
Abstract
Non-oxidative methane activation over particle-size adjusted alumina catalysts loaded with metal oxide (Al2O3, MgO/Al2O3, and TiO2/Al2O3) was investigated with a dielectric barrier discharge reactor using 10 % CH4 in Ar at plasma induced temperature. Plasma-assisted catalytic activity for direct conversion of methane over the catalysts was compared with that using plasma only. Catalyst hybrid reaction in a non-thermal discharge showed that MgO/Al2O3 had the highest activity for methane conversion. C2, C3, and C4 hydrocarbons were formed as products; ethane, ethylene, and acetylene were predominant over all catalysts. The effect of varying particle size of the MgO/Al2O3 catalyst was also examined. The conversion of methane over MgO/Al2O3 dramatically increased with decreasing catalyst particle size from 1.70 to 0.25 mm. It is interesting to note that distribution of C2 hydrocarbons was tuned by changing the particle size of the catalyst. It was also observed that the gas flow rate, frequency, and power supplied affected direct conversion of methane and selectivity of products significantly.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Methane is a major component of fossil fuels such as natural gas, shale gas, methane hydrates, and biomass derived resources. However, it is important to reduce the release of methane into the environment because methane has 25 times more warming potential as a greenhouse gas than carbon dioxide [1, 2]. Methane has the potential for transformation into other chemicals for further utilization in energy sources or feed stocks, but methane activation is very challenging. Methane has been used for production of synthesis gas (CO and H2) by steam reforming under extreme conditions (700–1,100 °C and 20–30 bar) using a metal-based catalyst [3, 4]. Since the 1980s, a lot of efforts has been put into methane transformation; including direct oxidation to methanol and formaldehyde, oxidative coupling, and non-oxidative coupling. The focus has been to overcome the difficulty of breaking C–H bonds and to form the desired C–C and C–O bonds from the perspectives of both basic chemistry and engineering [5, 6].
Direct coupling of methane in the presence of oxidants has attracted much attention, because the thermodynamics of this reaction are more favorable than for non-oxidative coupling. Unfortunately, undesirable by-products (COx) were also formed in the oxidative coupling process at high temperatures [7]. Moreover, it is difficult to control the reaction temperature because the overall reaction is exothermic and the actual catalyst bed temperature could be much higher than the set temperature [8]. Therefore, non-oxidative conversion of methane at low temperature is preferable to the oxidative process due to its high selectivity for desirable hydrocarbons. However, a major difficulty in the catalytic and thermal direct conversion of methane remains, because the strength of its C–H bonds is greater than that of hydrocarbons that might be produced [3]. Another problem associated with thermal catalytic conversion of methane is carbon or coke deposition causing catalyst deactivation.
Non-thermal plasma technology converts the major part of electrical energy to energetic electrons rather than using it to heat gases, thus it can supersede conventional thermal catalytic technology for the direct methane coupling reaction. However, the gas-phase non-catalytic plasma reaction usually occurs via a free radical mechanism which is unfavorable for controlling the selectivity of products [9–11]. To enhance the performance of methane conversion, intense effort has been devoted to hybrid plasma-heterogeneous catalyst system by using various types of non-thermal plasma, including dielectric barrier discharge (DBD), corona, glow discharge, and gliding arc [12–15]. It has been reported that control of electron energy with a suitable design of discharge reactor as well as a proper catalyst can strongly affect product selectivity in the methane conversion reaction, particularly in DBD and corona discharge systems [16].
Dielectric barrier discharge reactors have an advantage for developing a plasma-catalytic process because the catalyst particles are placed inside the discharge zone, and it is possible to effectively transfer the active species generated by plasma, onto the catalyst surface. However, the plasma-catalyst hybrid system makes for extra complexity of the discharge state. If some solid particles exist in the discharge zone, the discharge structure and its character will vary due to changing electrical parameters (electric field strength and current density) because of shifts in the current flow near the surface. Therefore, the properties of the catalyst can be used to tune the electrical parameters in the discharge zone. Other factors, such as the hydrodynamics of the gas stream and temperature distributions, may also change according to the kind of material used in the reactor.
Several catalysts have been used for methane activation in the presence of plasma. It has been reported that NaY zeolite showed high yield for C2 hydrocarbons [17]. Pt/γ-Al2O3 catalyst was also used for methane conversion in a DBD reactor and showed high selectivity for light alkanes [18]. In the presence of catalyst particles, molecular processes include adsorption onto the catalyst surface, reaction between the adsorbed species, and desorption of the products. During this phase, the catalyst can increase the lifespan of the high-energy radicals by adsorbing their excess energy [19]. One of the key roles of the catalyst in this hybrid system is conducting reactions between short-lived active species [20]. Mlotek et al. [12] showed that when the active species generated in plasma took a relatively long time to reach the catalyst surface, most of the fast reactions were terminated in the gas phase. In this case, the catalyst could have no influence on the reaction result, indicating that the reaction result depends on the discharge condition and the configuration of the catalyst. A few literature works concentrates on studying the effect of catalyst-bed porosity, controlled by packing different size of catalyst pellets in the discharge zone. Introducing catalyst of different size into the discharge gap generates void fraction leading to significant transition of the discharge behavior [21–23]. Studies suggest that relative contribution of filamentary and surface discharges is likely to depend on many factors including particle size, particle shape, packing location, and the void fraction of the electrode gap [24, 25].
Wan et al. [26] studied the effect of catalyst size on methane conversion in the presence of microwaves and reported that activity and selectivity for aromatics varied with changing particle size of the catalyst. It was also found that micro-discharge on the surface of the catalyst particles affected conversion and selectivity.
In this work, we investigated the effect of catalyst size on plasma-assisted catalytic reaction at ambient temperature and atmospheric pressure; to selectively produce C2 hydrocarbons from methane. We also examined the catalytic performance of various metal-oxide-loaded catalysts for this reaction.
Experimental
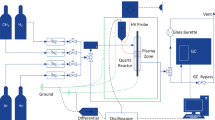
The plasma-assisted catalytic reaction was carried out in a packed-bed DBD reactor with a volume of 56 × 24 × 3 mm; between two dielectric plates attached to stainless steel electrodes. The gas distance between the electrodes was 3 mm. Figure 1 shows the scheme of the plasma reaction system. Balls (1.7 mm) of Al2O3, MgO/Al2O3 (7.1 wt%) and TiO2/Al2O3 (9.7 wt%) were provided from Sasolalumina; with BET surface areas of 201, 184, and 212 m2/g, respectively; were crushed to different mesh sizes and placed in the reactor. These materials were prepared by sol–gel method (www.Salsoalumina.com) and the surface area was recorded using Micrometric Tristar II 3000 by N2 adsorption isotherm at 77 K after dehydration of sample at 423 K. To examine the effect of catalyst mesh size, the ball shaped catalyst of 1.75 mm was crushed to different mesh size and filled the reactor volume. 1.21 g of 1.75 mm ball shaped catalyst was used to fill the entire volume of the reactor. When the smaller mesh size of the catalyst was used, the more amount of the catalyst was needed to occupy the same reactor volume. Argon and methane (10 mol%) flowed through this reactor at a flow-rate aimed at performing the reaction at ambient temperature and atmospheric pressure. In the non-thermal plasma reactions, the gas discharge was initiated at room temperature and the gas was heated by the plasma. A sinusoidal AC continuous supply of power (TTI, TGA 1244; Trek, 20/20C); with 0–6 kV and 1–5 kHz, was applied across the electrode. A 500 pf capacitor was connected to measure the applied power using an oscilloscope (Tektronix, DPO 5054) by integrating the single period power and multiplying the frequency. A 100 ohm resistor was also connected to monitor the micro-discharge plasma current.

Scheme of the plasma-assisted catalytic reaction system
A high speed ICCD camera (Hamamatsu, ORCA-05G) was mounted near the reactor to record images of the plasma generated during a single wave interval. The reactor temperature induced during the plasma reaction was measured by an IR temperature detector (FLIR, E50). TGA experiments were carried out using a SDTQ600 Thermogravimetric Analyzer to confirm the coke amount formed. For TGA analysis, 10 mg of catalyst after plasma reaction was used. TGA began by heating up the sample in air at a heating rate of 5 °C/min (100 ml/min, atmospheric pressure). The weight change of the catalyst was recorded up to 800 °C. The weight loss of catalyst was obtained by subtracting the weight loss of the catalyst from total weight loss of mixed sample. TPO was also run in the micrometrics Autochem II (20 % O2/He, 50 cm3/min, heating rate of 5 °C/min) for CO2 by an TCD detector. The gaseous product species after heating the sample was also monitored by the mass spectrometer (Pfeiffer Vacuum) to analyze the composition of CO2 and H2O.
The gas outlet was connected to a mass spectrometer (Pfeiffer Vacuum) to identify each product. Reaction products, such as hydrogen and hydrocarbons; were analyzed using a micro gas chromatograph (Agilent, 490 Micro GC) with four columns. Then, the conversion of methane and selectivity for each product were calculated. The conversion of CH4, selectivity for hydrocarbon products, and yield for H2, and carbon balance were calculated using Eqs. (1), (2), (3), and (4), respectively.
Result and Discussion
Effects of the Catalyst
Experiments were carried out in the presence of catalysts at atmospheric pressure and plasma heated temperature, with 10 % CH4/Ar mixed gas; at a total flow rate of 200 ml/min. Choice of these conditions was based on evidence that the methane activation reaction occurs well in response to micro-discharge induced by electric fields on the catalyst surfaces [27]. The mass spectroscope attached to the outlet of the reactor detected only light alkanes (C2H6, C2H4, C2H2, C3H8, C3H6, and C4H10) and H2. Other hydrocarbons obtained were negligible. To investigate the effect of catalyst in a DBD reactor, a non-catalytic plasma process was also conducted with variation of applied voltage from 3 to 6 kV at 3 kHz (Fig. 2a). No conversion of methane was observed until 3.5 kV, but the plasma was initiated at 4 kV and low conversion of methane was obtained. Conversion of methane increased steadily to 7.2 % with increasing voltage (to 6 kV). The temperature in the reactor recorded by the IR thermal detector was 53 °C without catalyst and 56 °C with catalyst. The temperature of the reactor increased slightly, as increased applied voltage and frequency. Gas temperature should be regarded as approximate values reflecting however the trend of the temperature increasing with time. When Al2O3 was introduced to the reactor, the plasma was also generated at 4 kV. Conversion of methane over Al2O3 was higher than with no catalyst, and reached 10.4 % at 6 kV. Conversion of methane over TiO2/Al2O3 was higher than that over Al2O3, and reached 14.1 % at 6 kV. The MgO/Al2O3 catalyst induced even better conversion of methane (16.2 % at 6 kV). SiO2 of same mesh size was also used as a catalyst, which gave low activity even compared to the plasma itself.

Conversion of methane over metal-oxide loaded alumina catalysts (0.5 mm) with plasma, as a function of a applied voltage and b power
The energy required to generate plasma was calculated from the corresponding Q–V Lissajous plots (Fig. 3) [28, 29]. The area of the Q–V parallelogram, obtained using a 500 pf capacitor, equals the discharge-energy dissipated during one cycle of the AC voltage. Figure 2b shows the conversion of methane over Al2O3, TiO2/Al2O3, and MgO/Al2O3 catalysts; as a function of the discharge energy. TiO2/Al2O3 required less energy to obtain the same conversion of methane than Al2O3. Moreover, MgO/Al2O3 needed less energy than TiO2/Al2O3. When the applied voltage fixed at 6 kV, generated discharge power was decreased from 4.18 W for Al2O3, 3.36 W for TiO2/Al2O3, and 3.60 W MgO/Al2O3, respectively. This indicates that metal-oxide doping affects the energy efficiency of the system. Figure 2b also clearly shows that the energy required for DBD to convert methane is lower when catalyst is used. The onset voltage of catalysts in a packed-bed DBD reactor is higher than that for plasma only, because of changes in electric filed and filamentary discharge densities [30].

Q–V patterns of a w/o catalyst, b Al2O3, c TiO2/Al2O3, and d MgO/Al2O3 catalysts (0.5 mm of particle size) at 6 kV and 3 kHz
Figure 4 shows the patterns of plasma-current generated in the DBD reactor; which consisted of a sine function wave and sharp peaks of high-energy electrons. A sufficiently high density of high-energy electrons in the plasma current; characterized by a large number of micro-discharge filaments, is able to activate hydrocarbons at room temperature and atmospheric pressure. In general, it is believed that DBD involves a large number of micro-discharges (about 106 micro-discharges per cm2 per second) [31]. The short lifetime of the current spikes may help to minimize excessive local heating.

Micro-discharge current patterns of a w/o catalyst, b Al2O3, c TiO2/Al2O3, and d MgO/Al2O3 catalysts (0.5 mm of particle size) at 6 kV and 3 kHz
Figure 5 shows the catalytic performance of Al2O3, TiO2/Al2O3, and MgO/Al2O3 for methane activation with plasma at 6 kV and 3 kHz. As mentioned earlier, conversion of methane was enhanced when catalyst was used. Among the catalysts tested, MgO/Al2O3 showed the highest methane conversion. No matter which catalyst (Al2O3, TiO2/Al2O3, or MgO/Al2O3) was used, selectivity for ethane and propane dropped; while selectivity for ethylene, acetylene, and propylene rose. From a modeling study of methane activation using only DBD, it was found that ethane and propane are the most favored products [32]. Therefore, this means that these early products acted as catalysts in this reaction. Each catalyst showed selectivity for different products. The activation of methane to CHX fragments is mainly caused by electric discharge. At this time, electric conductivity and surface charge of the catalyst would affect the type of radical formed. Barrier heating on the surface of the catalyst can be caused by a vibrational de-excitation process, or by a recombination of ions and electrons. The barrier is charged locally by the micro-discharge filaments, and this causes the barrier to heat strongly [33]. This local barrier heating on the surface of the catalyst increases the activity of the reaction. In this case, the dielectric constant and band gap of the catalyst may affect the occurrence of micro-discharge filaments [34]. Micro-discharge efficiency on the surface of MgO, TiO2, and Al2O3 differs, and this causes a difference in the temperature on the surface of the catalyst; resulting in different catalytic activity. Also, polymerization of radicals on the surface of the catalyst would depend on the nature of the catalyst, quite evident when the metal oxide catalysts were compared with SiO2 [16, 35, 36]. Therefore, the selectivity for products can be varied using the qualities of the catalyst. To determine thoroughly the reaction factors in this kind of reaction system, further studies are needed.

Catalytic performance of Al2O3, TiO2/Al2O3, and MgO/Al2O3 catalysts (0.5 mm of particle size) in a methane-conversion reaction assisted with plasma at 6 kV and 3 kHz
Effect of Catalyst Particle Size
Previous research has not been focused on the study of catalyst particle-size in plasma-assisted reactions. Therefore, balls of MgO/Al2O3 catalyst were crushed to different sizes and placed in the DBD reactor. The reaction was performed at 3 kHz and 6 kV at a flow rate of 200 ml/min; with 10 % CH4 in Ar. Table 1 shows the effect of catalyst particle size on plasma- assisted methane activation. When 1.75 mm balls of MgO/Al2O3 were used in the reactor, conversion of methane was 9.5 % and the energy utilized was 3.5 W (1,050 kJ/l). Conversion of methane increased with decreasing particle size. Selectivity for ethane and propane decreased significantly with decreasing particle size; while selectivity for acetylene increased with decreasing particle size. Selectivity for ethylene and propylene also increased slightly when smaller MgO/Al2O3 catalyst was used. These results indicate that filling the reactor with smaller catalyst particles increased the surface interaction between the catalyst and reaction species by stabilizing the radical; resulting in increased conversion of methane and changes in selectivity for products.
Q–V plots for MgO/Al2O3 of different particle sizes show a steady decrease in the capacitor voltage; leading to a lower energy in the system with decreasing particle-size of the catalyst (Fig. 6). Light emitted from excited molecules or radicals during the relaxation were captured by a high speed camera. Figure 7 displays the plasma-catalyst images recorded during the reaction. Figure 7a shows the uniform plasma pattern between the DBD electrodes when catalyst was not used. The smaller the catalyst particle used, the lighter the plasma observed on the catalyst surface. This indicates that the enhanced activity of the smaller catalyst is due to improved interaction between the surface of the catalyst and the plasma.

Q–V patterns of MgO/Al2O3 at 6 kV and 3 kHz; in relation to different catalyst particle size: a w/o catalyst, b 1.75 mm, c 1.00 mm, d 0.50 mm, and e 0.25 mm

ICCD images of MgO/Al2O3 in a plasma; with different catalyst particle sizes: a w/o catalyst, b 1.75 mm, c 1 mm, and d 0.5 mm
Carbon species deposited on the catalyst was confirmed by TGA analysis for the used catalyst. Figure 8 shows TGA profile of used MgO/Al2O3 catalyst with different particle sizes. TGA profile in Fig. 8 shows that as-synthesized catalyst MgO/Al2O3 gives the least weight loss, while the weight loss increases as the particle size decreases indicating that carbon deposition was increased. The deposited amount of carbon was calculated from the weight loss in the TGA experiments. The carbon balance obtained from the TGA profiles were similar to that obtained from Micro GC. A lower carbon balance was obtained when catalyst of smaller particle size was used. This indicates that the formation of solid carbon was favored by increasing the interaction between reactants and the catalyst surface. The TPO and mass spectrometer results from used catalyst also shows high amount of CO2 and low amount of H2O indicating very low composition of H in the deposited carbon (data not shown). The low amount of H content in the coke indicates major amount of solid product being formed by the agglomeration of monomolecular, active carbon species rather than polymerization of acetylene, ethylene, and propylene. Acetylene and propylene are well-known solid-carbon precursors during pyrolysis (combustion process). The presence of H species does specify that a similar phenomenon occurred in this plasma process. Further studies are required to explain the reasons for this result.

TGA profiles of MgO/Al2O3 catalysts with different particle sizes: a Fresh 1.75 mm, b 1.75 mm, c 1 mm, d 0.5 mm and e 0.25 mm used, respectively
Stability Check
To check the stability of catalyst, plasma reaction was performed using 0.5 mm of MgO/Al2O3 for 400 min at 6 kV and 3 kHz with 200 ml/min flow of 10 % CH4 in Ar. The result is represented in Fig. 9. Conversion of methane was 16 % and it remained relatively uniform during the entire reaction time. Formation of C2 and C3 hydrocarbons was predominant; with negligible production of other hydrocarbons. However, the total carbon balance of the products was only about 70 %, indicating that a lot of methane was converted to solid carbon species during the reaction. Carbon deposition on the reactor wall was also observed and carbon species deposited on the catalyst was confirmed by TGA analysis of the used catalyst. A lower carbon balance was obtained when catalyst of smaller particle size was used. This means that the formation of solid carbon was favored by increasing the interaction between reactants and the catalyst surface. We have not yet determined whether the solid product was coming from the polymerization of acetylene, ethylene, and propylene; or was formed by the agglomeration of monomolecular, active carbon species. Acetylene and propylene are well-known solid-carbon precursors during pyrolysis (combustion process). It seems that a similar phenomenon occurred in this plasma process. Further studies are required to explain the reasons for this result.

Time-on-stream over MgO/Al2O3 catalyst (0.5 mm particle size) for plasma-assisted methane conversion at 6 kV and 3 kHz generating 3.4 W of discharge power
Effect of Feed Flow Rate
Figure 10 shows the influence of the feed gas flow-rate on the reaction result. MgO/Al2O3 was used as a catalyst in plasma. Variation of the flow rate changed the conversion of methane; as well as selectivity for products. At high flow rates, the conversion of methane was very low due to the short contact time of gas with the catalyst. This indicates that higher conversion of methane can be achieved by lowering the flow rate. At lower flow rates, the molecules stayed in the plasma longer and had more chances to collide with energetic species such as electrons. This caused an increase in methane conversion [37]. Formation of CH3 radicals is initiated from the reaction of methane and electrons in the early stage of the plasma reaction; leading to abundant amounts of methyl radicals and to formation of ethane. It has been reported that CHX fragments are formed first by dissociation of CH4 via collision with metastable Ar, and then ethylene and acetylene can be formed via recombination [38]. Moreover, the reaction of CH3 with an ‘H’ or electron is also able to produce other radical species (e.g., CH2 and H2) [37, 39]. The homologous reaction of CH2 leads to the formation of ethylene. Because electron collisions with molecules are faster than collisions with ions or radicals [40, 41], longer residence time of molecules in the plasma will produce larger amounts of ethane and will inhibit formation of ethylene.

Effect of feed flow-rate on the catalytic performance of MgO/Al2O3 (0.5 mm particle size) for plasma-assisted, methane conversion at 6 kV and 3 kHz
Effect of Frequency of the Alternating Current
Figure 11 shows the effect of the frequency of the alternating current, on the methane conversion reaction using MgOAl2O3 catalyst. The frequency of the alternating current had a significant effect on the conversion of methane. Methane conversion increased with increasing frequency. High frequency implies application of a large amount of energy. When large amounts of power are supplied, the concentration of high-energy species increases in the plasma zone. These are easily able to break the chemical bonds of methane. Therefore, conversion of methane may be increased by increasing the power applied. Selectivity for products also varied according to the frequency. Selectivity for acetylene and ethylene was high at a low frequency. As frequency increased, selectivity for ethane increased; while selectivity for acetylene and ethylene decreased.

Effect of frequency on the catalytic performance of MgO/Al2O3 (0.5 mm particle size) for plasma-assisted methane conversion at 6 kV
Conclusions
Methane activation over Al2O3, MgO/Al2O3, and TiO2/Al2O3 catalysts was carried out in a DBD reactor. The conversion of methane over the catalysts was greater than in the absence of catalyst. In all cases, C2 hydrocarbons were the dominant products. The loading of MgO or TiO2 on Al2O3 increased the conversion of methane; as well as the selectivity for C3 hydrocarbons. The particle size of the catalyst used had a considerable effect on the conversion of methane; as well as on the selectivity for products. Higher conversion of methane, and higher selectivity for acetylene, ethylene, and propylene; was obtained with a smaller particle size of the catalyst, but more deposition of solid carbon species also occurred. Greater conversion of methane was achieved by supplying more energy to the DBD reactor, by lowering the flow rate, and by increasing the frequency of the alternating current. Selectivity for acetylene and ethylene was enhanced by supplying less energy, by increasing the flow rate, and by decreasing the frequency of the alternating current.
References
Brook EJ, Harder S, Severinghaus J, Steig EJ, Sucher CM (2000) Global Biogeochem Cycles 14:559–571
Lashof DA, Ahuja DR (1990) Nature 344:529–531
Lunsford JH (2000) Catal Today 63:165–174
Ross JRH (1975) In: Roberts MW, Thomas JM (eds) Surface and defect properties of solids, vol 4. The Chemical Society, London
Holmen A (2009) Catal Today 142:2–8
Rostrup-Nielsen JR (1993) Catal Today 18:305–324
Chou L, Cai Y, Zhang B, Niu J, Ji S, Li S (2002) J Nat Gas Chem 11:131–136
Pak S, Lunsford JH (1998) Appl Catal A 168:131–136
Kogelschatz U (2003) Plasma Chem Plasma Process 23:1–46
Kado S, Sekine Y, Fujimoto K (1999) Chem Commun 24:2485–2486
Alvarez-Galvan MC, Mota N, Ojeda M, Rojas S, Navarro RM, Fierro JLG (2011) Catal Today 171:15–23
Młotek M, Sentek J, Krawczyk K, Schmidt-Szałowski K (2009) Appl Catal A Gen 366:232–241
Liu C-J, Mallinson R, Lobban L (1998) J Catal 179:326–334
Wang Z-J, Zhao Y, Cui L, Du H, Yao P, Liu C-J (2007) Green Chem 9:554–559
Kim SH, Jung C-H, Sahu N, Park D, Yun JY, Ha H, Park JY (2013) Appl Catal A Gen 454:53–58
Istadi I, Amin NAS (2006) Fuel 85:577–592
Xu Y, Bao X, Lin L (1998) J Catal 216:386–395
Kim S-S, Lee H, Song HK, Na B-K (2006) J Ind Eng Chem 12:558–565
Arif M, Malik SA, Xuaen-Zhen J (1998) J Gas Chem 2:166
Sentek J, Krawczyk K, Młotek M, Kalczewska M, Kroker T, Kolb T, Schenk A, Gericke K-H, Schmidt-Szałowski K (2010) Appl Catal B Environ 94:19–26
Tu X, Whitehead JC (2012) Appl Catal B Environ 125:439–448
Jo S, Lee DH, Kang SW, Song HY (2013) Phys Plasma 20:123507–123514
Gallon HJ, Tu X, Whitehead JC (2012) Plasma Process Polym 9:90–97
Tu X, Gallon JH, Whitehead JC (2011) J Phys D Appl Phys 44:482003–482007
Tu X, Gallon HJ, Twigg MV, Gorry PA, Whitehead JC (2011) J Phys D Appl Phys 44:274007–274011
Wan JKS, Chen YG, Lee YJ, Derew MC (2000) Res Chem Intermed 26:599–619
Nozaki T, Hattori A, Okazaki K (2004) Catal Today 98:607–616
Kostov KG, Honda RY, Alves LMS, Kayama ME (2009) Braz J Phys 39:322–325
Manley TC (1943) Trans Electrochem Soc 84:83–96
Nozaki T, Tsukijihara H, Fukui W, Okazki K (2007) Energy Fuels 21:2525–2530
Coogan JJ, Sappey AD (1996) IEEE Trans Plasma Sci 24:91–92
Yang Y (2003) Plasma Chem Plasma Process 23:327–346
Hammer T, Kappes T, Baldauf M (2004) Catal Today 89:5–14
Robertsona J (2004) Eur Phys J Appl Phys 28:265–291
Chen HL, Lee HM, Chen SH, Chang MB, Yu SJ, Li SN (2009) Environ Sci Technol 43:2216–2227
Trionfetti C, Agıral A, Gardeniers JGE, Lefferts L, Seshan K (2008) J Phys Chem C 112:4267–4274
Indarto A, Choi JW, Lee H, Song HK (2006) J Nat Gas Chem 15:87–92
Riccardi C, Barni R, Fontanesi M, Tosi P (2000) Chem Phys Lett 329:66–70
Indarto A, Choi JW, Lee H, Song HK (2005) J Nat Gas Chem 14:13–21
Lieberman MA, Lichtenberg AJ (1994) Principles of plasma discharges and material processing, 1st edn. Wiley, New York
Lee DH, Kim K-T, Song Y-H, Kang WS, Jo S (2013) Plasma Chem Plasma Process 33:249–269
Acknowledgments
The authors acknowledge the financial support received from the Korea Research Council for Industrial Science and Technology (ISTK) of the Republic of Korea, Grant Number B551179-11-03-00.
Author information
Authors and Affiliations
Corresponding authors
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kasinathan, P., Park, S., Choi, W.C. et al. Plasma-Enhanced Methane Direct Conversion over Particle-Size Adjusted MOx/Al2O3 (M = Ti and Mg) Catalysts. Plasma Chem Plasma Process 34, 1317–1330 (2014). https://doi.org/10.1007/s11090-014-9574-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11090-014-9574-9