
Abstract
Preclinical studies suggest that mesenchymal stem cells (MSCs) may represent a potential therapeutic option for the treatment of chronic lung diseases including Idiopathic Pulmonary Fibrosis (IPF). IPF is an inexorably progressive lung disease of unknown origin. Despite the recent availability of two approved treatment options, median survival remains poor at 3–5 years.
While there remains a pressing need for further exploration of interval endpoints and biomarkers, promising results of Phase 1 studies of MSCs have reduced safety concerns and encouraged further interest in the potential applicability of cell-based therapeutic approaches for chronic lung diseases like IPF. This chapter will summarize the current state of knowledge regarding the use of stem cells for the treatment of IPF, present important safety and efficacy issues highlighting current and future challenges, and address the need for large, multicenter clinical trials coupled with realistic end-points, including biomarkers, to assess treatment efficacy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Interstitial lung fibrosis may develop as a consequence of occupational or drug exposures, lung injury, or as the end stage of chronic intersitital lung disease. The pathogenesis of lung fibrosis remains elusive and controversial, but prevailing hypotheses assume an ineffective wound healing response to alveolar epithelial cell injury [1, 2] Injury magnitude and susceptibility appears to be related to aging and genetic predisposition, with subsequent innate immune system and fibroblast activation [1, 3, 4].
In the right clinical picture, and in the absence of other known causes of lung fibrosis, the diagnosis of IPF may be made by typical radiologic findings on high-resolution computed tomography (subpleural and basilar predominance of honeycomb cysts and reticulation). In cases where the diagnosis is not clear, lung biopsy may be necessary. Histologically, IPF is identified by the presence of the usual interstitial pneumonia (UIP) pattern with extracellular matrix deposition, phenotypic alterations of fibroblasts and alveolar epithelial cells, formation of fibroblastic foci, and regional and temporal heterogeneity characterized by scattered areas of aberrant wound healing interspersed with normal lung parenchyma [1, 2, 5,6,7,8,9,10,11,12,13].
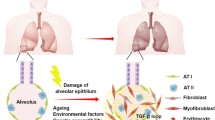
Evidence suggests that areas of fibrosis seen in the lungs of patients with IPF share features associated with normal aging lung, such as genomic instability, telomere attrition, mitochondrial dysfunction, cellular senescence, and immune dysregulation [9, 14, 15] (Fig. 7.1). Because of this overlap, connections between IPF and diseases of premature aging have been postulated.

Aged mesenchymal stem cells in aging lungs and IPF
Partly due to the inefficacy of immunomodulatory and immunosuppressive agents in the treatment of IPF, the role of the immune system in the pathogenesis of IPF remains poorly understood [16,17,18,19,20,21]. However, a link between IPF and immune dysregulation is suggested by the presence of highly activated and proliferative CD4+ cells and functional impairment of T-regulatory cells in patients with IPF [9, 22, 23]. Pathologic features of the epithelium suggest that a dysregulation of progenitor cells may contribute to the IPF phenotype, with abnormal cell cycling resulting in dysfunctional repair [24].
Currently, pirfenidone and nintedanib are the only two FDA approved compounds for the treatment of IPF. Pirfenidone, an antifibrotic compound with an unknown mechanism of action, targets several molecules including transforming growth factor-β (TGF-β), tumor necrosis factor-α (TNF-α), and interleukin-6 [25]. Nintedanib, a tyrosine kinase inhibitor, targets vascular endothelial growth factor receptor (VEGFR), fibroblast growth factor receptor (FGFR), and platelet-derived growth factor receptor (PDGFR) [21]. Pirfenidone was shown to slow the rate of decline in forced vital capacity (FVC) and 6-min walking distance in patients with IPF, and may improve mortality in select patients [3]. Nintedanib was also shown to slow the rate of decline in FVC with a trend toward reduced mortality [4]. However, neither of these compounds has been shown to ameliorate respiratory symptoms or improve acute exacerbation rates, and lung function has continued to decline in all trials completed to date. In addition, though pirfenidone and nintedanib have been shown to slow the progression of IPF [26,27,28], both compounds are associated with significant side effects [28,29,30]. The only definitive treatment for IPF at this time is lung transplantation. Morbidity and mortality from IPF remains high and thus there is a pressing need for alternative therapeutic options for this complex and devastating disease.
Ongoing clinical trials of other potential therapeutic targets include mofiers of connective tissue growth factor, IL-4 and IL-13, galectin-3, lysophosphatidic acid, the phosphoinositide 3-kinase pathway, and finally, mesenchymal stromal cells (MSCs).
2 Animal Models of Pulmonary Fibrosis
Spontaneous pulmonary fibrosis does occur in nonhuman animals, including ferrets, dogs, horses, donkeys, and cats. While the fibrotic lungs of these animals share many characteristics with the lungs of humans with IPF, current veterinary classifications of fibrotic lung disease are not equivalent. The field of comparative oncology has set the stage for collaborations utilizing spontaneous models of progressive fibrotic lung diseases of mutual interest to veterinary and human medicine. The results of these kinds of studies promise to enhance the understanding of common factors important to disease development in a variety of species and to refine treatments for both humans and animals. Moreover, they may provide insights into unanswered questions involving naturally occurring models of pulmonary fibrosis. However, further studies in veterinary models of lung fibrosis are needed to define their relation to human disease and their potential use as models for the development of effective treatments.
Because no reliable spontaneous animal model exists, understanding the pathogenesis of IPF and other fibrotic lung disorders has primarily relied on research using animal models of induced lung fibrosis. Unfortunately, although some of these animal models exhibit progressive disease, none fully recapitulates the histological pattern of UIP. Traditional animal models of lung fibrosis have generated important insights into the pathobiology of lung injury, inflammation, and fibroproliferation [8]. Although it is appreciated that the spontaneous development of fibrosis in other species (e.g., ferrets, donkey, sheep, cats, horses, and dogs) [9,10,11] can be instructive, the most tractable models for studies of pathogenesis involve rodents. Traditionally, preclinical trials have utilized mouse models of bleomycin (BLM)-induced pulmonary fibrosis and studies of BLM-induced fibrosis in aged male mice remain the most clinically relevant model for preclinical studies of IPF because young mice treated with BLM may show recovery from pulmonary fibrosis, an event not appreciated in human fibrotic lung disease.
BLM is a chemotherapeutic antibiotic first identified as a pro-fibrotic agent after the development of pulmonary fibrosis in patients being treated for lymphoma. BLM has been studied in multiple species including mice, rats, sheep, guinea pigs, hamsters, dogs, and primates and in various modes of administration [20, 21], but the consensus view at this time is that the intratracheal murine BLM model is “the best-characterized animal model available for preclinical testing” of IPF [31, 32].
BLM acts by causing single- and double-strand DNA breaks thereby inducing apoptosis. BLM hydrolase, a BLM-inactivating enzyme, influences drug effects on a tissue-specific basis. Becuase the lungs maintain low levels of this enzyme, lung tissue is highly susceptible to BLM-induced injury. An overproduction of reactive oxygen species, due to chelation of metal ions and reaction of the formed pseudoenzyme with oxygen, leads to epithelial cell death (days 1–3), excessive inflammatory infiltrates (days 3–9, neutrophils found in the bronchoalveolar lavage fluid at day 3 and lymphocytes at day 6), and ultimately to fibroblast activation, extracellular matrix deposition, and development of fibrosis (days 10–21 with a peak around day 14). These changes are seen at both the molecular [23, 25, 26] and histologic [20, 23, 25, 27] levels.
The early molecular signature of BLM-induced injury appears to be most similar to the accelerated acute phase of IPF in humans [28]. Measurements of alveolar septal thickening, intra-alveolar fibrosis, increases in alveolar macrophages, and dilation of bronchioles and alveolar ducts demonstrates fairly uniform fibrosis [29]. Nevertheless, the BLM-induced lung fibrosis model is not perfectly representative of IPF. The rapidity of development of BLM-induced fibrosis, the marked inflammation preceding fibrosis, and the possibility of spontaneous resolution are signignificnt differences between the BLM model and human IPF.
C57BL/6J mice have been the predominant animal model, as this particular strain is highly susceptible to lung injury following intratracheal BLM administration [30, 33]. Conversely, the BALB/c or SV129 strains confer resistance to BLM-induced pulmonary fibrosis, presumably due to alterations in transforming growth factor (TGF)-β expression [33].
While BLM-induced lung injury has been studied via intratracheal, intraperitoneal, subcutaneous, intravenous, and inhalational delivery methods, the intratracheal route is most commonly used because it best recapitulates the human phenotype which is limited to the lungs [20, 23, 26, 28, 29, 34,35,36,37,38,39]. Another issue identified in studies using the BLM mouse model is the wide range of dosing regimens used [40]. In mouse studies, weight-based dosing is most common [35, 36, 39] and slightly lower doses (2.0–2.5 U/kg) appear to provide the most effective model of lung fibrosis, while reducing sample loss due to high mortality [28]. With regards to the frequency of dosing, repetitive dosing in young mice was found to promote persistent fibrosis as evidenced by measures of hydroxyproline content and inflammatory cell infiltrates. In contrast, single-dose experiments have demonstrated spontaneous resolution in young mice [23, 41].
Animal models of IPF have yielded valuable insights, but studies using these models also have important limitations. Most studies of therapeutic interventions use a single dose of intratracheal BLM followed shortly thereafter by the administration of the therapy under investigation [35,36,37,38]. Because these thrapuetic agents are often administered within the first 1–7 days following BLM exposure, their observed effects may be due to prevention of the inflammatory cascade rather than reversal of fibrosis, thus limiting their applicability to human IPF [40]. More recent studies have begun to explore administration of drugs after 7 days [42, 43] and, to our knowledge, only two studies to date have evaluated repetitive BLM injury [44, 45]. Intriguingly, pirfenidone and nintedanib received approval to proceed to clinical trials based on preventive protocols or even therapeutic protocols targeting the inflammatory or the early-fibrotic phase of the BLM model [46,47,48].
In addition, most studies investigating BLM-induced pulmonary fibrosis have used young male mouse models, aged 8–12 weeks [28, 29, 34]. Young mice, however, have been shown to undergo spontaneous resolution of BLM-induced pulmonary fibrosis, a phenomenon not observed in aged mice [23, 41, 49]. Whether sex differences in mice parallel human IPF, which exhibits a tendency toward male predominance, has not been fully determined. However, the use of aged male mice may provide a more clinically relevant model of IPF [49]. Many of the hallmarks of aging, including genomic instability, telomere attrition, epigenetic alterations, deregulated cellular bioenergetics, and cellular senescence, are also seen in fibrotic lung disease [50,51,52]. Studies have shown that older mice are more susceptible than younger mice to pro-fibrotic stimuli including BLM [26]. This is of particular interest given that IPF is predominantly seen in older individuals. Transgenic deletion of senescence-related genes including RAGE and relaxin has been associated with spontaneous age-dependent development of lung fibrosis indicating a role for aging in disease susceptibility [31, 53, 54].
The preclinical efficacy of the majority of antifibrotic agents tested in animal models has utilized a single model, (most often BLM) and has measured histologic, not clinical, endpoints. In addition, the lack of blinding in most preclinical animal studies may contribute to evaluation bias in outcomes. Reproducibility issues arising from different experimental settings could also account for discrepancies in treatment effects and lack of generalizability. As with all studies, when using animal models, sample size must be balanced with the statistical power needed to generate robust data. Finally, insufficient reporting of experimental animal data or unpublished negative therapeutic results severely hamper the validity of experimental studies.
3 Rationale for Stem Cell Therapy
A stem cell is defined as an undifferentiated cell capable of self-renewal and multipotent differentiation potential. To achieve this remarkable task, stem cells undergo asymmetric cell division whereby one daughter cell is maintained as a self-renewing stem cell and the other becomes a precursor or progenitor cell capable of giving rise to futher differentiated cells. A progenitor cell shares the potential for differentiation into different tissue lineages, but has limited self-renewal capacity [6, 9].
Due to ethical issues and ecumenical directives, embryonic stem cells are not used in the research of human disease. Other stem cells used in research and disease treatment include tissue-specific stem cells, mesenchymal stem cells (MSCs), fetal stem cells, and cord blood stem cells. In addition, research has been directed at tissue-specific stem cells which reside in and give rise to mature parenchymal cells within a particular tissue or organ.
Mesenchymal stem cells (MSCs) have been shown to have immunomodulatory, antiproliferative, and anti-inflammatory effects. In addition, because of their migratory ability and immune-privileged state, much research has been aimed at understanding the therapeutic poetntial of MSCs. Among others, the therpuetic role of MSCs has been explored in cardiac ischemia, autoimmune disorders [10], severe graft-versus-host disease [11], chronic lung diseases [12,13,14,15], and acute lung injury [16,17,18,19,20].
While researchers, biologists, and bioethicists have rallied the medical community to improve our understanding of the biology and mechanism of action of stem cells, confusion over what exactly is a “stem cell” has led to a lack of standardization in production processes [7] and the indiscriminate commercialization of various cell products as purported treatments for patients [9].
4 The Role of Stem Cells in Lung Repair
Various stem/progenitor cells that function in lung repair reside in other areas of the body and are recruited in times of injury and inflammation. Cells from the bone marrow, blood, adipose tissue, placenta, and umbilical cord have been shown to structurally engraft in the airway [34, 49] as well as the pulmonary vasculature. In addition, evidence suggests that bone marrow-derived cells such as MSCs are recruited to areas of lung injury and exert their regenerative effects via a paracrine function [35, 41].
MSCs are multipotent and have a diverse but restricted differentiation capability. They appear to function in a paracrine manner with minimal engraftment, interact with the innate and adaptive immune systems [36, 37], and aid in lung repair and regeneration via secretion of cytokines and growth factors to restore alveolar epithelial and endothelial permeability [38,39,40, 42].
Other sources of stem cells have also been studied in lung disease. Endothelial progenitor cells (EPCs) appear to exert their therapeutic effects via direct differentiation and engraftment into the vasculature of the lung and secretion of factors that mobilize endothelial and progenitor cells and represent a promising source for pulmonary vascular regeneration [55]. Amniotic fluid stem cells (AFSCs), multipotent fetal-associated cells that can be easily and ethically obtained from amniocentesis specimens, have been shown to improve lung density and function in models of diaphragmatic hernia [56]. AFSCs have also been shown to integrate into areas of distal lung epitheleial injury with expedited repair and expression of NKX2-1 and SFPTC (markers of alveolar type 2 cell diffrernitation) [24, 49]. Human amnion epithelial cells (hAECs), found in the lining of the placenta, display low immunogenicity and also appear to possess regenerative and anti-inflammatory properties [57,58,59,60].
5 Endogenous Stem Cells
Recent work has called into question the existence of stem cells in slowly renewing tissues like the lung [44, 45]. The epithelium lining pulmonary airways turns over slowly during the normal process of tissue maintenance and is replaced far more slowly than specialized post-mitotic cell types of the gut or the epidermis, a property that is reflected in the functional characteristics of airway progenitor cells in their resting versus proliferative states. Therefore, it is not surprising that progenitor cell hierarchies of the lung and other slowly renewing tissues do not fit the classical stem cell hierarchies described in tissues of rapid turnover [42]. In fact, Hu and colleagues, through use of in vivo injury models, have recently described the existence of a stem cell capable of renewing the endocrine pancreas, a tissue that until recently was thought to be maintained solely through self-duplication of differentiated β cells [61]. This, and previous work in lung to be discussed in more detail below, illustrate a deviation from the classical stem cell hierarchy marked by lack of an obligate transit-amplifying progenitor cell in the steady state. Rather, slowly repairing tissues such as lung are maintained at steady state by an abundant facultative transit-amplifying progenitor that fulfils characteristics of a differentiated cell type in the quiescent state, yet retains proliferative capacity and the ability to generate daughter cells capable of generating other specialized lineages [61]. Therefore, the endogenous stem cell at steady state likely remains quiescent. However, studies utilizing in vivo injury models have revealed stem cells that can be functionally distinguished from facultative progenitors, based upon their resistance to environmental stimuli and spatial localization in the conducting airway.
6 Clara Cells
Within the normal lung, Clara cell proliferation maintains the facultative progenitor cell pool (self-renewal) and restores terminally differentiated cells of the conducting airway epithelium (ciliated cells). This vast reparative reservoir distinguishes lung epithelia from tissues such as the intestine that are maintained through proliferation and differentiation of tissue-specific stem cells. The unique features of lung epithelial maintenance and repair suggest that chronic lung disease could be treated through interventions that stabilize the Clara cell pool or by cell replacement strategies that restore this abundant cell type [62].
-
Clara cells are non-ciliated secretory cells in the small airways and trachea. Their morphology and biochemical composition display amazing heterogeneity within the airway epithelium of a single species, among different species, and in response to injury.
-
Clara cells have several lung protective functions. They detoxify xenobiotics and oxidant gasses, control the extent of inflammation, participate in mucociliary clearance of environmental agents, and proliferate/differentiate to maintain the ciliated cell population.
-
Clara cells are secretory and the source of Clara cell secretory protein (CCSP) and contribute surfactant apoproteins A, B, and D, proteases, antimicrobial peptides, several cytokines and chemokines, and mucins to the extracellular fluid lining the airspaces.
-
In humans, many forms of lung cancer may originate from Clara cells, including adenocarcinoma, the most frequently diagnosed form of lung cancer. Whether Clara cells have a similar etiologic function in mouse models of adenocarcinoma is more controversial [62].
7 Mesenchymal Stem Cells
MSCs, first desccibed by Friedenstein et al. in 1968, are a class of multipotent stem cells with self-proliferative and differentiation potential [40, 42]. MSCs may be isolated from bone marrow as well as other tissues including gingival, adipose, umbilical cord, and placenta. Isolation of MSCs requires that cells (1) exhibit fibroblastic morphology, clonogenicty, and plastic adherence when cultured in standard tissue culture conditions; (2) differentiate into adipocytes, osteoblasts, and chondrocytes in vitro; and (3) express certain cell surface markers such as CD44, Sca-1, CD29, and CD90 but not CD45, CD34, CD14, and CD11b [43,44,45].
In addition to their capacity for multipotent differentiation and their ease of isolation, MSCs are characterized by a number of features that make them attractive subjects for research in regenerative medicine (Fig. 7.2). MSCs lack immunogenicity, home to areas of injured tissue, and have anti-inflammatory and immunomodulatory effects [26, 61]. Because MSCs have linited expression of MHC class I and II molecules, both autologous and allogeneic administration are easily achieved (Fig. 7.2) [40, 63]. In addition, MSCs can be genetically modified using viral vectors to enhance their therapeutic potential [64,65,66]. Because of these favorable characterstics, the therapeutic potential of MSCs has been investigated not only for lung diseases [61] but also for a wide variety of other conditions, including hepatic failure [55], myocardial infarction [67], diabetes [68], sepsis [56], and acute renal failure [24].

Properties of MSCs
8 MSCs in the Treatment of IPF
A number of preclinical studies have examined the therapeutic potential of stem and progenitor cell populations in animal models of pulmonary fibrosis and MSCs have been studied in human phase I clinical trials [2]. In addition to those discussed above, a number of additional features suggest that MSCs may be beneficial in the treatment of IPF. MSCs inhibit cytotoxic T-cells and natural killer cells, are known to secrete growth factors including KGF, HGF, VEGF, and Ang-1, and play an important role in the repair of alveolar epithelium through mitochondrial transfer [51, 69].
Because epithelial injury underlies the pathogenesis of pulmonary fibrosis, the delivery of exogenous stem or progenitor cells capable of participating in alveolar reepithelialization may have therapeutic potential to break the cycle of aberrant epithelial–mesenchymal crosstalk and halt disease progression. It is possible that, due to changes in the lung microenvironment and the continued presence of injurious stimuli, exogenous progenitor cells, like endogenous cells, may simply participate in the characteristic pathological repair process. On the other hand, intrinsic factors are thought to play a role in alveolar epithelial cell injury, and preclinical studies suggest that stem cells can exert profound effects through the secretion of soluble mediators [31, 53, 54].
Systematic reviews reveal numerous preclinical studies of MSCs in the treatment of BLM-induced lung injury [5]. To date, these studies suggest that MSCs are effective in improving histopathology, Ashcroft scores of lung fibrosis, lung collagen deposition, and survival in animal models of BLM-induced lung injury. However, most of these studies used young animals and examined the initial inflammatory phase rather than the chronic fibrotic phase. As previously discussed, spontaneous reversal of BLM-induced lung fibrosis may occur spontaneously in young mice [70], but does not occur in aged mouse models [70, 71]. A more recent study utilizing an aged mouse model of BLM-induced lung fibrosis found that treatment with adipose-derived MSCs may promote a systemic acute repair phenotype to prevent fibrosis in multiple organs and enhance wound healing by modulating pro-fibotic factors such as miR-199 and its downstream target, CAV1 [1].
Preclinical studies have shown MSCs to be efficacious in the treatment and prevention of lung fibrosis [53, 72]. Nonetheless, concerns remain regarding the activity of MSCs within a pro-fibrotic microenvironment [73,74,75,76]. While some preclinical studies suggest that MSCs might promote fibrosis, to date, no human studies have found a similar pro-fibrotic effect [37, 43, 63, 70, 74, 75, 77,78,79,80,81,82,83,84,85].
9 Clinical Trials of MSCs for the Treatment of IPF
Twenty years ago, Lazarus conducted the first clinical trial using bone marrow cell injection in patients with hematologic malignancies [52]. Since then, numerous clinical trials have been conducted to test the feasibility and efficacy of MSC-based therapy, and more than 2000 patients have recieved allogeneic or autologous MSCs for the treatment of various diseases [52].
Early clinical studies of MSCs in patients with IPF have shown promising safety profiles [30, 78, 86]. A phase Ib study of endobronchially administered autologous adipose-derived MSCs showed not only acceptable safety outcomes but also improvements in quality of life parameters [78]. Longitudinal outcomes of this study also demonstrated an acceptable safety profile with a 100% survival rate at 2 years after the first administration of MSCs and a median overall progression-free survival of 26 months [87].
Studies of intravenously administered placental-derived MSCs [79, 83] have found that administration of up to 2 × 106 cells per kilogram is safe in subjects with moderately severe IPF [83]. Importantly, only minor and transient alterations in peri-infusion hemodynamics and gas exchange were reported, ameliorating concerns regarding the potential embolization of stem cells to an already compromised pulmonary vasculature. At 6 months, there were no observable declines in forced vital capacity (FVC), diffusing lung capacity for carbon monoxide (DLCO), six-minute walk test (6MWT), or CT fibrosis score [88].
The AETHER trial also showed favorable safety outcomes for the single-dose intravenous delivery of up to 2 × 108 allogeneic MSCs in patients with IPF [79]. Although this study was underpowered for the detection of significant changes in functional indices, the mean decline in % predicted FVC and DLCO were below the thresholds for disease progression [1, 46].
At this time, the only study actively recruiting IPF patients for treatment with stem cells is a Phase 1/2 clinical trial (NCT02745184) taking place at two sites in China. Researchers intend to isolate autologous lung stem cells, MSCs from the patient’s own bronchi, expand them in the laboratory, and then deliver the expanded cell population via a single injection directly into an area affected by IPF. Safety parameters will be monitored for 1 year and efficacy will be measured by changes in lung function and exercise ability tests.
Looking ahead, ReCell, an FDA-approved phase 1b trial, is planned but has not yet begun enrollment. In this multidose, randomized, double-blind trial, 10 × 106 MSCs will be delivered intravenously to patients with IPF.
A team of scientists from the UNC School of Medicine and North Carolina State University demonstrated that they could harvest lung stem cells from people using a noninvasive office procedure. They snipped tiny, seed-sized samples of airway tissue using a bronchoscope, this method involves far less risk to the patient than does a standard, chest-penetrating surgical biopsy of lung tissue. Cheng and his colleagues cultured lung spheroid cells from these tiny tissue samples until they multiplied to the thousands and were enough to be therapeutically injected.
Once the stem cells were harvested they were able to multiply these lung cells in the lab. The results yielded enough cells sufficient for human therapy. In 2017 these researchers were working with the FDA for preparation of clinical trials in patients with IPF. Cheng, Lobo, and their teams are now planning an initial study of therapeutic lung spheroid cells in a small group of IPF patients.
Currently this study is still undergoing data collection.
10 Moving Forward
Stem cells have been used in medicine since the 1950s when bone marrow transplants were first used to treat leukemia. Congressional involvement in stem cell policy started as early as 1974. The first major amendment related to the use of federal funds for research involving embryonic stem (ES) cells occurred in 1996. In 2016 President Obama signed into effect, the 21st Century Cures Act, which includes provisions intended to assure timely regulatory review of regenerative therapies, including cell therapies enabled by stem cell therapy.
While preclinical trials suggest that MSCs may be effective in the treatment of IPF, and early clinical trials support their safety, currently the data to support their efficacy for the treatment of IPF is insufficient. Despite this lack of evidence, cell-based therapies are being aggressively marketed to vulnerable patient populations. A review carried out in November 2018 of the FDA website lists over 1000 stem cell-related businesses registered. These clinics offer unproven, experimental treatments for a wide variety of conditions [89, 90].
In the case of IPF, desperate patients and their physicians continue to succumb to an onslaught of marketing and branding of as yet unproven “stem cell” treatments. Unfortunately, these businesses are also almost wholly unregulated [91]. A review of unapproved stem cell interventions by Turner and Knoepfler and the harm arising from the misuse of unproven treatments support increased government oversight in the interest of patient safety [92]. A sense of urgency exists to establish solid evidence regarding the efficacy of stem cell therapies for the treatment of chronic lung disease.
References
Tashiro J, Rubio GA, Limper AH, et al. Exploring animal models that resemble idiopathic pulmonary fibrosis. Front Med (Lausanne). 2017;4:118.. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5532376
Karampitsakos T, Woolard T, Bouros D, Tzouvelekis A. Toll-like receptors in the pathogenesis of pulmonary fibrosis. Eur J Pharmacol. 2017;808:35–43. https://doi.org/10.1016/j.ejphar.2016.06.045.
Tzouvelekis A, Toonkel R, Karampitsakos T, et al. Mesenchymal stem cells for the treatment of idiopathic pulmonary fibrosis. Front Med (Lausanne). 2018;5:142. https://doi.org/10.3389/fmed.2018.00142.
Karampitsakos T, Tzilas V, Tringidou R, Steiropoulos P, Aidinis V, Papiris SA, et al. Lung cancer in patients with idiopathic pulmonary fibrosis. Pulm Pharmacol Ther. 2017;45:1–10. https://doi.org/10.1016/j.pupt.2017.03.016.
Srour N, Thebaud B. Mesenchymal stromal cells in animal bleomycin pulmonary fibrosis models: a systematic review. Stem Cells Transl Med. 2015;4(12):1500–10. https://doi.org/10.5966/sctm.2015-0121.
Herazo-Maya JD, Sun J, Molyneaux PL, et al. Validation of a 52-gene risk profile for outcome prediction in patients with idiopathic pulmonary fibrosis: an international, multicentre, cohort study. Lancet Respir Med. 2017;5(11):857–68.. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5677538
Richeldi L, Costabel U, Selman M, Kim DS, Hansell DM, Nicholson AG, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365:1079–87. https://doi.org/10.1056/NEJMoa1103690.
Moore Bethany B, Lawson William E, Oury Tim D, et al. Animal models of fibrotic lung disease. Am J Respir Cell Mol Biol. 2013;49:2. https://doi.org/10.1165/rcmb.2013-0094TR.
Wuyts WA, Agostini C, Antoniou KM, Bouros D, Chambers RC, Cottin V, et al. The pathogenesis of pulmonary fibrosis: a moving target. Eur Respir J. 2013;41:1207–18. https://doi.org/10.1183/09031936.00073012.
Kolb M, Bonella F, Wollin L. Therapeutic targets in idiopathic pulmonary fibrosis. Respir Med. 2017;131:49–57. https://doi.org/10.1016/j.rmed.2017.07.062.
Fletcher S, Jones MG, Spinks K, Sgalla G, Marshall BG, Limbrey R, et al. The safety of new drug treatments for idiopathic pulmonary fibrosis. Expert Opin Drug Saf. 2016;15:1483–9. https://doi.org/10.1080/14740338.2016.1218470.
King TE, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. Phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–92. https://doi.org/10.1056/NEJMoa1402582.
Tzouvelekis A, Karampitsakos T, Kontou M, Granitsas A, Malliou I, Anagnostopoulos A, et al. Safety and efficacy of nintedanib in idiopathic pulmonary fibrosis: a real-life observational study. Pulm Pharmacol Ther. 2018;49:61–6. https://doi.org/10.1016/j.pupt.2018.01.006.
Tzouvelekis A, Karampitsakos T, Ntolios P, et al. Longitudinal “real-world” outcomes of pirfenidone in idiopathic pulmonary fibrosis in Greece. Front Med. 2017;4:213. https://doi.org/10.3389/fmed.2017.00213.
Chagastelles PC, et al. Biology of stem cells: an overview kidney international supplements. Kidney Int Suppl (2011). 2011;1(3):63–7. https://doi.org/10.1038/kisup.2011.15.
Sueblinvong V, Loi R, Eisenhauer PL, Bernstein IM, Suratt BT, Spees JL, et al. Derivation of lung epithelium from human cord blood-derived mesenchymal stem cells. Am J Respir Crit Care Med. 2008;177(7):701–11. https://doi.org/10.1164/rccm.200706-859OC.
Carraro G, Perin L, Sedrakyan S, Giuliani S, Tiozzo C, Lee J, et al. Human amniotic fluid stem cells can integrate and differentiate into epithelial lung lineages. Stem Cells. 2008;26(11):2902–11. https://doi.org/10.1634/stemcells.2008-0090.
Pierro M, Ionescu L, Montemurro T, Vadivel A, Weissmann G, Oudit G, et al. Short-term, long-term and paracrine effect of human umbilical cord-derived stem cells in lung injury prevention and repair in experimental bronchopulmonary dysplasia. Thorax. 2013;68(5):475–84. https://doi.org/10.1136/thoraxjnl-2012-202323.
Ionescu L, Byrne RN, van Haaften T, Vadivel A, Alphonse RS, Rey-Parra GJ, et al. Stem cell conditioned medium improves acute lung injury in mice: in vivo evidence for stem cell paracrine action. Am J Physiol Lung Cell Mol Physiol. 2012;303(11):L967–77. https://doi.org/10.1152/ajplung.00144.2011.
Sueblinvong V, Weiss DJ. Cell therapy approaches for lung diseases: current status. Curr Opin Pharmacol. 2009;9(3):268–73. https://doi.org/10.1016/j.coph.2009.03.002.
Griffin MD, Ritter T, Mahon BP. Immunological aspects of allogeneic mesenchymal stem cell therapies. Hum Gene Ther. 2010;21(12):1641–55. https://doi.org/10.1089/hum.2010.156.
Wong AP, Keating A, Lu WY, Duchesneau P, Wang X, Sacher A, et al. Identification of a bone marrow-derived epithelial-like population capable of repopulating injured mouse airway epithelium. J Clin Invest. 2009;119(2):336–48. https://doi.org/10.1172/JCI36882.
Bustos ML, Mura M, Marcus P, Hwang D, Ludkovski O, Wong AP, et al. Bone marrow cells expressing clara cell secretory protein increase epithelial repair after ablation of pulmonary clara cells. Mol Ther. 2013;21(6):1251–8. https://doi.org/10.1038/mt.2013.53.
Katrina MN, Janes Sam M. Stem cells and pulmonary fibrosis: cause or cure? Proc Am Thorac Soc. 2012;9(3):164–71. https://doi.org/10.1513/pats.201201-010AW.
Chang YS, Oh W, Choi SJ, Sung DK, Kim SY, Choi EY, et al. Human umbilical cord blood-derived mesenchymal stem cells attenuate hyperoxia-induced lung injury in neonatal rats. Cell Transplant. 2009;18(8):869–86. https://doi.org/10.3727/096368909X47118.
Zhang H, Fang J, Wu Y, Mai Y, Lai W, Su H. Mesenchymal stem cells protect against neonatal rat hyperoxic lung injury. Expert Opin Biol Ther. 2013;13(6):817–29. https://doi.org/10.1517/14712598.2013.778969.
Hansmann G, Fernandez-Gonzalez A, Aslam M, Vitali SH, Martin T, Mitsialis SA, et al. Mesenchymal stem cell-mediated reversal of bronchopulmonary dysplasia and associated pulmonary hypertension. Pulm Circ. 2012;2(2):170–81. https://doi.org/10.4103/2045-8932.97603.
O’Reilly M, Thebaud B. Animal models of bronchopulmonary dysplasia. The term rat models. Am J Physiol Lung Cell Mol Physiol. 2014;307(12):L948–58. https://doi.org/10.1152/ajplung.00160.2014.
Rehman J, Li J, Orschell CM, March KL. Peripheral blood “endothelial progenitor cells” are derived from monocyte/macrophages and secrete angiogenic growth factors. Circulation. 2003;107(8):1164–9. https://doi.org/10.1161/01.CIR.0000058702.69484.A0.
Kung EF, Wang F, Schechner JS. In vivo perfusion of human skin substitutes with microvessels formed by adult circulating endothelial progenitor cells. Dermatol Surg. 2008;34(2):137–46. https://doi.org/10.1111/j.1524-4725.2007.34030.x.
Jenkins RG, Moore BB, Chambers RC, Eickelberg O, Konigshoff M, Kolb M, et al. An official American Thoracic Society workshop report: use of animal models for the preclinical assessment of potential therapies for pulmonary fibrosis. Am J Respir Cell Mol Biol. 2017;56(5):667–79. https://doi.org/10.1165/rcmb.2017-0096ST.
Bonniaud P et al. Optimising experimental research in respiratory diseases: an ERS statement. Eur Respir J. 2018;51(5). pii: 1702133. doi:https://doi.org/10.1183/13993003.02133-2017. Print 2018.
Shepherd BR, Enis DR, Wang F, Suarez Y, Pober JS, Schechner JS. Vascularization and engraftment of a human skin substitute using circulating progenitor cell-derived endothelial cells. FASEB J. 2006;20(10):1739–41. https://doi.org/10.1096/fj.05-5682fje.
DeKoninck P, Toelen J, Roubliova X, Carter S, Pozzobon M, Russo FM, et al. The use of human amniotic fluid stem cells as an adjunct to promote pulmonary development in a rabbit model for congenital diaphragmatic hernia. Prenat Diagn. 2015;35(9):833–40. https://doi.org/10.1002/pd.4621.
Vosdoganes P, Hodges RJ, Lim R, Westover AJ, Acharya RY, Wallace EM, et al. Human amnion epithelial cells as a treatment for inflammation-induced fetal lung injury in sheep. Am J Obstet Gynecol. 2011;205(2):e26–33. https://doi.org/10.1016/j.ajog.2011.03.054.
Moodley Y, Ilancheran S, Samuel C, Vaghjiani V, Atienza D, Williams ED, et al. Human amnion epithelial cell transplantation abrogates lung fibrosis and augments repair. Am J Respir Crit Care Med. 2010;182(5):643–51. https://doi.org/10.1164/rccm.201001-0014OC.
Murphy S, Lim R, Dickinson H, Acharya R, Rosli S, Jenkin G, et al. Human amnion epithelial cells prevent bleomycin-induced lung injury and preserve lung function. Cell Transplant. 2011;20(6):909–23. https://doi.org/10.3727/096368910X543385.
Murphy SV, Lim R, Heraud P, Cholewa M, Le Gros M, de Jonge MD, et al. Human amnion epithelial cells induced to express functional cystic fibrosis transmembrane conductance regulator. PLoS One. 2012;7(9):e46533. https://doi.org/10.1371/journal.pone.004653.
Borthwick DW, Shahbazian M, Krantz QT, Dorin JR, Randell SH. Evidence for stem-cell niches in the tracheal epithelium. Am J Respir Cell Mol Biol. 2001;24:662–70.
Reynolds SD, Giangreco A, Power JH, Stripp BR. Neuroepithelial bodies of pulmonary airways serve as a reservoir of progenitor cells capable of epithelial regeneration. Am J Pathol. 2000;156:269–78.
Hodges RJ, Jenkin G, Hooper SB, Allison B, Lim R, Dickinson H, et al. Human amnion epithelial cells reduce ventilation-induced preterm lung injury in fetal sheep. Am J Obstet Gynecol. 2012;206(5):e8–15. https://doi.org/10.1016/j.ajog.2012.02.038.
Hong KU, Reynolds SD, Giangreco A, Hurley CM, Stripp BR. Clara cell secretory protein-expressing cells of the airway neuroepithelial body microenvironment include a label-retaining subset and are critical for epithelial renewal after progenitor cell depletion. Am J Respir Cell Mol Biol. 2001;24:671–81.
Rawlins EL, Hogan BL. Ciliated epithelial cell lifespan in the mouse trachea and lung. Am J Physiol Lung Cell Mol Physiol. 2008;295:L231–4.
Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of β-cells in aged adult mice. Diabetes. 2005;54:2557–67.
Barker N, van de Wetering M, Clevers H. The intestinal stem cell. Genes Dev. 2008;22:1856–64.
Sime PJ, Marr RA, Gauldie D, Xing Z, Hewlett BR, Graham FL, et al. Transfer of tumor necrosis factor-alpha to rat lung induces severe pulmonary inflammation and patchy interstitial fibrogenesis with induction of transforming growth factor-beta1 and myofibroblasts. Am J Pathol. 1998;153(3):825–32. https://doi.org/10.1016/S0002-9440(10)65624-6.
Xing Z, Tremblay GM, Sime PJ, Gauldie J. Overexpression of granulocyte-macrophage colony-stimulating factor induces pulmonary granulation tissue formation and fibrosis by induction of transforming growth factor-beta 1 and myofibroblast accumulation. Am J Pathol. 1997;150(1):59–66.
Lee CG, Homer RJ, Zhu Z, Lanone S, Wang X, Koteliansky V, et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1). J Exp Med. 2001;194(6):809–21. https://doi.org/10.1084/jem.194.6.809.
Buckley S, Shi W, Carraro G, Sedrakyan S, Da Sacco S, Driscoll BA, et al. The milieu of damaged alveolar epithelial type 2 cells stimulates alveolar wound repair by endogenous and exogenous progenitors. Am J Respir Cell Mol Biol. 2011;45(6):1212–21. https://doi.org/10.1165/rcmb.2010-0325OC.
Agostini C. Stem cell therapy for chronic lung diseases: hope and reality. Respir Med. 2010;104(Suppl 1):S86–91.
Álvarez D, Levine M, Rojas M. Regenerative medicine in the treatment of idiopathic pulmonary fibrosis: current position. Stem Cells Cloning. 2015;8:61–5. https://doi.org/10.2147/SCCAA.S49801.
Anna S-M. Cell therapy in idiopathic pulmonary fibrosis. Med Sci. 2018;6(3):64. https://doi.org/10.3390/medsci6030064.
Moore BB, Hogaboam CM. Murine models of pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;294(2):L152–60. https://doi.org/10.1152/ajplung.00313.2007.
Organ L, Bacci B, Koumoundouros E, Barcham G, Kimpton W, Nowell CJ, et al. A novel segmental challenge model for bleomycin-induced pulmonary fibrosis in sheep. Exp Lung Res. 2015;41(3):115–34. https://doi.org/10.3109/01902148.2014.985806.
Stripp BR, Reynolds SD. Maintenance and repair of the bronchiolar epithelium. Proc Am Thorac Soc. 2008;5:328–33.
Xu X, D'Hoker J, Stange G, Bonne S, De Leu N, Xiao X, et al. β cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell. 2008;132:197–207.
Friedenstein AJ, Petrakova KV, Kurolesova AI, Frolova GP. Heterotopic of bone marrow analysis of precursor cells for osteogenic and hematopoietic tissues. Transplantation. 1968;6:230–47.
Okamoto R, Yajima T, Yamazaki M, Kanai T, Mukai M, Okamoto S, Ikeda Y, Hibi T, Inazawa J, Watanabe M. Damaged epithelia regenerated by bone marrow-derived cells in the human gastrointestinal tract. Nat Med. 2002;8:1011–7.
Caplan AI. Mesenchymal stem cells: time to change the name. Stem Cells Transl Med. 2017;6(6):1445–51. https://doi.org/10.1002/sctm.17-0051.
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop D, Horwitz E. Minimal criteria for defining multipotent mesenchymal stromal cells. Cytotherapy. 2006;8:315–7.
Stripp BR. Hierarchical organization of lung progenitor cells: is there an adult lung tissue stem cell? Proc Am Thorac Soc. 2008;5:695–8.
Reynolds SD, Malkinson AM. Clara cell: progenitor for the bronchiolar epithelium. Int J Biochem Cell Biol. 2009;42(1):1–4. https://doi.org/10.1016/j.biocel.2009.09.002.
Davis GS, Pfeiffer LM, Hemenway DR. Interferon-gamma production by specific lung lymphocyte phenotypes in silicosis in mice. Am J Respir Cell Mol Biol. 2000;22(4):491–501. https://doi.org/10.1165/ajrcmb.22.4.3599.
Naik PK, Moore BB. Viral infection and aging as cofactors for the development of pulmonary fibrosis. Expert Rev Respir Med. 2010;4(6):759–71. https://doi.org/10.1586/ers.10.73.
Peng R, Sridhar S, Tyagi G, Phillips JE, Garrido R, Harris P, et al. Bleomycin induces molecular changes directly relevant to idiopathic pulmonary fibrosis: a model for “active” disease. PLoS One. 2013;8(4):e59348. https://doi.org/10.1371/journal.pone.0059348.
Izbicki G, Segel MJ, Christensen TG, Conner MW, Breuer R. Time course of bleomycin-induced lung fibrosis. Int J Exp Pathol. 2002;83(3):111–9. https://doi.org/10.1046/j.1365-2613.2002.00220.x.
Dor Y, Melton DA. How important are adult stem cells for tissue maintenance? Cell Cycle. 2004;3:1104–6.
Teta M, Rankin MM, Long SY, Stein GM, Kushner JA. Growth and regeneration of adult β cells does not involve specialized progenitors. Dev Cell. 2007;12:817–26.
Ma S, Xie N, Li W, Yuan B, Shi Y, Wang Y. Immunobiology of mesenchymal stem cells. Cell Death Differ. 2013;21(2):216–25. https://doi.org/10.1038/cdd.2013.158.
Tashiro J, Elliot SJ, Gerth DJ, et al. Therapeutic benefits of young, but not old, adipose-derived mesenchymal stem cells in a chronic mouse model of bleomycin-induced pulmonary fibrosis. Transl Res. 2015;166(6):554–67.
Hecker L, Logsdon NJ, Kurundkar D, Kurundkar A, Bernard K, Hock T, et al. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci Transl Med. 2014;6(231):231ra47. https://doi.org/10.1126/scitranslmed.3008182.
Degryse AL, Tanjore H, Xu XC, Polosukhin VV, Jones BR, McMahon FB, et al. Repetitive intratracheal bleomycin models several features of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2010;299(4):L442–52. https://doi.org/10.1152/ajplung.00026.2010.
Roggli V, Gibbs AR, Attanoos R, Churg A, Popper H, Corrin B, et al. Pathology of asbestosis: an update of the diagnostic criteria response to a critique. Arch Pathol Lab Med. 2016;140(9):950–2. https://doi.org/10.5858/arpa.2015-0503-SA.
Kim SJ, Cheresh P, Jablonski RP, Williams DB, Kamp DW. The role of mitochondrial DNA in mediating alveolar epithelial cell apoptosis and pulmonary fibrosis. Int J Mol Sci. 2015;16(9):21486–519. https://doi.org/10.3390/ijms160921486.
Li J, Poovey HG, Rodriguez JF, Brody A, Hoyle GW. Effect of platelet-derived growth factor on the development and persistence of asbestos-induced fibroproliferative lung disease. J Environ Pathol Toxicol Oncol. 2004;23(4):253–66. https://doi.org/10.1615/JEnvPathToxOncol.v23.i4.20.
Selman M, Buendia-Roldan I, Pardo A. Aging and pulmonary fibrosis. Rev Investig Clin. 2016;68(2):75–83.
Sueblinvong V, Neujahr DC, Mills ST, Roser-Page S, Ritzenthaler JD, Guidot D, et al. Predisposition for disrepair in the aged lung. Am J Med Sci. 2012;344(1):41–51. https://doi.org/10.1097/MAJ.0b013e318234c132.
Thannickal VJ, Murthy M, Balch WE, Chandel NS, Meiners S, Eickelberg O, et al. Blue journal conference. Aging and susceptibility to lung disease. Am J Respir Crit Care Med. 2015;191(3):261–9. https://doi.org/10.1164/rccm.201410-1876PP.
Englert JM, Hanford LE, Kaminski N, Tobolewski JM, Tan RJ, Fattman CL, et al. A role for the receptor for advanced glycation end products in idiopathic pulmonary fibrosis. Am J Pathol. 2008;172(3):583–91. https://doi.org/10.2353/ajpath.2008.070569.
Samuel CS, Zhao C, Bathgate RA, Bond CP, Burton MD, Parry LJ, et al. Relaxin deficiency in mice is associated with an age-related progression of pulmonary fibrosis. FASEB J. 2003;17(1):121–3. https://doi.org/10.1096/fj.02-0449fje.
Naik PN, Horowitz JC, Moore TA, Wilke CA, Toews GB, Moore BB. Pulmonary fibrosis induced by gamma-herpesvirus in aged mice is associated with increased fibroblast responsiveness to transforming growth factor-beta. J Gerontol A Biol Sci Med Sci. 2012;67(7):714–25. https://doi.org/10.1093/gerona/glr211.
Torres-Gonzalez E, Bueno M, Tanaka A, Krug LT, Cheng DS, Polosukhin VV, et al. Role of endoplasmic reticulum stress in age-related susceptibility to lung fibrosis. Am J Respir Cell Mol Biol. 2012;46(6):748–56. https://doi.org/10.1165/rcmb.2011-0224OC.
Egan JJ, Adamali HI, Lok SS, Stewart JP, Woodcock AA. Ganciclovir antiviral therapy in advanced idiopathic pulmonary fibrosis: an open pilot study. Pulm Med. 2011;2011:240805. https://doi.org/10.1155/2011/240805.
Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J Clin Invest. 1997;100(4):768–76. https://doi.org/10.1172/JCI119590.
Vicencio AG, Lee CG, Cho SJ, Eickelberg O, Chuu Y, Haddad GG, et al. Conditional overexpression of bioactive transforming growth factor-beta1 in neonatal mouse lung: a new model for bronchopulmonary dysplasia? Am J Respir Cell Mol Biol. 2004;31(6):650–6. https://doi.org/10.1165/rcmb.2004-0092OC.
Kolb M, Margetts PJ, Anthony DC, Pitossi F, Gauldie J. Transient expression of IL-1beta induces acute lung injury and chronic repair leading to pulmonary fibrosis. J Clin Invest. 2001;107(12):1529–36. https://doi.org/10.1172/JCI12568.
Lee CG, Cho SJ, Kang MJ, Chapoval SP, Lee PJ, Noble PW, et al. Early growth response gene 1-mediated apoptosis is essential for transforming growth factor beta1-induced pulmonary fibrosis. J Exp Med. 2004;200(3):377–89. https://doi.org/10.1084/jem.20040104.
Richeldi L, Ryerson CJ, et al. Relative versus absolute change in forced vital capacity in idiopathic pulmonary fibrosis. Thorax. 2012;67:407–11. https://doi.org/10.1136/thoraxjnl-2011-201184.
Ikonomou L, Panoskaltsis-Mortari A, Wagner DE, Freishtat RJ. Weiss DJ unproven stem cell treatments for lung disease-an emerging public health problem. Am J Respir Crit Care Med. 2017;195:P13–4. https://doi.org/10.1164/rccm.201607-1461ED.
Turner L, Knoepfler P. Selling stem cells in the USA: assessing the direct-to-consumer industry. Cell Stem Cell. 2016;19:154–7. https://doi.org/10.1016/j.stem.2016.06.007.
Charo RA, Sipp D. Rejuvenating regenerative medicine regulation. N Engl J Med. 2018;378:504–5. https://doi.org/10.1056/NEJMp1715736.
Leigh T, Paul K. Selling stem cells in the USA: assessing the direct to consumer-industry. Cell Stem Cell. 2016;19(2):154–7. https://doi.org/10.1016/j.stem.2016.06.007.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Patete, C.L., Toonkel, R.L., Glassberg, M. (2019). Stem Cell Based Therapy for Lung Disease Preclinical evidence for the role of stem/stromal cells Clinical application of stem/stromal cells in lung fibrosis. In: Burgess, J., Heijink, I. (eds) Stem Cell-Based Therapy for Lung Disease. Springer, Cham. https://doi.org/10.1007/978-3-030-29403-8_7
Download citation
DOI: https://doi.org/10.1007/978-3-030-29403-8_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-29402-1
Online ISBN: 978-3-030-29403-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)