogenic IL-18 have been found in bronchoalveolar lavage fluid (BALF) samples from IPF patients (231). Thus, therapy directed at attenuation of angiogenic or enhancement of angiostatic CXC chemokines might present a novel therapeutic option for the treatment of IPF (225–231).
MATRIX METALLOPROTEINASES (MMPS) INHIBITOR
Both intra-alveolar and interstitial fibroblasts secrete ECM proteins, including collagens, fibronectin, elastic fibers and proteoglycans. IPF is characterized by an excessive amount of collagen deposited in the lung. IPF fibroblasts produce a number of ECM protein and integrin molecules. Abundant Col-1 can be identified in areas of mature fibrosis, while Col-III is detected in areas of early fibrosis (232). The mechanism of tissue remodeling involves proteolysis by metalloproteinases. Matrix metalloproteinases (MMPs) have been implicated in the remodeling of ECM. The control of MMPs is achieved by tissue inhibitors of metalloproteinases (TIMPs). The importance of the roles of MMPs and their inhibitors in the pathogenesis of lung disease has been reviewed elsewhere (233). Recent studies have shown a higher expression of TIMP compared with MMPs in IPF patients (234). An imbalance between MMPs and their inhibitors seems to play an important role in remodeling the ECM during fibrotic process (235–239).
In addition, batimastal (BB-94), a synthetic inhibitor of MMPs, has been shown to significantly reduce lung fibrosis in an animal model (240). Relaxin alone or in combination with IFN-γ reduces collagen synthesis by scleroderma-derived fibroblasts. Importantly, a randomized, double-blind, placebo-controlled trial has been performed in patients with systemic sclerosis (241). Thus, a combination of MMP inhibitors and anti-fibrotic agents may help to reduce fibrosis.
ANTIOXIDANT AGENTS
Some evidence suggests that IPF might represent a state of increased oxidative stress, with increased release of oxidants from neutrophils, macrophages, and fibroblasts, and decreased levels of antioxidants, all contributing to impaired ECM and cellular function. An imbalance between oxidants and anti-oxidants seems to play an important role in the pathogenesis of IPF. Although the roles of neutrophils in the pathogenesis of IPF is unknown, these cells, when migrating into tissue, release reactive oxygen species (ROS), including superoxide anion \({\left( {O^{ - }_{2} } \right)}\), hydrogen peroxide (H2O2) and hypochlorous acid (HOCL). ROS may directly convert pro-metalloproteinases into active enzymes, mediate cross-linking of ECM proteins, alter the balance MMPs and their inhibitors, and contribute to the remodeling process.
N-acetyl Cystine (NAC)
NAC, a derivative of the cysteine amino acid, has been demonstrated to augment the synthesis of anti-oxidant glutathione (GSH) (242,243). GSH plays an important role as a defense mechanism against intra and extracellular oxidative stress. It scavenges free radicals and contributes to the reduction of hydrogen peroxide and lipid hyperoxide. IPF is characterized by GSH deficiency (244). A phase III multiple center, randomized, double-blind, placebo-controlled 1 trial was designed to examine the efficacy of NAC in IPF (245).
Nitric Oxide Synthase Inhibitors (Aminoguanidine)
Activated macrophages produce both nitric oxide (NO) and peroxynitrite, contributing to the cellular injury mediated by ROS (246). It has been shown that a high level of NO exists in BALF, and over-expression of inducible nitric oxide synthase (iNOS) also exists in BIPF in animal models (247). The combined treatment of taurine and niacin significantly reduces lung fibrosis in animal models and blocks the production of NO in BALF (247). Thus, this strategy which minimizes the production of abnormal levels of NO could be promising, since aminoguanidine, a specific inhibitor of iNOS, abrogated BIPF in mice without systemic toxicity.
Cysteine Pro-drugs
The depletion of GSH in IPF patients may place the alveolar space at increased risk of additional injury caused by ROS. Since exogenous administration of GSH is relatively ineffective and toxic, cysteine pro-drugs have been used to achieve this goal. A new cysteine pro-drug, Z2196 (2RS, 4R-2-methylthiazolidine carboxylic acid), attenuated BIPF successfully, providing readily bioavailability of cysteine (246,248).
TISSUE-RE-GENERATOR
The important role of epithelial-mesenchymal induction in the repair process in lung fibrosis has been discussed above. Recent studies suggest that AEC may serve as a source of myofibroblasts through EMT, and TGF-β induces an EMT to a myofibroblast-like phenotype in AEC (249,250).
The procoagulant/fibrinolytic balance in the broncheoalveolar space is important in the lung repair process, and AEC are key contributors to that balance. Recently, there has been focus on the role of HGF in the lung repair process. HGF is a multipotent growth factor produced by pulmonary fibroblasts that stimulates the migration and morphogenesis of various epithelial cells, and has an antiapoptotic effect on alveolar and bronchial cells (251). HGF plays an important role in regulating the growth of lung epithelium, and in regeneration of the lung as a paracrine or endocrine factor in lung injury and fibrosis. in vivo studies have shown that either continuous systemic injection or intratracheal administration of recombinant human HGF (rhHGF) attenuated lung fibrosis in BIPF in animal models (251,252). From the therapeutic point of view, because HGF has both antiapoptotic and fibrinolytic potential, it is one of the ideal therapeutic agents for lung fibrosis (253). Thus, HGF gene therapy would be ideal from the clinical point of view (253).
FIBROBLAST DIFFERENTIATION INHIBITORS
Myofibroblasts arise from the differentiation of fibroblasts under the effect of TGF-β, PDGF, and other fibrogenic cytokines. Recently, there has been interest in the role of the phosphatase and tensin homolog which is mutated in multiple advanced cancers (PTEN) in the lung repair process. PTEN inhibits cell proliferation and induces apoptosis (254,255).
Recent studies have shown that PTEN expression is downregulated in fibroblasts isolated from IPF patients, compared with those obtained from control subjects (256,257). There is an inverse correlation between PTEN and α-SMA expression in fibroblast foci of lung tissue from IPF patients. Additionally, inhibition of PTEN increases α-SMA expression both in fibroblasts in vitro and PF in vivo. Importantly, the inhibition or loss of PTEN is both necessary and sufficient to induce myofibroblast differentiation, including proliferation, α-SMA expression, and collagen production.
In contrast, reconstitution of PTEN into PTEN knocked-out cells inhibits myofibroblast differentiation, whereas the overexpression of PTEN suppresses any myofibroblast differentiation that may be induced by TGFβ (257). These results suggest that PTEN may play a crucial role in myofibroblast differentiation both in vitro and in vivo (258). Therefore, from the therapeutic point of view, targeting PTEN may well prove to be a novel treatment strategy for IPF.
THERAPEUTIC POTENTIAL OF STEM CELLS IN LUNG DISEASES
Recently, BM is becoming a recognized source of progenitor cells for several cell types, including endothelial cells, epithelial cells, fibroblasts, myocytes and neurons. BM-derived endothelial stem/reparative cells can circulate in the peripheral blood and track to other organs (259). Recently a plethora of studies have suggested that BM-derived cells have the potential to differentiate into a variety of cell types (260). Additionally, studies suggest that BM-derived stem/reparative cells are required for repairing epithelium damaged by LPS-induced inflammation in the lung (261). Other investigators reported that BM-derived circulating fibrocytes were recruited to the wound and contributed to tissue repair during wound healing in asthma and fibrosis (18–20,22–24,28,135).
In addition, several studies reported that BM cells adopt the phenotype of other kinds of cells by cell fusion rather than by differentiation, and that one hematopoietic cell can serve as a progenitor for both blood and functional hepatocytes. It was also reported that BM-derived hepatocytes are primarily derived from hepatopoietic cells of the myeloid rather than the lymphoid lineage, and the mature myeloid cells spontaneously fuse with hepatocytes (262–265). These findings raise the possibility that localized administration of fusogenic cells such as myeloid cells could be a new strategy of cellular therapy for multiple tissues. The role of stem/reparative cells in lung injury and fibrosis has been discussed above. Recently we and other investigators have shown that most collagen-expressing lung fibroblasts in a model of lung injury and fibrosis were derived from BM precursor cells (18,20,22). In addition, recent study results suggest that fibrocytes comprise a population of cells that may play a functional role in angiogenesis (266). These results lead us to reconsider the complexity of BM stem/reparative cell therapy for the IPF patients in greater detail.
Block Bone Marrow-derived Fibroblast Recruitment
There is ample evidence that suggests mobilization and recruitment of BM stromal cells, especially circulating fibrocytes, to the injured lung, contributes to the development of fibrosis. Circulating fibrocytes have been shown to express several chemokine receptors, and to respond chemotactically to cognate ligands (18,20,142), thus suggesting their possible roles in recruitment of BM stem/reparative cells. Chemokine ligands CCL21 and CXCL12 are found to be induced in murine lungs in BIPF models, and their respective cognate receptors are up-regulated in fibroblasts isolated from these lungs (20). The chemotactic activity of these chemokines was confirmed using these isolated lung fibroblasts. Thus, BM derived fibrocytes can be recruited to the lung in response to chemokine signals released from the injured lung. This evidence suggests significant contribution of extrapulmonary cells to the fibroblast population in PF. Delineation of the function or pathophysiologic importance of fibrocytes to the development of fibrosis is at an early stage.
Current data suggests conflicting roles (both a protective and a harmful effect) for these cells. Recent studies have shown that myelosuppression enhanced injury and fibrosis, whereas transfer of BM-derived MSCs protected, and was accompanied by the presence of MSCs-derived fibroblasts in the lung (28). In contrast, neutralization of CXCL12 resulted in reduced recruitment of fibrocytes to the injured lung and attenuation of fibrosis (20). More studies are necessary to determine the potential roles for the BM-derived fibrocytes in lung injury and fibrosis.
Use of Stem Cells for Cellular Therapy in Lung Injury
The purpose of cellular therapy is to restore the function of damaged tissue by the transplantation of cells. Recent studies provided evidence that ES or adult stem cells can be used for cellular therapy in lung repair (65). Adult stem cells can be purified from the cells of BM, umbilical cord, and skin, and then stored and injected back into patients with lung injury to enhance lung repair (267). ES cells derived by somatic nuclear transfer are genetically identical to the donor, and thus potentially useful for cellular therapy. It is also possible to use stem cells to reconstitute more complex tissues and organs in vitro, and then transplant them to replace any failed organs. However, only a few stem cell generated products have entered clinical trials. It is also important to mention that the process of cell fusion leading to hybrid cells should be excluded as true stem cells integrating into tissue. Cell fusion may be important for delivery of healthy genes to specialized cells to correct genetically defective cells.
LUNG TRANSPLANT
Lung transplant remains the only option for end-stage IPF and for patients who fail to respond to medical treatment. Many patients show improvement with single-lung transplantation (268). This procedure can prolong life and may improve the quality of the patient’s life. However, many patients die because of the shortage of donated organs, rejection, infections, other complications, and the high cost associated with organ transplant.
CONCLUSION
Over the past two decades, it has been evident that IPF, a disease more complex than previously thought, constitutes a challenge for clinicians and researchers. Recent advances with regard to the molecular mechanisms of lung fibrosis concerning endothelial and epithelial cell injury, fibroblast proliferation, myofibroblast differentiation, impaired ECM remodeling, and abnormal repair process have resulted in an increased recognition of its complexity.
However, recent efforts have helped to bring some light into the complex pathogenesis of lung fibrosis, leading to a paradigm shift, and suggesting that IPF is not as much a chronic inflammatory disease as a disorder associated with abnormal wound healing involving the breakdown of reepithalization of AEC and the persistence of fibroblasts and myofibroblasts. Therefore, IPF represents a syndrome rather than one specific disease, and that it is the end stage of a fibrosing process. In addition, with the recent use of stem cells in lung injury, we seem to be entering a new area in the understanding of the pathogenesis of IPF.
An integral understanding of the sequence of the pathogenic mechanisms of lung fibrosis will improve therapeutic strategies in treating patients with this disease. Although no drug therapy has clearly been demonstrated to benefit patients with IPF, a number of novel investigational agents hold promise for further study. New strategies should be specific, effective, and safe. In the process of lung injury and fibrosis, various growth factors, cytokines, and other mediators are involved. Therefore, it is unlikely that any single treatment would be sufficiently effective in cases of lung fibrosis.
Current therapeutic approaches target apoptosis, epithelial replacement, fibroblast/myofibroblasts, angiogenesis, growth factors, cytokines, and other mediators. New therapeutic strategies should target the different pathways of this disease. In addition, stem cell-related therapies are widely viewed as offering promise for people suffering from various types of pulmonary diseases, and gender mismatched BM transplant recipients serve as natural populations in which to study the role of BM-derived stem cells in recovery from pulmonary injury. The current study results indicate a dominant role for resident stem cells in alveolar repair and regeneration.
The appropriate repair of the alveolar epithelium at an early stage after injury seems to be a major determinant of lung recovery. In this context, there is some evidence that BM-derived MSCs contribute to regeneration of AEC. This protective effect of BM-derived cells appears to minimize the extent of lung injury in humans as well as in animal models. In addition, it is now known that circulating fibrocytes, which are derived from BM, migrate to sites of lung injury and fibrosis. The protective effect of fibrocytes has been noted by in vivo studies showing that myelosuppression causes increased severity of fibrosis in animal models, whereas transfer of BM-derived MSCs diminishes fibrosis and is accompanied by the presence of fibrocytes in the lung.
However, another conflicting study has shown that treatment with neutralizing antibodies to the CXCR4 ligand CXCL12 not only reduced recruitment of fibrocytes to the site of injury in BIPF, but also diminished lung fibrosis.
In summary, a continuing accumulation of data in both human and animal models suggests that exogenous stem/reparative cells play a significant role in lung development, lung repair, and remodeling after injury. Nonetheless, with regard to lung repair, the literature is sparse. Initial promising reports showing that adult BM-derived cells participated in homing, engraftment, and lung phenotype adoption are now challenged. This highlights the need for more reliable techniques and assays to evaluate adult BM-derived cell engraftment and transdifferentiation. Other challenges to consider before moving to the clinical application with this promising therapeutic strategy include: improvement of purification and characterization of BM-derived stem/reparative cells, determination of mechanisms of homing, and determination of the long term safety use of these cells. Advances in stem cells biology and technology will eventually turn cell transplantation into a useful treatment with variety of diseases.
Abbreviations
- α-SMA:
-
alpha smooth muscle actin
- AEC:
-
alveolar epithelial cell
- AT1:
-
alveolar epithelial type-1
- AT2:
-
alveolar epithelial type-2
- BIBF:
-
bleomycin induced pulmonary fibrosis
- BM:
-
bone marrow
- BMT:
-
bone marrow transplantation
- BALF:
-
bronchoalveolar lavage fluid
- CCRs:
-
CC chemokine receptors
- Col-1:
-
collagen-1
- CTGF:
-
connective tissue growth factor
- ES:
-
embryonic stem cell
- ECPC:
-
endothelial cell precursor cells
- EPC:
-
endothelial progenitor cell
- ET-1:
-
endothelin-1
- EMT:
-
endothelial mesenchymal transition
- GFP:
-
enhanced green fluorescent protein
- ECM:
-
extracellular matrix
- FGF:
-
fibroblast growth factor
- HSCs:
-
hematopoietic stem cells
- HGF:
-
hepatocyte growth factor
- IPF:
-
idiopathic pulmonary fibrosis
- INOS:
-
inducible nitric oxide synthase
- IFN:
-
interferon
- IL:
-
interleukin
- KGF:
-
keratinocyte growth factor
- MMPs:
-
matrix metalloproteinases
- MSCs:
-
mesenchymal stem cells
- MAPC:
-
multiple adult progenitor cells
- NHLBI:
-
National Heart Lung Blood Institute
- NO:
-
nitric oxide
- PDGF:
-
platelet derived growth factor
- PTEN:
-
phosphatase and tensin homolog mutated in multiple advanced cancers 1
- PF:
-
pulmonary fibrosis
- rhHGF:
-
recombinant human hepatocyte growth factor
- ROS:
-
reactive oxygen spices
- SDF-1:
-
stromal-derived factor-1
- TIMPs:
-
tissue inhibitors of metalloproteinases
- TGF:
-
transforming growth factor
- TNF:
-
tumor necrosis factor
References
N. Hiwatari, S. Shimura, T. Sasaki, T. Aikawa, Y. Ando, H. Ishihara, K. Sekizawa, H. Sasaki, and T. Takishima. Prognosis of idiopathic pulmonary fibrosis in-patients with mucous hypersecretion. Am. Rev. Respir. Dis. 143:182–185 (1991).
M. Gharaee-Kermani and S. H. Phan. Molecular mechanisms of and possible treatment strategies for idiopathic pulmonary fibrosis. Curr. Pharm. Des. 11:3943–3971 (2005).
G. Raghu, D. Weycker, J. Edelsberg, W. Z. Bradford, and G. Oster. Incidence and prevalence of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 174:810–816 (2006).
K. Ask, G. E. M. Martin, M. Kolb, and J. Gauldie. Targeting genes for treatment in idiopathic pulmonary fibrosis. Proc. Am. Thorac. Soc. 3:389–393 (2006).
R. M. Strieter. Pathogenesis and natural history of usual interstitial pneumonia: the whole story or the last chapter of a long novel. Chest 128:526S–532S (2005).
T. J. Gross and G. W. Hunninghake. Idiopathic pulmonary fibrosis. N. Engl. J. Med. 345:517–525 (2001).
P. E. Piguet, M. A. Collart, G. E. Grau, A. P. Sappino, and P. Vassalli. Requirement of tumor necrosis factor for development of silica-induced pulmonary fibrosis. Nature 344:245–247 (1990).
M. Gharaee-Kermani and S. H. Phan. Role of cytokines and cytokine therapy in wound healing and fibrotic diseases. Curr. Pharm. Des. 7:1083–1103 (2001)
M. Gharaee-Kermani, Y. Nozaki, K. Hatano, and S. H. Phan. Lung interlukin-4 gene expression in a murine model of bleomycin-induced pulmonary fibrosis. Cytokine 5:138–147 (2001).
F. Huaux, T. Liu, B. McGarry, M. Ullenbruch, and S. H. Phan. Dual roles of interleukin-4 (IL-4) in lung injury and fibrosis. J. Immunol. 170:2083–2092 (2003).
M. Gharaee-Kermani and S. H. Phan. Lung Interleukin-5 expression in murine bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 16:438–447 (1997).
K. Zhang, M. Gharaee-Kermani, B. McGarry, and S. H. Phan. In situ hybridization analysis of rat lung al (I), and a2 (I) collagen gene expression in pulmonary fibrosis induced by endotracheal bleomycin injection. Lab. Invest. 70:192–202 (1994).
K. Zhang, M. Gharaee-Kermani, M. L. Jones, J. S. Warren, and S. H. Phan. Lung monocyte chemoattractant protein-1 gene expression in bleomycin-induced pulmonary fibrosis. J. Immunol. 153:4733–4741 (1994).
K, Zhang, M. Gharaee-Kermani, B. McGarry, D. Remick, and S. H. Phan. TNFα mediated lung cytokine networking and eosinophil recruitment in pulmonary fibrosis. J. Immunol. 158:954–959 (1997).
K. Zhang, K. C. Flanders, and S. H. Phan. Cellular localization of transforming growth factor-β expression in bleomycin-induced pulmonary fibrosis. Am. J. Pathol. 147:352–361 (1995).
V. N. Lama and S. H. Phan. The extrapulmonary origin of fibroblasts. Proc. Am. Thorac. Soc. 3:373–376 (2006).
F. Zuo, N. Kaminski, E. Eugui, J. Allard, Z. Yakhini, A. Ben-Dor, L. Lollini, D. Morris, Y. Kim, B. Delustro, D. Sheppard, A. Pardo, M. Selman, and R. A. Heller. Gene expression analysis reveals matrilysin as a key regulator of pulmonary fibrosis in mice and humans. Proc. Natl. Acad. Sci. U. S. A. 99:6292–6297 (2002).
N. Hashimoto, H. Jin, T. Liu, S. W. Chensue, and S. H. Phan. Bone marrow-derived progenitor cells in pulmonary fibrosis. J. Clin. Invest. 113:243–252 (2004).
S. E. Dunsmore and S. D. Shapiro. The bone marrow leaves its scar: new concepts in pulmonary fibrosis. J. Clin. Invest. 113:180–182 (2004).
R. J. Phillips, M. D. Burdick, K. Hong, M. A. Lutz, L. A. Murray, Y. Y. Xue, J. A. Belperio, M. P. Keane, and R. M. Strieter. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J. Clin. Invest. 114:438–446 (2004).
S. Garantziotis, M. P. Steele, and D. A. Schwartz. Pulmonary fibrosis: thinking outside of the lung. J. Clin. Invest. 114:319–321 (2004).
M. W. Epperly, H. Guo, J. E. Gretton, and J. S. Greenberger. Bone marrow origin of myofibroblasts in irradiation pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 29:213–224 (2003).
T. V. Haaften and B. Thébaud. Adult bone marrow-derived stem cells for the lung: implications for pediatric lung diseases. Pediatr. Res. 59:94R–99R (2006).
G. Ishii, T. Sangai, K. Sugiyama, T. Ito, T. Hasebe, Y. Endoh, J. Magae, and A. Ochiai. in vivo characterization of bone marrow-derived fibroblasts recruited into fibrotic lesions. Stem Cells 23:699–706 (2005).
D. J. Weiss, M. A. Berberich, Z. Borok, D. B. Gail, J. K. Kolls, C. Penland, and D. J. Prockop. Adult stem cells, lung biology, and lung disease. Proc. Am. Thorac. Soc. 3:193–207 (2006).
B. P. Stripp and S. D. Shapiro. Stem cells in lung disease, repair, and the potential for therapeutic interventions. Am. J. Respir. Cell Mol. Biol. 34:517–522 (2006).
C. C. Yen, S. H. Yang, C. Y. Lin, and C. M. Chen. Stem cells in the lung parenchyma and prospects for lung injury therapy. Eur. J. Clin. Investig. 36:310–319 (2006).
M. Rojas, J. Xu, C. R. Woods, A. L. Mora, W. Spears, J. Roman, and K. L. Brigham. Bone marrow-derived mesenchymal stem cells in repair of the injured lung. Am. J. Respir. Cell Mol. Biol. 33:145–152 (2005).
M. J. D. Griffiths, D. Bonnet, and S. M. Janes. Stem cells of the alveolar epithelium. Lancet 366:249–260 (2005).
M. J. Evans and M. H. Kaufman. Establishment in culture of pluripotential cells from mouse embryos. Nature 292:154–156 (1981).
A. G. Smith. Embryo-derived stem cells: of mice and men. Annu. Rev. Cell Dev. Biol. 17:435–462 (2001).
J. H. Kim, J. M. Auerbach, J. A. Rodriguez-Gomez, I. Velasco, D. Gavin, N. Lumelsky, S. H. Lee, J. Nguyen, R. Sanchez-Pernaute, K. Bankiewicz, and R. Mckay. Dopamine neurons derived from embryonic stem cells function in an animal model of Parkinson’s disease. Nature 418:50–56 (2002).
A. McLaren. Ethical and social considerations of stem cell research. Nature 414:129–131 (2001).
A. J. Wagers, and I. L. Weissman. Plasticity of adult stem cells. Cell 116:639–648 (2004).
C. Coraux, B. Nawrocki-Raby, J. Hinnrasky, C. Kileztky, D. Gaullard, C. Dani, and E. Puchelle. Embryonic stem cells generate airway epithelial tissue. Am. J. Respir. Cell Mol. Biol. 32:87–92 (2005).
H. J. Rippon, N. N. Ali, J. M. Polak, and A. E. Bishop. Initial observations on the effect of medium composition on the differentiation of murine embryonic stem cells to alveolar type II cells. Cloning Stem Cells 6:49–56 (2004).
I. P. Neuringer and S. H. Randell. Lung stem cell update: promise and controversy. Monaldi Arch. Chest Dis. 65:47–51 (2006).
T. Asahara, T. Takahashi, H. Masuda, C. Kalka, D. Chen, H. Iwaguro, Y. Inai, M. Silver, and J. M. Isner. VEGF contributes to postnatal neovascularization by mobilizing bone marrow-derived endothelial progenitor cells. EMBO J. 18:3964–3972 (1999).
M. J. Evans, L. J. Cabral-Anderson, and G. Freeman. Role of the Clara cell in renewal of the bronchiolar epithelium. Lab. Invest. 38:648–653 (1978).
M. J. Evans, N. P. Dekker, L. J. Cabral-Anderson, and G. Freeman. Quantitation of damage to the alveolar epithelium by means of type 2 cell proliferation. Am. Rev. Respir. Dis. 118:787–790 (1978).
M. J. Evans, S. G. Shami, L. J. Cabral-Anderson, and N. P. Dekker. Role of nonciliated cells in renewal of the bronchial epithelium of rats exposed to NO2. Am. J. Pathol. 123:126–133 (1986).
A. Giangreco, S. D. Reynolds, and B. R. Stripp. Terminal bronchioles harbor a unique airway stem cell population that localizes to the bronchoalveolar duct junction. Am. J. Pathol. 161:173–182 (2002).
K. U. Hong, S. D. Reynolds, A. Giangreco, C. M. Hurley, and B. R. Stripp. Clara cell secretory protein-expressing cells of the airway neuroepithelial body microenvironment include a label-retaining subset and are critical for epithelial renewal after progenitor cell depletion. Am. J. Respir. Cell Mol. Biol. 24:671–681 (2001).
C. F. Kim, E. L. Jackson, A. E. Woolfenden, S. Lawrence, I. Babar, S. Vogel, D. Crowley, R. T. Bronson, and T. Jacks. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 121:823–835 (2005).
K. Dahlin, E. M. Mager, L. Allen, Z. Tigue, L. Goodglick, M. Wadehra, and L. Dobbs. Identification of genes differentially expressed in rat alveolar type I cells. Am. J. Respir. Cell Mol. Biol. 31:309–316 (2004).
K. H. W. M. Albertine, and D. M. Hyde. Anatomy and development of the respiratory tract. In J. F. Murray, R. J. Mason, and H. A. Boushey (eds.), Textbook of Respiratory Medicine, Saunders, Philadelphia, PA, 2000, pp. 1–33.
W. Olivera, K. Ridge, L. D. Wood, and J. L. Sznajder. Active sodium transport and alveolar epithelial Na-K-ATPase increase during subacute hyperoxia in rats. Am. J. Physiol. 266:L577–L584 (1994).
J. F. Haskell, G. Yue, D. J. Benos, and S. Matalon. Up-regulation of sodium conductive pathways in alveolar type II cells in sublethal hyperoxia. Am. J. Physiol. 266:L30–37 (1994).
S. L. Kauffman. Cell proliferation in the mammalian lung. Int. Rev. Exp. Pathol. 22:131–191 (1980).
M. J. Evans, L. J. Cabral, R. J. Stephens, and G. Freeman. Renewal of alveolar epithelium in the rat following exposure to NO2. Am. J. Pathol. 70:175–198 (1973).
M. J. Evans, L. J. Cabral, R. J. Stephens, and G. Freeman. Transformation of alveolar type II cells to type I cells following exposure to NO2. Exp. Mol. Pathol. 22:142–150 (1975).
W. R. Otto. Lung epithelial stem cells. J. Pathol. 197:527–535 (2002).
C. W. Wuenschell, M. E. Sunday, G. Singh, P. Minoo, H. C. Slavkin, and D. Warburton. Embryonic mouse lung epithelial progenitor cells co-express immunohistochemical markers of diverse mature cell lineages. J. Histochem. Cytochem. 44:113–123 (1996).
A. Meneghetti, W. V. Cardoso, J. S. Brody, and M. C. Williams. Epithelial marker genes are expressed in cultured embryonic rat lung and in vivo with similar spatial and temporal patterns. J. Histochem. Cytochem. 44:1173–1182 (1996).
B. D. Uhal. Cell cycle kinetics in the alveolar epithelium. Am. J. Physiol. 272:L1031–L1045 (1997).
H. Fehrenbach. Alveolar epithelial type II cell: defender of the alveolus revisited. Respir. Res. 2:33–46 (2001).
S. I. Danto, J. M. Shannon, Z. Borok, S. M. Zabski, and E. D. Crandall. Reversible transdifferentiation of alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 12:497–502 (1995).
K. C. Bui, S. Buckley, F. Wu, B. Uhal, I. Joshi, J. Liu, M. Hussain, I. Makhoul, and D. Warburton. Induction of A-and D-type cyclins and cdc2 kinase activity during recovery from short-term hypertoxic lung injury. Am. J. Physiol. 268: L625–L635 (1995).
R. H. Simon. Alveolar epithelial cells in pulmonary fibrosis. In S. H. Phan and R. S. Thrall (eds.), Pulmonary Fibrosis, Dekker, New York, 1995, pp. 511–539.
B. D. Uhal, I. Joshi, A. L. True, S. Mundle, A. Raza, A. Pardo, and M. Selman. Fibroblast isolated after fibrotic lung injury induce apoptosis of alveolar epithelial cells in vitro. Am. J. Physiol. 269:L819–L828 (1995).
J. F. Tomashefski Jr. Pulmonary pathology of acute respiratory distress syndrome. Clin. Chest Med. 21:435–466 (2000).
W. R. Anderson, and K. Thielen. Correlative study of adult respiratory distress syndrome by light, scanning, and transmission electron microscopy. Ultrastruct. Pathol. 16:615–628 (1992).
P. L. Keeling, I. S. Pratt, W. N. Aldridge, and L. L. Smith. The enhancement of paraquat toxicity in rats by 85% oxygen: lethality and cell-specific lung damage. Br. J. Exp. Pathol. 62:643–654 (1981).
D. N. Kotton, B. Y. Ma, W. V. Cardoso, E. A. Sanderson, R. S. Summer, M. C. Williams, and A. Fine. Bone marrow-derived cells as progenitors of lung alveolar epithelium. Development 128:5181–5188 (2001).
D. S. Krause, N. D. Theise, M. I. Collector, O. Henegariu, S. Hwang, R. Gardner, S. Neutzel, and S. J. Sharkis. Multi-organ, multi-lineage engraftment by a single bone marrow-derived stem cell. Cell 105:369–377 (2001).
L. A. Ortiz, F. Gambelli, C. McBride, D. Gaupp, M. Baddoo, N. Kaminski, and D. G. Phinney. Mesenchymal stem cell engraftment in lung is enhanced in response to bleomycin exposure and ameliorates in fibrotic effects. Proc. Natl. Acad. Sci. U. S. A. 108:8407–8411 (2003).
W. Kleeberger, A. Versmold, T. Rothamel, S. Glockner, M. Bredt, A. Haverich, U. Lehmann, and H. Krepe. Increased chimerism of bronchial and alveolar epithelium in human lung allografts undergoing chronic injury. Am. J. Pathol. 162:1487–1494 (2003).
J. Mattsson, M. Jansson, A. Wernerson, and M. Hassan. Lung epithelial cells and type II pneumocytes of donor origin after allogeneic hematopoietic stem cell transplantation. Transplantation 78:154–157 (2004).
B. T. Suratt, C. D. Cool, A. E. Serls, L. Chen, M. Varella-Garcia, E. J. Shpall, K. K. K. Brown, and G. S. Worthen. Human pulmonary chimerism after hematopoietic stem cell transplantation. Am. J. Respir. Crit. Care Med. 168:318–322 (2003).
A. J. Wagers, R. Sherwood, J. L. Christensen, and I. L. Weisssman. Little evidence for developmental plasticity of adult hematopoietic stem cells. Science 297:2256–2259 (2002).
R. Voswinckel, T. Ziegelhoeffer, M. Heil, S. Kostin, G. Brier, T. Mehling, R. Haberberger, M. Clauss, A. Gaumann, W. Schaper, and W. Seeger. Circulating vascular progenitor cells do not contribute to compensatory lung growth. Circ. Res. 93:372–379 (2003).
R. J. Panos, P. M. Bak, W. S. Simonet, J. S. Rubin, and L. J. Smith. Intratracheal instillation of keratinocyte growth factor decreases hyperoxia-induced mortality in rats. J. Clin. Invest. 96:2026–2033 (1995).
T. Yano, R. R. Deterding, W. S. Simonet, J. M. Shannon, and R. J. Mason. Keratinocyte growth factor reduces lung damage due to acid instillation in rats. Am. J. Respir. Cell Mol. Biol. 15:433–442 (1996).
C. F. Kim, E. L. Jackson, A. E. Woolfenden, S. Lawrence, I. Babar, S. Vogel, D. Crowley, R. I. Bronson, and T. Jacks. Identification of bronchio-alveolar stem cells in normal lung and lung cancer. Cell 121:823–835 (2005).
U. Bartram and C. P. Speer. The role of transforming growth factor β in lung development and disease. Chest 125:754–765 (2004).
S. Flecknoe, R. Harding, G. Maritz, and S. B. Hooper. Increased lung expansion alters the proportions of type I and type II alveolar epithelial cells in fetal sheep. Am. J. Physiol. Lung Cell. Mol. Physiol. 278:L1180–L1185 (2000).
Z. Borok, R. L. Lubman, S. I. Danto, X. L. Zhang, S. M. Zabski, L. S. King, D. M. Lee, P. Agre, and E. D. Crandall. Keratinocyte growth factor modulates alveolar epithelial cell phenotype in vitro: expression of aquaporin 5. Am. J. Respir. Cell Mol. Biol. 18:554–561 (1998).
E. Nabeyrat, S. Corroyer, V. Besnard, V. Cazals-Laville, J. Bourbon, and A. Clement. Retinoic acid protects against hyperoxia-mediated-cell-cycle arrest of lung alveolar epithelial cells by preserving late G1 cyclin activities. Am. J. Respir. Cell Mol. Biol. 25:507–514 (2001).
G. D. Massaro and D. Massaro. Retinoic acid treatment abrogates elastase-induced pulmonary emphysema in rats. Nat. Med. 3:675–677 (1997).
E. A. Ozer, A. Kumral, E. Ozer, N. Duman, O. Yilmaz, S. Ozkal, and H. Ozkan. Effect of retinoic acid on oxygen-induced lung injury in the newborn rat. Pediatr. Pulmonol. 39:35–40 (2005).
S. Shoji, R. F. Ertl, J. Linder, D. J. Romberger, and S. I. Rennard. Bronchial epithelial cells produce chemotactic activity for bronchial epithelial cells. Possible role for fibronectin in airway repair. Am. Rev. Respir. Dis. 141:218–225 (1990).
R. H. Bardales, S. S. Xie, R. F. Schaefer, and S. M Hsu. Apoptosis is a major pathway responsible for the resolution of type II pneumocytes in acute lung injury. Am. J. Pathol. 149(3):845–852 (1996).
B. D. Uhal, I. Joshi, W. F. Hughes, C. Ramos, A. Pardo, and M. Selman. Alveolar Epithelial cell death adjacent to underlying myofibroblasts in advanced fibrotic human lung. Am. J. Physiol. 275:L1192–L1199 (1998).
W. M. Haschek, and H. Witschi. Pulmonary fibrosis—a possible mechanism. Toxicol. Appl. Pharmacol. 51:475–487 (1979).
Y. Adamson, L. Young, and D. H. Bowden. Relationship of alveolar epithelial injury and repair to the induction of pulmonary fibrosis. Am. J. Pathol. 130(2):377–383 (1988).
R. F. Pereira, K. W. Halford, M. D. O’Hara, D. B. Leeper, B. P. Sokolov, M. D. Pollard, O. Bagasra, and D. J. Prockop. Cultured adherent cells from marrow can serve as long-lasting precursor cells for bone, cartilage, and lung in irradiated mice. Proc. Natl. Acad. Sci. U. S. A. 92:4857–4861 (1995).
Y. Jiang, B. N. Jahagirdar, R. L. Reinhardt, R. E. Schwartz, C. D. Keene, X. R. Ortiz-Gonzalez, M. Reyes, T. Lenvik, T. Lund, M. Blackstad, J. Du, S. Aldrich, A. Lisherg, W. C. Low, D. A. Largaespada, and C. M. Verfaillie. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 418:41–49 (2002).
A. J. Wagers, J. L. Christensen, and I. L. Weissman. Cell fate determination from stem cells. Gene Ther. 9:606–612 (2002).
M. Korbling and Z. Estrov. Adult stem cells for tissue repair: a new therapeutic concept? N. Engl. J. Med. 349:570–582 (2003).
D. J. Prockop. Further proof of the plasticity of adult stem cells and their role in tissue repair. J. Cell Biol. 160:807–809 (2003).
E. L. Herzog, L. Chai, and D. S. Krause. Plasticity of marrow-derived stem cells. Blood 102:3483–3493 (2003).
F. Anjos-Afonso, E. K. Siapati, and D. Bonnet. in vivo contribution of murine mesenchymal stem cells into multiple cell types under minimal damage conditions. J. Cell Sci. 117:5655–5664 (2004).
I. P. Neuringer and S. H. Randell. Stem cells and repair of lung injuries. Respir. Res. 5:6 (2004).
F. D. Camargo, R. Green, Y. Capetanaki, K. A. Jackson, and M. A. Goodell. Single hematopoietic stem cells generate skeletal muscle through myeloid intermediates. Nat. Med. 9:1520–1527 (2003).
D. S. Krause, N. D. Theise, M. I. Collector, O. Henegariu, S. Hwang, R. Gardner, S. Neutzel, and S. J. Sharkis. Multi-organ, multi-lineage engraftment by a single bone marrow-derived stem cell. Cell 105:369–377 (2001).
D. N. Kotton, B. Y. Ma, W. V. Cardoso, E. A. Sanderson, R. S. Summer, M. C. Williams, and A. Fine. Bone marrow-derived cells as progenitors of lung alveolar epithelium. Development 128:5181–5188 (2001).
N. D. Theise, O. Henegariu, J. Grove, J. Jagirdar, P. N. Kao, J. M. Crawford, S. Badve, R. Saxena, and D. S. Krause. Radiation pneumonitis in mice: a severe injury model for pneumocyte engraftment from bone marrow. Exp. Hematol. 30:1333–1338 (2002).
J. E. Grove, C. Lutzko, J. Priller, O. Henegariu, N. D. Theise, D. B. Kohn, and D. S. Krause. Marrow-derived cells as vehicles for delivery of gene therapy to pulmonary epithelium. Am. J. Respir. Cell Mol. Biol. 27:645–651 (2002).
L. A. Ortiz, F. Gambelli, C. McBride, D. Gaupp, M. Baddoo, N. Kaminski, and D. G. Phinney. Mesenchymal stem cell engraftment in lung is enhanced in response to bleomycin exposure and ameliorates in fibrotic effects. Proc. Natl. Acad. Sci. U. S. A. 100:8407–8411 (2003).
S. Abe, G. Lauby, C. Boyer, S. I. Rennard, and J. G. Sharp. Transplanted BM and BM side population cells contribute progeny to the lung and liver in irradiated mice. Cytotherapy 5:523–533 (2003).
K. Ishizawa, H. Kudo, M. Yamada, S. Kobayashi, M. Numasaki, S. Ueda, T. Suzuki, and H. Sasaki. Bone marrow-derived cells contribute to lung regeneration after elastase-induced pulmonary emphysema. FEBS Lett. 556:249–252 (2004).
M. Yamada, H. Kubo, S. Kobayashi, K. Ishizawa, M. Numasaki, S. Ueda, T. Suzuki, and H. Sasaki. Bone marrow-derived progenitor cells are important for lung repair after lipopolysaccharide-induced lung injury. J. Immunol. 172:1266–1272 (2004).
S. Abe, C. Boyer, X. Liu, F. Q. Wen, T. Kobayashi, Q. Fang, X. Wang, M. Hashimoto, J. G. Sharp, and S. I. Rennard. Cells derived from the circulation contribute to the repair of lung injury. Am. J. Respir. Crit. Care Med. 170:1158–1163 (2004).
M. Dooner, J. Cerny, G. Colvin, D. Demers, J. Pimentel, D. Greer, M. Abedi, C. McAuliffe, and P. Quesenberry. Homing and conversion of murine hematopoietic stem cells to lung. Blood Cells Mol. Diseases 32:47–51 (2004).
T. Beckett, R. Lori, R. Prenovitz, M. Poynter, K. K. Gonez, B. T. Suratt, and D. J. Weiss. Acute lung injury with endotoxin or NO2 does not enhance development of airway epithelium from bone marrow. Molec. Ther. 12:680–686 (2005).
H. Macpherson, P. Keir, S. Webb, K. Samuel, S. Boyle, W. Bickmore, L. Forrester, and J. Dorin. Bone marrow-derived SP cells can contribute to the respiratory tract of mice in vivo. J. Cell Sci. 118:2441–2450 (2005).
A. Schoeberlein, W. Holzgreve, L. Dudler, S. Hahn, and D. V. Surbek. Tissue-specific engraftment after in utero transplantation of allogeneic mesenchymal stem cells into sheep fetuses. Am. J. Obstet. Gynecol. 192:1044–1052 (2005).
R. Loi, T. Beckett, K. K. Goncz, B. T. Suratt, and D. J. Weiss. Limited restoration of defective cystic fibrosis lung epithelium in vivo with adult marrow derived cells. Am. J. Respir. Crit. Care Med. 173:171–179 (2006).
B. T. Suratt, C. D. Cool, A. E. Serls, L. Chen, M. Varella-Garcia, E. J. Shpall, K. K. Brown, and G. S. Worthen. Human pulmonary chimerism after hematopoietic stem cell transplantation. Am. J. Respir. Crit. Care Med. 168:318–322 (2003).
J. Mattsson, M. Jansson, A. Wernerson, and M. Hassan. Lung epithelial cells and type II pneumocytes of donor origin after allogeneic hematopoietic stem cell transplantation. Transplantation 78:154–157 (2004).
N. C. Direkze, S. J. Forbes, M. Brittan, T. Hunt, R. Jeffery, S. L. Preston, R. Poulson, K. Hodivala-Dilke, M. R. Alison, and N. A. Wright. Multiple organ engraftment by bone-marrow-derived myofibroblasts and fibroblasts in bone-marrow-transplanted mice. Stem Cells 21:514–520 (2003).
M. Schmidt, G. Sun, M. A. Stacey, L. Mori, and S. Mattoli. Identification of circulating fibrocytes as precursors of bronchial myofibroblasts in asthma. J. Immunol. 171:380–389 (2003).
B. B. Moore, J. E. Kolodsick, V. J. Thannickal, K. Cooke, T. A. Moore, C. Hogaboam, C. A. Wilke, and G. B. Toews. CCR2-mediated recruitment of fibrocytes to the alveolar space after fibrotic injury. Am. J. Pathol. 166:675–684 (2005).
G. Ishii, T. Sangai, K. Sugiyama, T. Ito, T. Hasebe, Y. Endoh, J. Magae, and A. Ochiai. in vivo characterization of bone marrow-derived fibroblasts recruited into fibrotic lesions. Stem Cells 23:699–706 (2005).
B. N. Gomperts, J. A. Belperio, M. D. Burdick, and R. M. Streiter. Circulating progenitor cells traffic via CXCR4/CXCL12 in response to airway epithelial injury. J. Immunol. 176:1916–1927 (2006).
E. R. Weibel, P. Gehr, D. Haies, J. Gill, and M. Bachofen. The cell population of the normal lung. In A. Bouhuys (ed.), Lung Cells in Disease, Elsevier, Amsterdam, The Netherlands, 1976, pp. 3–16.
O. Skalli, W. Schürch, T. Seemayer, R. Lagacé, D. Montandon, B. Pittett, and G. Gabbiani. Myofibroblasts from diverse pathologic settings are heterogeneous in their content of actin isoforms and intermediate filament proteins. Lab. Invest. 60:275–285 (1989).
S. H. Phan. Role of the myofibroblast in pulmonary fibrosis. Kidney Int. 49(54):S-46–S-48 (1996).
S. G. Roy, Y. Nozaki, and S. H. Phan. Regulation of α-smooth muscle actin gene expression in myofibroblast differentiation from rat lung fibroblasts. Int J. Biochem. Cell. Biol. 33:723–734 (2001).
L. S. Schissei, and D. L. Matthew. Telomerase, myofibroblasts, and pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 34:520–522 (2006).
S. H. Phan. The myofibroblast in pulmonary fibrosis. Chest 122:286S–289S (2002).
M. Gharaee-Kermani and S. H. Phan. Role of fibroblasts and myofibroblasts in idiopathic pulmonary fibrosis. In J. P. Lynch (ed.), Idiopatic Pulmonary Fibrosis, Marrcel Dekker, New York, 2004, pp. 509–561.
Y. Nozaki, T. Liu, K. Hatano, M. Gharaee-Kermani, and S. H. Phan. Induction of telomerase activity in fibroblasts from bleomycin-induced lungs. Am. J. Respir. Cell. Mol. Biol. 23:460–465 (2000).
T. Liu, Y. Nozaki, and S. H. Phan. Regulation of telomerase activity in rat lung fibroblasts. Am. J. Respir. Cell. Mol. Biol. 26:534–540 (2002).
S. H. Phan. The myofibroblast in pulmonary fibrosis. Chest 122:286S–289S (2002).
H. Y. Zhang, M. Gharaee-Kermani, K. Zhang, S. Karmiol, and S. H. Phan. Lung fibroblast alpha-smooth muscle actin expression and contractile phenotype in bleomycin-induced pulmonary fibrosis. Am. J. Pathol. 148:527–537 (1996).
H. Y. Zhang, M. Gharaee-Kermani, and S. H. Phan. Regulation of lung fibroblast alpha-smooth muscle actin expression, contractile phenotype, and apoptosis by IL-1beta. J. Immunol. 158:1392–1399 (1997).
T. Liu, B. Hu, M. J. Chung, M. Ullenbruch, H. Jin, and S. H. Phan. Telomerase regulation of myofibroblast differentiation. Am. J. Respir. Cell Mol. Biol. 34:625–633 (2006).
G. M. Tremblay, M. Jordana, J. Gauldie, and B. Sarnstrand. Fibroblasts as effector cells in fibrosis. In S. H. Phan, and R. S. Thrall (eds.) Pulmonary Fibrosis, Marcel Dekker, New York, 1995, pp. 541–577.
R. Abe, S. C. Donnelly, T. Peng, R. Bucala, and C. N. Metz. Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J. Immunol. 166:7556–7562 (2001).
R. Bucala, L. A. Spiegel, J. Chesney, M. Hogan, and A. Cerami. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol. Med. 1:71–81 (1994).
J. Chesney, M. Bacher, A. Bender, and R. Bucala. The peripheral blood. Fibrocyte is a potent antigen-presenting cell capable of priming naïve T cells in situ. Proc. Natl. Acad. Sci. U. S. A. 94:6307 (1997).
C. N. Metz. Fibrocytes: a unique cell population implicated in wound healing. Cell. Mol. Life Sci. 60:1342–1350 (2003).
J. Chesney, C. Metz, A. B. Stavitsky, M. Bacher, and R. Bucala. Regulated production of type I collagen and inflammatory cytokines by peripheral blood fibrocytes. J. Immunol. 160:419–425 (1998).
M. Schmidt, G. Sun, M. A. Stacey, L. Mori, and S. Mattoli. Identification of circulating fibrocytes as precursors of bronchial myofibroblasts in asthma. J. Immunol. 171:380–389 (2003).
S. Abe, C. Boyer, X. Liu, F. Q. Wen, T. Kobayashi, Q. Fang, X. Wang, M. Hashimoto, J. G. Sharp, and S. I. Rennard. Cells derived from the circulation contribute to the repair of lung injury. Am. J. Respir. Crit. Care Med. 170:1158–1163 (2004).
R. M. Strieter, J. A. Belperio, and M. P. Keane. Cytokines in innate host defense in the lung. J. Clin. Invest. 109:699–705 (2002).
K. Hattori, B. Heissig, K. Tashiro, T. Honjo, M. Tateno, J. H. Shieh, N. R. Hackett, M. S. Quitoriano, R. G. Crystal, S. Raffi, and M. A. Moore. Plasma elevation of stromal cell-derived factor-1 induces mobilization of mature and immature hematopoietic progenitor and stem cells. Blood 97:3354–3360 (2001).
S. Raffi, B. Heissig, and K. Hattori. Efficient mobilization and recruitment of marrow-derived endothelial and hematopoietic stem cells by adenoviral vectors expressing angiogenic factors. Gene Ther. 9:631–641 (2002).
S. P. Zielske and S. E. Braun. Cytokines: value-added products in hematopoietic stem cell gene therapy. Molec. Ther. 10:211–219 (2004).
P. S. Frenette, S. Subbarao, I. B. Mazo, U. H. von Andrian, and D. D. Wagner. Endothelial selectins and vascular cell adhesion molecule-1 promote hematopoietic progenitor homing to bone marrow. Proc. Natl. Acad. Sci. U. S. A. 95:14423–14428 (1998).
M. Selman and A. Pardo. Role of epithelial cells in idiopathic pulmonary fibrosis: from innocent targets to serial killers. Proc. Am. Thorac. Soc. 3:364–372 (2006).
B. C. Willis, J. M. Liebler, K. Luby-Phelps, A. G. Nicholson, E. D. Crandall, R. M. du Bois, and Z. Borok. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta 1: potential role in idiopathic pulmonary fibrosis. Am. J. Pathol. 166:1321–1332 (2005).
M. Chilosi, V. Poletti, A. Zamo, M. Lestani, L. Montagna, P. Piccoli, S. Pedron, M. Bertaso, A. Scarpa, B. Murer, A. Cancellieri, R. Maestro, G. Semenzato, and C. Doglioni. Aberrant Wnt/beta-catenin pathway activation in idiopathic pulmonary fibrosis. Am. J. Pathol. 162:1495–1502 (2003).
M. Selman, A. Pardo, L. Barrera, A. Estrada, S. R. Watson, K. Wilson, N. Aziz, N. Kaminski, and A. Zlotnik. Gene expression profiles distinguish idiopathic pulmonary fibrosis from hypersensitivity pneumonitis. Am. J. Respir. Crit. Care Med. 173:188–198 (2006).
D. S. Krause, N. D. Theise, M. I. Collector, O. Henegariu, S. Hwang, R. Gardner, S. Neutzel, and S. J. Sharkis. Multi-organ, multi-lineage engraftment by a single bone marrow-derived stem cell. Cell 105:369–377 (2001).
N. D. Theise, O. Henegariu, J. Grove, J. Jagirdar, P. N. Kao, J. M. Crawford, S. Badve, S. Saxena, and D. S. Krause. Radiation pneumonitis in mice: a severe injury model for pneumocyte engraftment from bone marrow. Exp. Hematol. 30:1333–1338 (2002).
Z. Zhu, R. J. Homer, Z. Wang, Q. Chen, G. P. Geba, J. Wang, Y. Zhang, and J. A. Elias. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J. Clin. Invest. 103:779–788 (1999).
M. Gharaee-Kermani, R. E. McCullumsmith, I. F. Charo, S. L. Kunkel, and S. H. Phan. CC-chemokine receptor 2 required for bleomycin-induced pulmonary fibrosis. Cytokine 24:266–276 (2003).
Y. Nozaki, T. Liu, K. Hatano, M. Gharaee-Kermani, and S. H. Phan. Induction of telomerase activity in fibroblasts from bleomycin-injured lungs. Am. J. Resp. Cell. Mol. Biol. 23:460–465 (2000).
M. Gharaee-Kermani, M. Ullenbruch, and S. H. Phan. Animal models of pulmonary fibrosis. Methods Mol. Med. 117:251–259 (2005).
M. Gharaee-Kermani, K. Hatano, Y. Nozaki, and S. H. Phan. Gender-based differences in bleomycin-induced pulmonary fibrosis. Am. J. Pathol. 166:1593–1606 (2005).
T. Liu, S. M. Dhanasekaran, H. Jin, B. Hu, S. A. Tomlins, A. M. Chinnaiyan, and S. H. Phan. FIZZI stimulation of myofibroblast differentiation. Am. J. Pathol. 164:1315–1326 (2004).
B. Hu, D. C. Tack, T. Liu, Z. Wu, M. R. Ullenbruch, and S. H. Phan. Role of Smad3 in the regulation of rat telomerase reverse transcriptase by TGFβ. Oncogene 25:1030–1041 (2006)
D. H. Bowden. Unraveling pulmonary fibrosis: the bleomycin model. Lab. Invest. 50:487–488 (1984).
L. A. Ortiz, F. Gambelli, C. McBride, D. Gaupp, M. Baddoo, N. Kaminski, and D. G. Phinney. Mesenchymal stem cell engraftment in lung is enhanced in response to bleomycin exposure and ameliorates its fibrotic effects. Proc. Natl. Acad. Sci. U. S. A. 100:8407–8411 (2003).
M. Gharaee-Kermani, B. McGarry, N. Lukacs, G. Huffnagle, R. W. Egan, and S. H. Phan. The role of IL-5 in bleomycin-induced pulmonary fibrosis. J. Leukoc. Biol. 64:657–666 (1998).
D. S. Krause, N. D. Theise, and M. I. Collector et al. Multi-organ, multi-lineage engraftment by a single bone marrow-derived stem cell. Cell 105:369–377 (2001).
N. D. Theise, O. Henegariu, J. Grove, J. Jagirdar, P. N. Kao, J. M. Crawford, S. Badve, R. Saxena, and D. S. Krause. Radiation pneumonitis in mice: a severe injury model for pneumocyte engraftment from bone marrow. Exp. Hematol. 30:1333–1338 (2002).
N. D. Theise, M. Nimmakayalu, R. Gardner, P. B. Illei, G. Morgan, L. Teperman, O. Henegariu, and D. S. Krause. Liver from bone marrow in humans. Hematology 32:11–16 (2000).
M. A. Laflamme, D. Myerson, J. E. Saffitz, and C. E. Murry. Evidence for cardiomyocyte repopultion by extracardiac progenitors in transplanted human hearts. Circ. Res. 90:634–640 (2002).
F. Quaini, K. Urbanek, A. P. Beltrami, N. Finato, C. A. Beltrami, B. Nadal-Ginard, J. Kagstura, A. Leri, and P. Anversa. Chimerism of the transplanted heart. N. Engl. J. Med. 346:5–15 (2002).
P. Muller, P. Pfeiffer, J. Koglin, H. J. Schafers, U. Seeland, I. Janzen, S. Urbschat, and M. Bohm. Cardiomyocyte of noncardiac origin in myocardial biopsies of human transplanted hearts. Circulation 106:31–35 (2002).
R. Okamoto, T. Yajima, M. Yamazaki, T. Kani, M. Mukai, S. Okamoto, Y. Ikeda, T. Hibi, J. Inazawa, and M. Watanabe. Damaged epithelial regenerated by bone marrow-derived cells in the human gastrointestinal tract. Nat. Med. 8:1011–1017 (2002).
M. Korbling, R. L. Katz, A. Khanna, A. C. Rurfrok, G. Rondon, M. Albitar, R. F. Champlin, and Z. Estrov. Hepatocytes and epithelial cells of donor origin in recipients of peripheral-blood stem cells. N. Engl. J. Med. 346:738–746 (2002)
E. I. Lagaaij, G. F. Cramer-Knijnenburj, F. J. Van kemenade, L. A Van Es, J. A. Bruijn, and G. H. Van Krieken. Endothelial cell chimerism after renal transplantation and vascular rejection. Lancet 357:33–37 (2001).
R. Poulsom, S. J. Forbes, K. Hodivala-Dilke, E. Ryan, S. Wyles, S. Navaratnarasah, R. Jeffery, T. Hunt, M. Allison, T. Cook, C. Busey, and N. A. Wright. Bone marrow contributes to renal parenchymal and regeneration. J. Pathol. 195:229–235 (2001).
R. Glaser, M. Lu, N. Narula, and J. A. Epstein. Smooth muscle cells, but not myocytes, of host origin in transplanted human hearts. Circulation 106:17–19 (2002).
R. H. Hruban, P. P. Long, E. J. Perlman, G. M. Hutchins, W. A. Baumgartner, K. L. Baughman, and C. A. Griffin. Fluorescence in situ hybridization for the Y-chromosome can be used to detect cells of recipient origin in allografted hearts following cardiac transplantation. Am. J. Pathol. 142:975–980 (1993).
B. T. Suratt, C. D. Cool, A. E. Serls, L. Chen, M. Varella-Garcia, E. J. Shpall, K. K. Brown, and G. S. Worthen. Human pulmonary chimerism after hematopoietic stem cells transplantation. Am. J. Respir. Crit. Care Med. 168:318–322 (2003).
S. A. Yousem and E. Sonmez-Alpan. Use of a biotinylated DNA probe specific for the human Y-chromosome in the evaluation of the allograft lung. Chest 99:275–279 (1991).
J. Chesney and R. Bucala. Peripheral blood fibrocytes: novel fibroblast-like cells that present antigen and mediate tissue repair. Biochem. Soc. Trans. 25:520 (1997).
J. Chesney, C. Metz, A. B. Stavitsky, M. Bacher, and R. Bucala. Regulated production of type 1 collagen and inflammatory cytokines by peripheral blood fibrocytes. J. Immunol. 160:419 (1998).
J. Chesney and R. Bucala. Peripheral blood fibrocytes: mesenchimal precursor cells and the pathogenesis of fibrosis. Curr. Rheumatol. Rep. 2:501 (2000).
M. Selman. From anti-inflammatory drugs through antifibrotic agents to lung transportation. A long road of research, clinical attempts, and failures in the treatment of idiopathic pulmonary fibrosis. Chest 123(3):759–761 (2002).
M. M. Abdelaziz, Y. S. Samman, S. O. Wali, and M. M. Hamad. Treatment idiopathic pulmonary fibrosis: Is there anything new. Respiratory 10:284–289 (2005).
M. J. Cushley, A. G. Davison, and R. M. du Bois. The diagnosis, assessment and treatment of diffuse parenchymal lung disease in adults. British Thoracic Society recommendations. Thorax 54:S1–S30 (1999).
R. M. du Bois. Is idiopathic pulmonary fibrosis now treatable? Am. J. Respir. Crit. Care Med. 171(9):939–940 (2005).
M. Selman, V. J. Thannickal, A. Pardo, D. A. Zisman, F. J. Martine, and J. P. Lynch. Idiopathic pulmonary fibrosis: pathogenesis therapeutic approaches. Drugs 64:405–430 (2004).
M. Selman, E. K. Talmadge Jr, and A. Pardo. Idiopathic pulmonary fibrosis: Prevailing and evolving hypotheses about its pathogenesis and implication for therapy. Ann. Intern. Med. 134:136–151 (2001).
J. P. Lynch 3rd, E. White, and K. Flaherty. Corticosteroids in idiopathic pulmonary fibrosis. Curr. Opin. Pulm. Med. 7(5):298–308 (2001).
D. Bouros and K. M. Antoniou. Current and future therapeutic approaches in idiopathic pulmonary fibrosis. Eur. Respir. J. 26:693–702 (2005).
L. Richeldi, H. R. Daviec, G. Ferrara, and F. Franco. Corticosteroids for idiopathic pulmonary fibrosis. Cochrane Database Syst. Rev. 3:CD002880 (2003).
J. J. Swigris, W. G. Kuschner, J. L. Kelsey, and M. K. Gould. Idiopathic pulmonary fibrosis: challenges and opportunities for the clinician and investigator. Chest 127:275–283 (2005).
R. J. Mason, M. I. Schwarz, G. W. Hunninghake, and R. A. Musson. Pharmacological therapy for idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 160:1771–1777 (1999).
R. Ganesh. Idiopathic pulmonary fibrosis: a need for treatment with drugs other than corticosteroids—a role for antifibrotic agents. Mayo Clin. Proc. 72(2):285–287 (1997).
K. M. Antoniou, A. Potaka, D. Bouros, and M. N. Siafakas. Pathogenetic pathways and novel pharmacotherapeutic targets in idiopathic pulmonary fibrosis. Pulm. Pharmacol. Ther. 19:Mar 3 E Pub (2006).
E. A. Bauer and K. J. Valle. Colchicine-induced modulation of collagenase in human skin fibroblast-cultures. J. Invest. Dermatol. 79:398–403 (1982).
C. M. Herbert, K. A. Lindberg, M. I. Jayason, and A. J. Bailey. Biosynthesis and maturation of skin collagen in scleroderma and effect of D-penicillamine. Lancet 1:187–192 (1974).
J. M. Lurton, T. Trejo, and A. S. Narayanan. Pirfenidone inhibits the stimulatory effects of profibrotic cytokines of human lung fibroblasts in vitro. Am. J. Respir. Crit. Care Med. 153:A403 (1996).
G. Gurujeyalakshmi, M. A. Hollinger, and S. N. Giri. Regulation of transforming growth factor-beta1 mRNA expression by taurine and niacin in the bleomycin hamster model of lung fibrosis. Am. J. Respir. Cell Mol. Biol. 18:334–342 (1998).
P. F. Piguet, and C. Vesin. Treatment by human recombinant soluble TNF receptor of pulmonary fibrosis induced by bleomycin or silica in mice. Eur. Respir. J. 7:515–518 (1994).
R. M. Du Bois. Interferon gamma-1b for the treatment of idiopathic pulmonary fibrosis. N. Engl. J. Med. 341(17):1302–1304 (1999).
T. Okada, I. Sugie, and K. Aisaka. Effects of gamma-interferon on collagen and histamine content in bleomycin-induced lung fibrosis in rats. Lymphokine Cytokine Res. 12:87–91 (1993).
E. K. Bajwa, N. T. Ayas, M. Schulzer, E. Mak, J. H. Ryu, and A. Malhotra. Interferon gamma-1b therapy in idiopathic pulmonary fibrosis: a meta analysis. Chest 128:203–206 (2005).
D. Bouros. Current classification of idiopathic interstitial pneumonias. Monaldi Arch. Chest Dis. 55:450–454 (2000).
V. J. Thannickal, K. R. Flaherty, R. C. Hyzy, and J. P. Lynch 3rd. Emerging drugs for idiopathic pulmonary fibrosis. Expert Opin. Emerg. Drugs 10:707–727 (2005).
A. Hildebrand, M. Romaris, L. M. Rasmussen, D. Heinegard, D. R. Twardzik, W. A. Border, and E. Ruoslahti. Interaction of the small interstitial proteoglycans biglycan, decorin and fibromodulin with transforming growth factor beta. Biochem. J. 302:527–534 (1994).
M. Kolb, P. J. Margetts, T. Galt, P. J. Sime, Z. Xing, M. Schmidt, and J. Gauldie. Transient transgene expression of decorin in the lung reduces the fibrotic response to bleomycin. Am. J. Respir. Crit. Care Med. 163:770–777 (2001).
E. Buchdunger, T. Oreilly, and J. Wood. Pharmacology of imatinib (STI571). Eur. J. Cancer. 38(Suppl.5):S28–S36 (2002).
D. Saleh, K. Furukawa, M. S. Tsao, A. Maghazchi, B. Corrin, N. Yanagisawa, P. G. Barnes, and A. Giaid. Elevated expression of endothlin-1 and endothelin-converting enzyme-1 in idiopathic pulmonary fibrosis: possible involvement of proinflammatory cytokines. Am. J. Respir. Cell Mol. Biol. 16:187–193 (1997).
D. Salani, G. Taraboletti, L. Rosano, V. Dicastro, P. Borsotti, R. Giavazzi, and A. Bagnato. Endothelin-1 induce an angiogenic phenotype in cultured endothelial cells and stimulate neovascularization in vivo. Am. J. Pathol. 157:1703–1711 (2000).
K. Kuwano, N. Hagimoto N, and N. Hara. Molecular mechanisms of pulmonary fibrosis and current treatment. Current Molecular Medicine 1:551–573 (2002).
Y. Zhao and D. A. Geverd. Regulation of Smad3 expression in bleomycin-induced pulmonary fibrosis: a negative feedback loop of TGF-beta signaling. Biochem. Biophys. Res. Commun. 294:319–323 (2002).
P. J. Sime and K. M. A. O. Reilly. Fibrosis of the lung and other tissues: new concepts in pathogenesis and treatment. Clin. Immunol. 99(3):308–319 (2001).
B. Hu, Z. Wu, and S. H. Phan. Smad3 mediates transformiong growth factor-beta-induced-alpha-smooth muscle actin expression. Am. J. Respir. Cell. Mol. Biol. 29:397–404 (2003).
M. Huang, S. Sharma, L. X. Zhu, M. P. Keane, J. Luo, L. Zhang, M. D. Burdick, Y. Q. Lin, M. Dohadwala, B. Gardner, R. K. Batra, R. M. Strieter, and S. M. Dubinett. IL-7 inhibits fibroblast TGF-β production and signaling in pulmonary fibrosis. J. Clin. Invest. 109:931–937 (2002).
A. Nakao, M. Fujii, R. Matsumura, K. Kumano, Y. Saito, K. Miyazono, and I. Iwamoto. Transient gene transfer and expression of Smad 7 prevents bleomycin-induced lung fibrosis in mice. J. Clin. Invest. 104:5–11 (1999).
P. Bonniaud, P. G. Margets, M. Kolb, J. A. Schroeder, A. M. Kapoun, D. Damm, A. Murphy, S. Chakravarty, S. Dugar, L. Higgins, A. A. Protter, and J. Gauldie. Progressive transforming growth factor (beta) 1 induced lung fibrosis is blocked by an orally active ALK5 kinase inhibitor. Am. J. Respir. Crit. Care Med. 171:889–898 (2005).
R. Derynck and E. Y. Zhang. Smad-dependent and smad independent pathways in TGF-beta family signaling. Nature 425:577–584 (2003).
G. Singh, L. E. Ling, G. S. Sawyer, W. C. Lee, F. Zhang, and G. N. Yingling. Transforming the TGF beta pathway: Pharmacophore for Tbeta R1 (ALK5). Curr. Opin. Drug Discov. Dev. 7:437–445 (2004).
R. Vittal, J. C. Horowitz, B. B. Moore, H. Zhang, F. G. Martinez, G. B. Toews, T. J. Standiford, and V. J. Thannickal. Modulation of prosurvival signaling in fibroblasts by a protein kinase inhibitor protects against fibrotic tissue injury. Am. J. Pathol. 166:367–375 (2005).
M. Selman and A. Pardo. Idiopathic pulmonary fibrosis: an epithelial fibroblastic cross-talk disorder. Rev. Respir. Res. 3:3 (2002).
B. D. Uhal, I. Joshi, A. L. True, S. Mundle, A. Raza, A. Pardo, and M. Selman. Fibroblasts isolated after fibrotic lung injury induced apoptosis of alveolar epithelial cells in vitro. Am. J. Physiol. 269:L819–L828 (1995).
M. Waghray, Z. Cui, J. C. Horowitz, I. M. Subramanian, F. G. Martinez, G. B. Toews, and V. J. Thannickal. Hydrogen peroxide is a diffusible paracrine signal for the induction of epithelial cell death by activated myofibroblasts. FASEB J. 19:854–856 (2005)
P. M. de Souza and M. A. Lindsay. Apoptosis as a therapeutic target for the treatment of lung disease. Curr. Opin. Pharmacol. 5:232–237 (2005).
K. Kuwno, R. Kunitake, T. Maeyama, N. Hagimoto, M. Kawasaki, T. Matsuba, M. Yoshimi, I. Inoshima, K. Yoshida, and N. Hara. Attenuation of bleomycin-induced peneumopathy in mice by a caspase inhibitor. Am. J. Physiol., Lung Cell. Mol. Physiol. 280:L316–L325. (2001).
R. Wang, O. Ibarra-Sunga, R. Pick, and B. D. Uhal. Abrogation of bleomycin-induced epithelial apoptosis and lung fibrosis by captopril or by a caspase inhibitor. Am. J. Physiol., Lung Cell. Mol. Physiol. 279:L143–L151 (2000).
H. Miyazaki, K. Kuwano, K. Yoshida, T. Maeyama, M. Yoshimi, M. Fujita, N. Hagimoto, R. Yoshida, and Y. Nakanishi. The perforin mediated apoptatic pathway in lung injury and fibrosis. J. Clin. Pathol. 57:1292–1298 (2004).
J. Domagala-Kulawik, P. Droszcz, I. Karszewska, and R. Chazan. Expression of fas antigen in the cells from bronchoalveolar lavage fluid (BALF). Folia Histochem. Cytobiol. 38:185–188 (2000).
N. Hagimoto, K. Kuwano, Y. Nomoto, R. Kunitake, and N. Hara. Apoptosis and expression of fas /fas ligand mRNA in bleomycin-inducd pulmonary fibrosis in mice. Am. J. Respir. Cell Mol. Biol. 16:91–101 (1997).
T. Tsuburai, M. Suzuki, Y. Nagashima, S. Suzuki, S. Inoue, T. Hashiba, A. Ueda, K. Ikehara, T. Matsuse, and Y. Ishigatsubo. Adenovirus-mediated transfer and overexpression of heme oxygenase 1 cDNA in lung prevents bleomycin-induced pulmonary fibrosis via a Fas-Fas ligand-independent pathway. Hum. Gene Ther. 13:1945–1960 (2002).
G. Raghu, S. Safrin, D. Weycker, K. Starko, and W. Z. Bradford. Association of statins and angiotensin converting enzyme inhibitor use with survival and diseases progression in patient with IPF. Am. J. Respir. Crit. Care Med. 169:A706 (2004).
M. P. Keane, D. A. Arenberg, J. P. Lynch, R. I. Whyte, M. D. Iannettoni, M. D. Burdick, C. A. Wilke, S. B. Morris, M. C. Glass, B. DiGiovine, S. L. Kunkel, and R. M. Strieter. The CXC chemokines IL-8 and IP-10 regulate angiogenic activity in idiopathic pulmonary fibrosis. J. Immunol. 159:1437–1443 (1997).
M. P. Keane. Angiogenesis and pulmonary fibrosis. Feast or famine? Editorial. Am. J. Respir. Crit. Care Med. 170:207 (2004).
R. M. Strieter, K. M. Starko, R. I. Enelow, I. Noth, and V. G. Valentine. Idiopathic pulmonary fibrosis. Biomarkers study group effects of interferon gamma-1b on biomarker expression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 170:133–140 (2004).
J. A. Belperio, M. P. Keane, D. A. Arenberg, C. L. Addison, J. E. Ehlert, M. D. Burdick, and R. M. Strieter. CXC chemokine in angiogenesis. J. Leukoc. Biol. 68:1–8 (2000).
M. P. Keane, J. A. Belperio, M. D. Brudick, J. P. Lynch III, M. C. Fishbein, and R. M. Srieter. ENA-78 is an important angiogenic factor in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 164:2239–2242 (2001).
J. A. Belperio, M. P. Keane, M. D. Burdick, V. Londhe, Y. Y. Xue, K. Li, R. J. Phillips, and R. M. Strieter. Critical role for CXCR2 and CXCR2 ligands during the pathogenesis of ventilator-induced lung injury. J. Clin. Invest. 110:1703–1716 (2002).
D. Bouros, K. M. Antoniou, and N. M. Siafakas. Does interferon gamma-1b therapy improve survival in IPF via an angiogenetic pathways? Chest (electronic letter to editor) (2005).
K. M. Antoniou, M. G. Alexandrakis, K. Sfirivaki, I. Tsiligialli, K. Perisinakis, F. G. Tzortzaki, N. M. Siafakas, and D. E. Bouros. Th1 cytokine pattern (IL-12 and IL-18) in BALF before and after treatment with interferon gamma-1b (IFN-γ-1b) or colchicine in patients with in idiopathic pulmonary fibrosis (IPF/UIP). Sarcoidosis Vasc. Diffuse Lung Dis. 21:105–110 (2004).
E. D. Bateman, N. Turner-Warwick, and B. C. Adelmann-Grill. Immunohistochemical study of collagen types in human fetal lung and fibrotic lung disease. Thorax 36:645–653 (1981).
M. M. Gueders, J. M. Foidart, A. Noel, and D. D. Cataldo. Matrix metalloproteinases (MMPs) and tissue inhibitors of MMPs in the respiratory track: potential implications in asthma and other lung diseases. Eur. J. Pharmacol. 533:133–144 (2006).
M. Selman, V. Ruiz, S. Cabrera, L. Segura, R. Ramirez, R. Barrios, and A. Pardo. Localization of tissue inhibitor of metalloproteinases (TIMPs) −1, −2, −3, and −4 in idiopathic pulmonary fibrosis. Am. J. Physiol. 279:L562–L574 (2000).
H. Nagase and J. F. Woessner Jr. Matrix metalloproteinases. J. Biol. Chem. 274:21491–21494 (1999).
W. C. Parks and S. D. Shapiro. Matrix metalloproteinases in lung biology. Respir. Res. 2:10–19 (2001).
J. F. Woessner, Jr. MMPs and TIMPs: an histological perspective. Methods Mol. Biol. 151:1–23 (2001).
W. Bode, C. Fernadez-Catalan, H. Tschesche, F. Grams, H. Nagase, and K. Maskos. Structural properties of matrix metalloproteinases. Cell. Mol. Life Sci. 55:639–652 (1999).
M. D. Sternlicht and Z. Werb. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 17:463–516 (2001).
J. Atkinson and M. Senior. Matrix metalloproteinase-9 in lung remodeling. Am. J. Respir. Cell Mol. Biol. 28:12–24 (2003).
J. R. Seibold, J. H. Korn, R. Simms, P. J. Clements, I. W. Moreland, M. D. Mayes, D. E. Furst, N. Rothfield, V. Steen, M. Weisman, D. Collier, F. M. Wigley, P. A. Merkel, M. E. Csuka, V. Shu, S. Rocco, M. Erikson, J. Hannigan, W. S. Harkonen, and M. E. Sanders. Recombinant human relaxin in the treatment of scleroderma. A randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 132:871–879 (2000).
N. Bhatt, C. P. Baran, J. Allen, C. Magro, and C. B. Marsh. Promising pharmacologic innovations in treating pulmonary fibrosis. Curr. Opin. Pharmacol. 6:284–292 (2006).
G. W. Hunninghaki. Antioxidant therapy for idiopathic pulmonary fibrosis. N. Engl. J. Med. 353:2285–2287 (2005).
R. G. Crystal. Oxidant and respiratory track epithelial injury pathogenesis and strategies for therapeutic intervention. Am. J. Med. 91:395–445 (1991).
M. Demedts, J. Behr, R. Buhl, U. Costabel, R. Dekhuijze, H. M. Jansen, W. Macnee, M. Thomeet, B. Wallaert, F. Laurent, A. G. Nicholson, F. K. Verbeken, J. Verschakelen, C. D. Flower, F. Capron, S. Petruzzelli, P. De Vuyst, G. M. Van den Bosch, E. Rodriguez-Becerra, G. Corvasce, I. Lankhorst, M. Sardina, and M. Montanari. IFIGENIA study group, high dose acetyl cysteine in idiopathic pulmonary fibrosis. N. Engl. J. Med. 353:2229–2242 (2005).
X. Y. Zhang, S. Shimura, T. Masuda, H. Saitoh, and K. Shirato. Antisense oligonucleotides to NF-kappaB improve survival in bleomycin-induced pneumopathy of the mouse. Am. J. Respir. Crit. Care Med. 162:1561–1568 (2000).
G. Guruleyalakshmi, Y. Wang, and S. N. Giri. Suppression of bleomycin-induced nitric oxide production in mice by taurin and nicin. Nitric Oxide 4:399–411 (2000).
D. M. Hyde, S. N. Giri, M. J. Schiedt, and G. A. Krishna. Effect of three cysteine pro-drugs on bleomycin-induced lung fibrosis in hamsters. Pathol. 22:93–101 (1990).
B. C. Willis, J. M. Liebler, K. Luby-Phelps, A. G. Nicholson, E. D. Crandall, R. M. du Bois, and Z. Borok. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-β1. Am. J. Pathol. 166:1321–1332 (2005).
H. Kasai, J. T. Allen, R. M. Mason, T. Kamimura, and Z. Zhang. TGF-β1 induced human alveolar epithelial to mesenchymal cell. Respir. Res. 6:56–71 (2005).
M. Yaekashiwa, S. Nakayama, K. Ohnuma, T. Sakai, T. Abe, K. Satoh, K. Matsumoto, T. Nakamura, T. Takahashi, and T. Nukiwa. Simultaneous or delayed administration of hepatocyte growth factor equally represses the changes in murine lung injury induced by bleomycin. Am. J. Respir. Crit. Care Med. 156:1937–1944 (1997).
M. Dohi, T. Hasegawa, K. Yamamoto, and B. C. Marshil. Hepatocyte growth factor attenuates collagen accumulation in murine model of pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 162:2302–2307 (2000).
Y. Umeda, T. Marui, Y. Matsuno, K. Shirahashi, H. Iwata, and H. Takagi. Skeletal muscle targeting in vivo electroporation-mediated HGF gene therapy of bleomycin-induced pulmonary fibrosis in mice. Lab. Invest. 84:836–844 (2004).
M.Tamura, J. Gu, T. Takino, and K. M. Yamada. Tumor suppressor PTEN inhibition of cell invasion, migration and growth: differential involvement of local adhesion kinase and p130Cas. Cancer Res. 59:442–449 (1999).
V. Stambolic, A. Suzuki, J. L. Delapompa, G. M. Brothers, C. Mirtsos, T. Sasali, J. Rulnd, J. M. Penninger, D. P. Siderovski, and T. W. Mak. Negative regulatin of PKB/AKt-dependent cell survival by the tumor suppressor PTEN. Cell 95:29–39 (1998).
E. S. White, V. J. Thnnickal, S. L. Carskadon, E. G. Dickie, D. L. Livant, S. Markwart, G. B. Toews, and D. A. Arenberg. Integrin α4β1 regulates migration across basement membranes by lung fibroblast: a role for phosphatase and tensin homologue deleted on chromosome 10. Am. J. Respir. Crit. Care Med. 168:436–442 (2003).
E. S. White, R. G. Atrasz, B. Hu, S. H. Phan, V. Stambolic, T. W. Mak, C. M. Hogabom, K. R. Flaherty, F. J. Martinz, C. D. Kontos, and G. B. Toews. Negative regulation of myofibroblast by PTEN (phosphatase and tensin homologue deleted on chromosome 10). Am. J. Respir. Crit. Care Med. 173:112–121 (2006).
K. Kuwano. PTEN as a new agent in the fight against firogenesis. Am. J. Respir. Crit. Care Med. 173:5–6 (2006).
J. M. Hill, G. Zalos, J. P. Halcox, W. H. Schenke, M. A. Waclawiw, A. A. Quyyumi, and T. Finkel. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N. Engl. J. Med. 347(7):593–600 (2003).
Y. Jiang, B. N. Jahagirdar, R. L. Reinhardt, R. E. Schwartz, C. D. Keene, X. R. Ortiz-Gonzalez, M. Reyes, T. Lenvik, T. Lund, M. Blackstand, J. Du, S. Aldrich, A. Lisberg, W. C. Low, D. A. Largaespada, and C. M. Verfaillie. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 418(6893):41–49 (2002).
M. Yamada, H. Kubo, S. Kobayashi, K. Ishizawa, M. Numasaki, S. Ueda, T. Suzuki, and H. Sasaki. Bone marrow-derived progenitor cells are important for lung repair after lipopolysaccharide-induced lung injury. J. Immunol. 172:1266–1272 (2004).
N. Terada, T. Hamazaki, M. Oka, M. Hoki, D. M. Mastalerz, Y. Nakano, E. M. Meyer, L. Morel, B. E. Petersen, and E. W. Scott. Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature 416(6880):542–545 (2002).
X. Wang, H. Willenbring, Y. Akkari, Y. Torimaru, M. Foster, M. Al-Dhalimy, E. Lagasse, M. Finegold, S. Olson, and M. Grompe. Cell fusion is the principal source of bone-marrow-derived hepatocytes. Nature 422(6934):897–901 (2003).
G. Vassilopoulos, P. R. Wang, and D. W. Russell. Transplanted bone marrow regenerates liver by cell fusion. Nature 422(6934):901–904 (2003).
F. D. Camargo, M. Finegold, and M. A. Goodell. Hematopoietic myelomonocytic cells are the major source of hepatocyte fusion partners. J. Clin. Invest. 113:1266–1270 (2004).
I. Hartlapp, R. Abe, R. W. Saeed, T. Peng, W. Voelter, R. Bucala, and C. N. Metz. Fibrocytes induce an angiogenic phenotype in cultured endothelial cells and promote angiogenesis in vivo. FASEB J. 15:2215–2224 (2001).
K. Ishizawa, H. Kubo, S. Kobayashi, K. Ishizawa, M. Numasaki, S. Ueda, T. Suzuki, and H. Sasaki. Bone marrow-derived cells contribute to lung regeneration after elastase-induced pulmonary emphysema. FEBS 556:249–252 (2004).
J. H. Ryu, T. V. Colby, and T. E. Hartman. Idiopathic pulmonary fibrosis: current concepts. Mayo Clin. Proc. 73:1085–1101 (1998).
Acknowledgements
This work was supported by grants from the National Institutes of Health (HL28737, HL31963, HL52285 and HL77297 to S.H.P.) and the Office of Veterans’ Affairs (VA Merit to M.R.G.). We would like to acknowledge Jana Jacobs for critical review of manuscript and the excellent artwork of Robin G. Kunkel and Elizabeth A. Horn.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gharaee-Kermani, M., Gyetko, M.R., Hu, B. et al. New Insights into the Pathogenesis and Treatment of Idiopathic Pulmonary Fibrosis: A Potential Role for Stem Cells in the Lung Parenchyma and Implications for Therapy. Pharm Res 24, 819–841 (2007). https://doi.org/10.1007/s11095-006-9216-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-006-9216-x