
Abstract
Plasma catalysis is a promising approach to further enhance the conversion of methane into value-added products such as methanol. In this work, the mechanisms enabling the conversion of methane to CO, CO2 and methanol enabled by plasma-enhanced catalysis were investigated. A catalyst reactor was incorporated downstream of the plasma jet to enable the separation between plasma generation and the catalyst bed. An enhancement in CH3OH and CO2 production was observed for the shortest distance between the plasma and catalyst compared to the plasma-only case. Plasma-enabled gas heating was shown not to be responsible for the observed synergy while a gas temperature increase as low as 30–40 K significantly impacted desorption rates of CH3OH/C2H5OH on alumina particles. Correlations between molecular beam mass spectrometry (MBMS) measurements at the inlet and outlet of the catalytic reactor suggest that the observed synergistic effect was caused by radical species most likely the CH3O2 radical. This study shows that surface reactions induced by radicals such as alkylperoxy radicals might play an important role in surface reactions in plasma-catalysis.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Catalytic partial oxidation of CH4 is a particularly interesting route for converting the abundantly available natural gas into higher energy density liquid fuels [1]. Methanol (CH3OH), one of the products of partial CH4 oxidation, is furthermore a clean and renewable fuel source and is also in great demand as an intermediate source of green energy to provide electric energy generation via fuel cell technology applications [2].
The conventional approach to convert CH4 into CH3OH is a two-step catalytic process. The first step is to convert CH4 into syngas (CO + H2) at high temperatures. The syngas is subsequently converted to methanol or other liquid fuels at high temperature and pressures (typically 60–100 bar and 250–280 ºC) over a Cu–ZnO-based catalyst. Due to the high cost of this process, alternative energy-efficient technologies to directly convert CH4 into CH3OH or other oxygenates are being developed based on the partial oxidation of methane.
Although significant efforts have been devoted to the investigation of direct conversion of methane to oxygenates via thermal catalysis, the reported product yields in terms of CH3OH to date are insufficient to replace the conventional two-step process [3]. Plasma-catalysis has been proposed as an innovative approach to enable methane to methanol conversion. This approach exploits the observed synergistic effect between plasma and thermal catalysis that potentially enables chemical conversion at lower temperatures and pressures as in conventional thermal catalytic processes [4].
Nozaki and Okazaki [5] experimentally and theoretically analyzed the energy efficiency of non-oxidative methane conversion using a dielectric barrier discharge (DBD), and showed an enhanced CH4 conversion efficiency attributed to vibrationally excited species. Kim et al. [6] performed a detailed kinetic study of CH4 activation in a DBD to quantify plasma-catalyst interactions via kinetic parameters. The results showed that the thermal catalysis had a typical Arrhenius behavior with an activation energy of ~ 73.5 kJ mol−1, while the plasma catalysis case exhibited non-Arrhenius behavior suggested to be due to the interaction between plasma and catalysts. In addition, the reported energy barrier for plasma catalysis was found to be much lower than the activation energy needed in thermal catalysis. The same group reported on the dry reforming of CH4 with CO2 using Ni/Al2O3 catalysts, and showed a significant enhancement of the activation of C-H bonds by the plasma when the gas temperature > 630 K [7]. The comparison between the CH4 conversions of thermal catalysis and plasma catalysis indicated a ~ 50 K shift in the activation energy. This 50 K gas temperature increase could be easily caused by localized plasma-induced gas heating. In addition to these synergistic effects, the use of non-thermal plasmas for catalyst regeneration was also reported [8]. The active species produced by the plasma can oxidize the coke formed on the catalyst and regenerate the catalyst at room temperature (293 K), requiring potentially lower energy than the common thermal regeneration requiring ~ 800 K.
Recently, Chawdhury et al. [9] investigated the plasma-catalytic partial oxidation of methane into oxygenates in a dielectric barrier discharge (DBD) reactor at room temperature. Three different supported transition metal catalysts (Ni/γ-Al2O3, Cu/γ-Al2O3, and Fe/γ-Al2O3) were tested, and the result showed that Fe/γ-Al2O3 provided the highest methanol selectivity (36%) and a methanol yield of 4.7%. The authors suggested that plasma-produced CHx species were critical for CH3OH production. De Bie et al. [10] established a one-dimensional fluid model of a DBD reactor to model the conversion of methane into oxygenates. The main underlying reaction pathways forming syngas, methanol and other oxygenates were determined for both partial oxidation and dry reforming of methane. The formation of methanol in the gas phase was shown to be due to radical reactions involving CH3, CH3O2 and CH3O as the main intermediate species. Yi et al. [11] investigated the selective oxidation of CH4 to CH3OH over Ni-based catalysts at low temperature and atmospheric pressure and achieved 81% oxygenate selectivity and 50% CH3OH selectivity. This work also reported kinetic modelling results with similar conclusions as De Bie et al. [10] The production of CH3OH on the catalyst was however attributed to chemisorbed oxygen species that react with the plasma-produced gas-phase CH3 radicals to form the intermediate species CH3Oad on the catalyst. The authors also hypothesized that H2O molecules produced by the plasma might be activated by the NiO/γ-Al2O3 catalyst and then further promote the desorption of CH3OH from the catalyst surfaces, according to the findings from [12].
Although researchers have successfully increased CH3OH yields in plasma catalysis reactors by adjusting the composition of catalysts and plasma conditions, such findings require trial-and-error parametric studies and our understanding of the underpinning process remains limited. This lack of understanding hampers further improvements in energy efficiencies, selectivity and yield for CH3OH production by plasma catalysis. Inspired by the work of Chawdhury et al. [9] we implemented a detailed study of plasma-produced species and conversion yields to investigate the plasma-catalyst interaction in the context of direct conversion of methane to oxygenates at room temperature. This is enabled by decoupling the catalyst and the plasma reactor similarly as in [13]. The products and plasma-produced species with or without catalysts were both quantified using molecular beam mass spectrometry (MBMS). Plasma-produced reactive species entering the catalytic reactor were correlated with the products downstream of the catalyst reactor to assess their importance in the plasma-catalyst interaction.
Experimental Setups
Plasma Jet and Catalyst Reactor
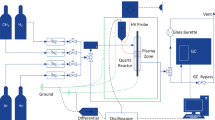
In this work, we coupled an atmospheric pressure plasma jet with a catalytic reactor downstream enabling the separation of the active plasma region and catalyst, as shown in Fig. 1. As the catalyst reactor can be detached from the plasma jet, this configuration also allowed us to measure the species densities both at the entrance and exit of the catalytic reactor. We used three different combinations of the plasma jet and the catalyst reactor, as shown in Fig. 1. These configurations allow for distinguishing effects of short-lived species and long-lived species by investigating two distances from the plasma jet electrode to the catalysis reactor (5 mm and 33 mm, respectively).

Schematic of the different configurations of the flow-through plasma-catalysis reactor used in this study
The plasma jet used in this work is identical to the one used in [13]. The plasma jet was driven by a radiofrequency voltage waveform modulated by a 20 kHz modulation with a 20% duty cycle. The main difference with the work in [13] is that CH4 is added into the feed gas instead of being mixed downstream in the plasma effluent. The composition of the feed gas was fixed at Ar + 1% CH4 + 0.5% O2 throughout this study. The Ar flow rates were kept constant at 200 sccm in this study unless otherwise stated. Higher CH4/O2 ratios as used in [9] were also attempted in the preliminary experiments but resulted in carbon deposition or gas-phase carbon particle formation, which was not further pursued to avoid clogging of the sampling orifice of the MBMS. A 200 mesh was implemented at the outlet of the catalyst reactor to prevent the catalyst particles from being blown out and similarly blocking the sampling orifice of the MBMS. The operation of the plasma jet and the plasma dissipated power calculation are described in detail elsewhere [14]. The gas temperature measurements in this study were performed with a K-type thermal couple or a temperature strip (OMEGA, RLC) with a temperature resolution of 5 K.
Catalyst Information and Preparation
Microsphere catalysts (20% wt% Fe/γ-Al2O3) and the control group γ-Al2O3 particles from Riogen, Inc. were used in this study. All the particles have the same size (~ 375 µm), and the entire catalytic reactor was loaded loosely without compressing the catalyst to ensure a low flow resistance. The weights of the loaded Fe/γ-Al2O3 and Al2O3 particles were 0.53 g and 0.39 g, respectively. The pretreatment consisted of an Ar + 4% H2 plasma treatment at atmospheric pressure for about 2 h after the catalyst reactor was installed in the reactor.
Molecular Beam Mass Spectrometry (MBMS)
MBMS was used to quantify the reactive species produced by the plasma as well as the downstream products of the catalyst reactor. The details of the MBMS system used can be found in [15]. The species measured in this work include the reactants (CH4 and O2), main products (CO, CO2, H2), minor products (oxygenates) and the short-lived species CH3. While attempted, we were unable to measure atomic oxygen and hydrogen radicals. Table 1 provides a summary of the electron energy and calibration gas used for each species measured by MBMS.
The measurements of stable species including CO, CO2, H2, O2, and CH4 have been performed using electron energy of 70 eV. Their absolute calibration is straightforward because it can be achieved with a known concentration of the particular gas. For the short-lived reactive species, threshold ionization mass spectrometry (TIMS) is used to exclude the contribution of dissociative ionization from their parent molecules. An additional 50 Hz RF plasma modulation is implemented on top of the 20 kHz RF modulation to enable the accurate subtraction of the background signals. Details regarding the TIMS and the rationale and implementation of the background subtraction approach can be found in [15]. We were only able to measure a radical signal at 15 amu. A detailed analysis in the result section shows that this cannot be due to the methyl radical as initially expected but is due to the CH3O2 radical. We have also measured m/z = 47 corresponding to CH3O2+ which yielded similar trends as CH3+ although with lower signals likely because CH3O2+ is an unstable ion [18]. We have for this reason used the CH3+ signal (see results section for more details).
The possible oxygenates produced in this work include methanol (CH3OH), ethanol (C2H5OH), formaldehyde (CH2O), formic acid (HCOOH), acetic acid (CH3COCH3), and acetone (CH3COCH3). Due to the possible existence of other hydrocarbons with the same masses such as C2H6 and the complex cracking pattern of these oxygenates in the mass spectrum, further analysis of the MBMS results is required. According to the mass spectrum of oxygenates and relative hydrocarbons, the masses (m/z = 60 amu and m/z = 58 amu) can represent formic acid and acetic acid, respectively. The mass (m/z = 46 amu) is a combination of ethanol and formic acid, and the mass (m/z = 30 amu) can represent both C2H6 and CH2O.
As for CH3OH with a mass of 32 amu, we tracked the fragment (m/z = 31 amu) which is the highest signal at 70 eV in the CH3OH mass spectrum, as this enables to exclude the contribution from the abundantly present O2 molecules which have the same mass as methanol. Nevertheless, ethanol (C2H5OH) might also contribute significantly to the mass (m/z = 31). Contributions of other oxygenates to the m/z = 31 is less likely as this fragment is less abundant (< 1%) for other oxygenates at 70 eV. Hence the measured species with mass of 31 represent CH3OH or C2H5OH. The partial cross-sections of CH3O+ from CH3OH and C2H5OH at 70 eV are 1.5 × 10–16 cm−3 and 2.5 × 10–16 cm−3, respectively [20] and require further analysis for distinguishing between the two species.
In this work, the percentage conversion (η) of CH4 and O2 are calculated as:
The production rates of species are calculated as:
with \({n}_{i}\) the measured species density of species i at the exit of the reactor and Φ the volumetric gas flow rate.
The conversion rate of CH4 and O2 are calculated as:
with \({n}_{j}\) the density of CH4 and O2 in the feed gas entering the reactor.
Results and Discussion
CH4 and O2 Conversion by Plasma and Product Identification
In this section, a general overview of the CH4 and O2 conversion as well as the main products for the plasma without catalyst case are provided as a reference for the plasma catalysis results reported in later sections. Figure 2(a) shows the CH4 and O2 conversion as a function of plasma dissipated power. The influence of gas temperature is minimized by performing the measurements with an extended quartz tube (30 mm) which keeps the gas temperature near room temperature (below 340 K) at the nozzle. As both CH4 and O2 are flowing through the ionizing plasma region, CH4 and O2 conversions up to 30% and 50% respectively are observed in the investigated power range. The conversion of both CH4 and O2 increases with increasing plasma dissipated power.

a CH4 and O2 conversions as a function of plasma dissipated power for the plasma only (case C in Fig. 1); b Conversion rate of CH4 and associated production rate of CO and CO2
Figure 2b compares the conversion rate of CH4 with the production rate of CO and CO2, the dominant carbon-containing products. Comparing the combined CO and CO2 production rate with the CH4 conversion rate indicates that more than 90% of the CH4 is converted into CO and CO2, with CO accounting for about ~ 80% of the CH4 conversion. As shown below, the H2 production rate is similar to the CO production rate, hence based on stoichiometric considerations the H2O production rate, which is more challenging to measure is also to be expected similar to the CO production. This also means that the selectivity of hydrocarbon to oxygenate conversion by the RF plasma jet is below ~ 10% and seems to be favored for higher plasma powers.
To quantify the oxygenates produced by the plasma, measurements of methanol/ethanol (CH3OH/ C2H5OH), formaldehyde (CH2O), formic acid (HCOOH), acetic acid (CH3COOH) and acetone (CH3COCH3) were performed with the MBMS. Figure 3a shows time-resolved measurements of the mass corresponding to several oxygenates with an electron energy of 70 eV. The partial ionization cross-sections at 70 eV of these measured fragments from oxygenates are all within a factor of 2 and on the order of 10–16 cm2 [20,21,22,23]. Nonetheless, smaller molecules would have larger losses during the supersonic expansion and molecular beam [24] than heavier molecules although their ionization cross-sections are generally smaller as well which will compensate to some extent for this difference. Hence, the relative mass spectrometry signals represent within approximately a factor 2 the concentration of each oxygenate. The MS signal is corrected for background contributions by subtracting the MS signal collected during ‘plasma off’ for the oxygenates.

a Time-resolved mass spectrometry signals for CO and different oxygenates; b comparison of mass spectrometry signals (the difference between plasma ON and plasma OFF) for CO and different oxygenates. The plasma power was 4.7 W and the distance between the electrode and the nozzle was 5 mm
Figure 3b shows that methanol/ethanol (m/z = 31) has the largest signal (difference between plasma ON and OFF) compared to the other measured oxygenates. Both CH3COOH and CH3COCH3 are approaching the detection limit and are not analyzed in detail. To further evaluate the contribution of C2H5OH to the measured signals (m/z = 31), we compared the signals at m/z = 45 and m/z = 46. At electron energy of 70 eV, the ratios of fragments at m/z = 45 and m/z = 46 for C2H5OH and HCOOH are about 2.5 and 0.8, respectively [25]. Based on this fragment ratio (~ 2.0 in our experiments), a rough estimation of the density ratio of C2H5OH and HCOOH can be obtained, which is about 7:3. Furthermore, with this density ratio of C2H5OH and HCOOH, the contribution from C2H5OH to the signal at m/z = 31 can be estimated, which is about 30%. In addition, the signals at m/z = 30 are about 70% of that at m/z = 31, which might be interpreted as formaldehyde (CH2O) or C2H6 but these two species are not distinguishable. The experimental results from [9] indicated that the CH2O density might be comparable to that of C2H6.
The above comparison shows that CH3OH is the most dominant oxygenate produced by the plasma. Therefore, we focus on m/z = 31 for the investigation of oxygenates in this study while recognizing that C2H5OH contributes to the MS signal.
Comparison of Plasma with and Without Catalyst
The observed synergy in plasma catalysis is recently becoming more and more attributed to radicals [11, 26] although this attribution is typically based on indirect observations. To assess the role of short-lived species in plasma-catalyst interactions, we investigated in this study the two configurations in Fig. 2 which enabled us to study plasma-catalyst interactions for a distance of 5 and 33 mm between the electrode and the entrance of the catalytic reactor.
As the plasma plume length for the maximum power case is less than 5 mm, the case of 33 mm has at least a 28 mm distance between the plasma and catalyst particles. Given that for the reference flow rate of 200 sccm, this distance corresponds to a gas residence time of 26 ms short-lived plasma-produced radicals will not reach the catalytic reactor although the impact of long-lived reactive plasma-produced species on the catalysts can be assessed. Indeed radicals such as CH3, O, H and CH3O2 have for the conditions and gas composition investigated in this work lifetimes of at most 1 ms (see also futher). In the case of 5 mm, the catalyst and the plasma remain separated (no direct coupling) but when the plasma power is increased the tip of the plasma plume can reach the entrance of the catalyst reactor. This might enable a more complex interaction between the plasma and the catalyst, and a fraction of the plasma-produced radicals will reach the catalyst particles before recombining. While Ar plasmas are known to produce excimer radiation and argon metastable species that have the potential to impact catalytic surface reactions, the presence of O2 and CH4 in excess of 1% of the total gas composition reduces the lifetimes of the Ar excited states by reactions with molecules. This leads to dissociation of O2 and CH4 and the production of radicals while the metastable density becomes very low and the excimer production is suppressed [27, 28]. Hence, CH4 and O2 chemistry will dominate under the investigated conditions.
CH4 and O2 Conversion
Figure 4 compares the CH4 and O2 conversion as a function of plasma power with catalyst particles or with alumina particles (configuration A and B in Fig. 1) to evaluate potential synergistic effects. Nonetheless, the CH4 conversion is identical within the experimental uncertainty (~ 5 × 1015 cm−3) for the four cases suggesting that the addition of a catalyst does not enhance the CH4 conversion for the investigated conditions. On the other hand, an enhancement of the O2 conversion was observed at higher plasma powers for the 5 mm case (configuration A in Fig. 1) where radicals are able to reach the catalyst (see further). Considering that the enhancement in CH4 conversion is negligible, it suggests an increase in oxygen-containing products, possible oxygenates or CO2.

Comparison of CH4 and O2 conversion as a function of plasma dissipated power for plasma only (configuration C in Fig. 1) and plasma-catalysts at a distance of 5 and 33 mm (configuration A and B in Fig. 1, respectively). A case where the catalyst was replaced with alumina pellets at a distance of 5 mm was also included as a reference
Products
Figure 5a–d show the densities of the dominant conversion products (H2, CO, CO2 and CH3OH/C2H5OH) as a function of plasma dissipated power for the same experimental conditions as the CH4 and O2 conversion shown in Fig. 4. The introduction of the catalyst does not impact the H2 and CO production significantly. Nonetheless, a significant increase in CO2 production is observed for the 5 mm case (configuration A in Fig. 1) in the presence of catalysts compared to the other cases for powers in excess of 4 W. This increase in CO2 density of ~ 0.7 × 1016 cm−3 correlates with the observed increase in O2 conversion at higher plasma power as shown in Fig. 4. As the increase is not found for alumina pellets without catalyst, the increase of CO2 is enabled by the presence of the catalyst in the plasma effluent, as discussed in detail [13].

Product densities as a function of plasma dissipated power for identical experimental conditions as reported in Fig. 4: a H2 density b CO density c CO2 density d CH3OH/C2H5OH density. The data encircled in the red dashed line in subfigure (d) is impacted by non-steady state effects and the blue arrow indicates the observed plasma-catalyst synergy
Figure 5d shows the methanol/ethanol production which interestingly, shows two obvious different trends for low and high plasma powers. The ‘plasma only’ case can be considered representative of the CH3OH/C2H5OH production by the plasma and CH3OH/C2H5OH species entering the catalytic reactor. While the CH3OH/C2H5OH densities do not show a strong dependence on the plasma dissipated power, at larger plasma powers, the CH3OH density decreases likely due to the dissociation of the formed CH3OH in the ionizing plasma. The following sections mostly focus on the explanation of these two changes.
At small plasma powers (1–2.5 W), the addition of catalyst and alumina particles leads to a remarkable reduction in the measured CH3OH/C2H5OH density. This reduction is caused by the fact that it takes up to 3–4 h for the system to reach a steady state. We studied this effect in the case of alumina in detail and showed that once the plasma-catalyst system reaches steady-state it yields, within the experimental accuracy, the same amount of CH3OH/C2H5OH as in the ‘plasma only’ case. It was further confirmed that at larger plasma powers the system reaches a steady state on a timescale ~ 10 min and the CH3OH/C2H5OH losses to the alumina causing this effect do not have an impact on the reported measurements. Hence, the increase found for the 5 mm case with catalyst suggests a plasma-catalyst synergy which is analyzed in more detail below. It should be noted that this synergy cannot be readily explained by conversion of plasma-produced syngas into CH3OH/C2H5OH as the 33 mm case (configuration B in Fig. 1) does not yield an enhancement of CH3OH/C2H5OH. In the next section, we assess why the time to reach steady state in the CH3OH/C2H5OH production is so strongly dependent on the plasma power.
Absorption of Methanol/Ethanol on Catalyst Substrate
The vapor pressure of methanol and ethanol at 20° is 94 and 44 mmHg [25] respectively corresponding to 6 and 12% which is larger than the original reactants concentration, hence there will not be significant condensation in the reactor even without additional heating. Examples of time-resolved mass spectrometer signals at m/z = 31 amu corresponding to methanol/ethanol after switching off the plasma are shown in Fig. 6. Figure 3a shows that this minutes-long decay is found for CH3OH/C2H5OH but not for CO. The ‘plasma only’ case in Fig. 6 shows that once the plasma is switched off, the production of CH3OH/C2H5OH stops and the MS signals decrease on a timescale of a few milliseconds suggesting negligible accumulation of CH3OH/C2H5OH on the quartz capillary wall. However, in the presence of catalyst and alumina particles, the MS signal decreases on a time scale of minutes, orders of magnitude larger than the gas residence time in the system, suggesting another source of CH3OH/C2H5OH is present after switching off the plasma.

Time-resolved normalized MS signal at 31 amu attributed to methanol/ethanol after switching off the plasma at t = 0 s for configuration A (Fig. 1). The accuracy of the power measurement is 0.1 W
This additional source of methanol/ethanol is due to desorption of previously adsorbed CH3OH/C2H5OH onto the particles in the reactor as the phenomenon is also observed for alumina particles which cannot serve as catalysts. This conclusion is further supported by several publications reporting the possibility of CH3OH/C2H5OH absorption on catalysts or Al2O3 support [29,30,31]. Therefore, the various decay times for different conditions are impacted by changes in desorption. In addition, the comparison between the decay curves for catalysts and alumina for the same plasma dissipated power in Fig. 6 suggests that it requires a shorter time to remove the absorbed CH3OH/C2H5OH molecules from alumina particles than from catalyst particles under the investigated experimental conditions.
The area under the decay curve in Fig. 6 is the total amount of CH3OH/C2H5OH molecules desorbed from the particles after the plasma is switched off and hence is a measure of the amount of surface adsorbed molecules during steady-state exposure by the plasma effluent. Figure 7 shows this total amount of absorbed molecules on the particle surface as a function of plasma dissipated power. The amount of absorbed molecules at small plasma powers (~ 1 W) is about 5 times larger than the higher plasma power (> 3 W). The 5 times larger amount of surface molecules for low plasma powers might contribute to the longer time it takes to reach a steady state surface coverage. Figure 7 also shows that the absorption abilities of catalysts and alumina particles are similar which might suggest the dominant contribution of the alumina substrate to the observed CH3OH/C2H5OH absorption.

Amount of absorbed methanol/ethanol molecules on the surface of the catalyst and alumina particles during steady-state plasma effluent exposure as a function of plasma dissipated power for a distance of 33 mm between plasma and catalyst. The measurements are conducted after steady-state operation of the reactor is achieved
The observed change in the absorption ability of particles might be due to the change in gas temperature or plasma-produced reactive species as both scales with the plasma dissipated power. Considering that the absorption ability is also significantly impacted for the plasma at 33 mm from the catalytic reaction (see Fig. 7), it is unlikely that radicals play a dominant role in this effect and hence it must be due to long-lived reactive species or gas heating.
The gas temperatures at the nozzle of the quartz capillary (corresponding to the entrance of the catalytic reactor) are shown for an electrode-nozzle distance of 5 and 33 mm as a function of plasma dissipated power in Fig. 8. While the gas temperature increases by more than 100 °C for the 5 mm distance case, the gas temperature for the 33 mm case does not increase more than 30 °C. Hence, the variation in surface coverage for the 33 mm case, if due to temperature must be caused by a mere increase in gas temperature of 30 °C.

Gas temperature as measured at the nozzle of the quartz capillary with a thermocouple as a function of plasma dissipated power for an electrode to nozzle distance of 5 and 33 mm
Figure 9 shows the evolution of m/z = 31 signals after the Ar-CH4-O2 plasma is switched off for different sequential plasma exposures to evaluate the influence of gas temperature and plasma-produced species on the desorption. All three cases shown in Fig. 9 have the same initial conditions, that is, the starting number of absorbed methanol molecules on the particle surface is the same. Note that the area under the curves is also the same within the experimental accuracy and hence no new sources of methanol are introduced by introducing the additional post plasma treatments during the desorption process.

Time-resolved MS signal at 31 amu attributed to methanol/ethanol after switching off the Ar + 1% CH4 + 0.5% O2 plasma at t = 0 s for different post plasma exposure conditions during the desorption phase: Ar plasma, Ar + 1% CH4 + 0.5% O2 gas flow (no plasma) and Ar + 0.5% O2 plasma yielding a gas temperature at the entrance of the catalytic reactor of 65 ºC, 25 ºC and 30 ºC respectively
As the MS measures the desorbed methanol and the net desorption rate will be fixed at a given temperature, the slopes of the MS signal are a measure of the surface coverage and hence the time constant (inverse of the initial slope) yields a measure of the methanol desorption time. As the argon plasma does not produce reactive species with a lifetime exceeding or similar to the gas residence time in the quartz capillary, it can be concluded from the enhanced CH3OH/C2H5OH signal the first 20 min after switching off the plasma that gas heating of 30 – 40 ºC is able to significantly impact methanol desorption. This is further confirmed by comparing the desorption of CH3OH/C2H5OH during the exposure of the catalyst to the effluent of a low temperature Ar + O2 plasma (Fig. 9) operating at 30 ºC which has a significantly smaller impact on the methanol desorption than the Ar plasma case at 65 ºC. The results shown in Fig. 9 suggest that small temperature variations are more dominantly contributing to the desorption of methanol than long-lived plasma-produced species such as O3 or O2(a1Δg). These results further suggest that small plasma-induced gas temperature increases even as low as 20 ºC (and corresponding surface temperature increase of the catalyst and substrate) can impact species desorption and hence potentially impact conversion rates in plasma catalysis.
Correlation Between Plasma-Produced Reactive Species and Methanol/Ethanol Production
In the previous section, we showed that the plasma-catalyst combination can impact desorption rates and CH3OH/C2H5OH yields particularly at low plasma powers. In this section, we analyze the synergistic enhancement in methanol production as reported in Fig. 5d, at high plasma powers for steady-state conditions in more detail. The largest increase in methanol production found for the investigated conditions in this study is 60% compared to the ‘plasma only’ case. This corresponds to an increase of the absolute density of methanol of ~ 2 × 1015 cm−3. While this synergistic effect is moderate and insufficient to be valuable from an application perspective, the controlled decoupled plasma-catalysis experiments in this study provide an excellent opportunity to contribute to a more detailed understanding of the observed synergistic effect in plasma catalysis applications based on detailed experiments.
We measured the reactive species by MBMS implementing another 50 Hz modulation onto the plasma to enable the accurate subtraction of the background [15]. While several radicals were probed, we were only able to measure a radical species at a mass of 15 amu corresponding to the mass of CH3+ and 47 amu corresponding to CH3O2+. Figure 10a presents a time-resolved measurement of CH3+ with the electron energy of the ionizer of 12.5 eV at a plasma power of 5 W for an electrode to nozzle distance of 5 mm. Significant differences can be found between the ‘plasma on’ and ‘plasma off’ periods, indicating the existence of a radical species. The electron energy of 12.5 eV was chosen to be sufficiently above the ionization energy of the CH3 radical (9.8 eV) and low enough to avoid the contribution of CH3+ formed by dissociative ionization of CH4 molecules which requires an electron energy of 14.0 eV [32]. In addition, the contributions of CH3+ from other stable products like CH3OH can be excluded as the measured signal dramatically dropped down below the detection limit when increasing the distance between the plasma to the MBMS orifice by only 1 mm.

a Time-resolved MS signals of CH3+ attributed to CH3O2 (see text for details) for a plasma power of 4.7 W and an electrode to nozzle-distance of 5 mm; b Correlation between the CH3O2 radical flux at the nozzle and the increase in CH3OH production in the catalyst reactor for variations in plasma dissipated power and gas flow rate
Assuming the measured species is CH3, an absolute calibration with CH4, results in an absolute density of the order of ~ 1014 cm−3. This seems however inconsistent with the kinetics of CH3 and the CH3 lifetime. The production reactions of CH3 in the plasma afterglow was previously shown to be negligible compared to the destruction reactions [10] and the lifetime of CH3 in the afterglow region can be estimated according to the dominant destruction reaction for the investigated experimental conditions [10]
Assuming the gas temperature is 400 K and the O2 density is depleted by 50% in the plasma, yields a lifetime for CH3 of ~ 1 µs. This lifetime is significantly smaller than the time needed to transition the suction region of the mass spectrometer (~ 18 µs) [15] and hence the CH3 density should drop below the MBMS detection limit before entering the molecular beam. The measurement hence suggests that the measured MS signal at 15 amu is due to a reactive species with a lifetime between 10 µs to a few ms at most (gas residence time corresponding to a gas plug of 1 mm in the reactor). We can further exclude the possibility of vibrationally excited CH4 because its lifetime is even shorter than CH3 [33]. CH3O2, which was detected at m/z = 41 amu, is identified as the most abundant radical in kinetic models of CH4-O2 plasma [10] and is the most likely candidate. Unfortunately, the data regarding the electron impact ionization (EII) cross-section of CH3O2 is not reported to our knowledge and a quantitative study would require chemical ionization or photoionization [18, 34] which is not a capability of the MBMS used. Fu et al. [18] reported that the CH3O2+ ion might be unstable and likely decomposes into CH3+ and O2 depending on the excess energy during the ionization process. The calculated ionization potential of CH3O2 was 10.8 eV and the extra energy available from the 12.5 eV electrons in the ionizer would dissociate CH3O2+ to yield CH3+. Meloni et al. [19] measured the photoionization efficiency curve for CH3O2, yielding adiabatic ionization energy of CH3O2 to be (10.33 ± 0.05 eV) and a CH3+– O2 bond energy of 0.83 ± 0.07 eV. The above information would suggest a threshold energy of (11.2–11.5 eV) to produce CH3+ from CH3O2. This is consistent with our experimental findings showing that the CH3+ signal was close to the detection limit for electron energy of 12.0 eV. The significant difference in CH3+ signals measured at 12.0 eV and 12.5 eV is more consistent with a species having a threshold ionization of 12.0 eV rather than 9.84 eV like CH3. The ratio of the electron-impact ionization cross sections of CH3 forming CH3+ at 12.0 eV and 12.5 eV is only 0.8 consistent with the interpretation of the MS signal at 15 amu to be due to CH3O2. As the CH3O2 signal is more than one order of magnitude smaller than m/z = 31, likely due to the reported unstable nature of the CH3O2+ ion [18], we estimate the CH3O2 density from the m/z = 31 signal.
While there is no partial EII cross-section for CH3+ formation from CH3O2 available in the literature, we can still make an approximate estimation of the CH3O2 density assuming the EII cross-section is the same as for CH3OH although with correction for the ionization threshold. The threshold energy to produce CH3+ from CH3OH is about 13.8 eV, and the EII cross-section is about 2 × 10–18 cm2 at 15.0 eV [20]. Assuming a similar EII cross-section for CH3+ formation from CH3O2 would yield the highest CH3O2 density in Fig. 10b to be 2 × 1015 cm−3 with a calibration using CO2 (m/z = 44). Although this density estimation has large uncertainties, it suggests that the measurement is consistent with a density of CH3O2 on the order of 1015 cm−3.
If CH3O2 is the dominant radical in the afterglow region, its lifetime can be estimated according to its self-recombination reaction since its reaction with CH4 or other long-lived molecules is very slow:
The lifetime is estimated to be 1.4 ms if assuming a CH3O2 density of 2 × 1015 cm−3 equal to the observed increase in the absolute density of methanol by the plasma catalyst interaction. This lifetime is likely an overestimation as we neglected its reaction with other radicals such as H, O or OH. All the above analysis suggests that a density of CH3O2 on the order of 1015 cm−3 with a lifetime of the order of ~ 1 ms are consistent with our MBMS measurements.
Figure 10b shows the MS signal at 15 amu attributed to CH3O2 at the inlet of the catalytic reactor and the increase in methanol/ethanol density at the outlet of the catalytic reactor as a function of plasma power. The trends of both the CH3+ MS signal attributed to CH3O2 and methanol/methanol correlate well. In addition, the CH3+ signal is below the detection limit for plasma powers less than 4 W, consistent with the lack of increase in methanol compared to the plasma only case suggesting that only for cases with a detectible amount of CH3O2 an enhancement of the methanol formation by the catalyst was found. This correlation suggests that CH3O2 might play a key role in the production of CH3OH on the surface of the catalyst.
Nonetheless, gas temperatures also vary with power as shown in Fig. 8. At a distance of 5 mm, the gas temperature can reach 160 °C at 5 W. The resulting elevation of the catalyst temperature might be able to trigger thermal catalysis which could enhance methanol production. To assess this effect, the impact of gas flow rates on the methanol production, gas temperature and radical density was measured. Figure 11a shows that while the methanol density remains constant as a function of the gas flow rate for the plasma only case (resulting in a linear increase in methanol production as a function of gas flow rate), a reduction in the methanol/ethanol density with increasing gas flow rate was observed when the catalyst is present. This decrease in methanol/ethanol density results in the lack of a significant observed synergistic effect at a flow rate of 600 sccm. However, this effect can be further enhanced by the reduced gas residence time.

Impact of the gas flow rate on a the CH3OH density and production rates and b the gas temperature and MS signal of CH3+ attributed to CH3O2 at a fixed plasma dissipated power of 4.7 W ± 0.1 W
Figure 11b shows the corresponding gas temperatures and MS signals of CH3O2. The gas temperature reduces from 160 °C to about 100 °C and the CH3O2 signal decreases about 70% with increasing gas flow rate from 200 to 600 sccm. The similar trends do not allow to make a conclusion of the cause of the reduction in methanol-based from this data alone but a comparison with another condition at the same gas temperature provides more insights. The 400 sccm case has a gas temperature of about 125 °C and the synergistic effect was observed. This case has the same gas temperature as the reported results between 3 and 4 W in Fig. 5d, which does not show any synergistic effect in methanol production. Hence, the observed synergistic effects can be attributed to plasma-produced species rather than gas heating.
The strong correlation both for the power and gas flow variation between the CH3O2 signal and the CH3OH (see Fig. 10b suggests the dominant role of radicals and in particular radicals with lifetimes between 100 µs and 1 ms like CH3O2.
Possible Reaction Pathways Underpinning Observed Synergy
Yi et al. [11] previously suggested that the production of CH3OH on plasma-exposed catalyst could be attributed to the reaction between chemisorbed oxygen species with the plasma-produced CH3 radicals to form CH3O adsorbed to the catalytic surface. In the present study, this proposed mechanism is not able to explain the observed results in view of the sub-microsecond lifetime of the CH3 radical. Nevertheless, another reaction pathway including the CH3O2 radicals is a more likely candidate to explain the increased CH3OH production. For example, in the gas-phase plasma, the self-recombination of CH3O2 would lead to the formation of CH3O radicals via R3-R4, which can chemisorb on the catalytic surface as CH3Oad yielding the same surface chemistry without the requirement of CH3 radicals.
Furthermore, surface chemistry to form methanol from CH3Oad was proposed to involve H radicals [11]. If the reaction between H + CH3Oad is indeed responsible for the production of CH3OH on the catalyst surface, the H density should be at least similar to the observed increase in methanol ~ 2 × 1015 cm−3, the detection limit of the MBMS [35]. This is highly unlikely as the lifetime of H is about ~ 2 µs (considering R2 and the three-body reaction between H and O2 with a CH3O2 density of at least 2 × 1015 cm−3). The diffusion timescale of H over a distance of 20 µm is on the order of 4 µs according to the diffusion coefficients taken from [22], which will lead to a large amount of gas phase recombination of H radicals before reaching the catalyst. This suggests that H cannot be responsible for the observed synergistic plasma-catalyst effect. One possibility is that CH3Oad can react with itself to produce CH3OH, like the equivalent gas-phase reaction:
Alternatively, reactions of CH3Oad with H2, similar to their gas-phase equivalent might be possible if they would proceed at a higher rate than in the gas phase.
Agarwal et al. [36] investigated aqueous selective CH4 catalytic oxidation to CH3OH and demonstrated that adding O2 to the reaction mixture would produce CH3O2 radicals by reacting with CH3 radicals. The proposed reaction pathway suggests the importance of CH3O2 radicals to enhance oxygenates’ production in surface reactions although for completely different reaction conditions. While the detailed mechanism is subject to further study, these results suggest a viable pathway through CH3O2 for methanol/ethanol production.
While recognizing that this study was performed in a reactor in which the catalyst particles were not in direct contact with the active plasma, an important outcome of this study is the large impact of transport limitations that might also play an important role for packed bed reactors where the catalyst is in ‘direct contact’ with the plasma. These transport limitations have been largely ignored by the research community when proposing surface reaction mechanisms in many plasma-catalysis studies. Even when a surface-hugging plasma might be in direct contact with the surface, a sheath will be present which depletes electrons near the interface of the catalyst and hence also radical production requiring radicals produced in the bulk plasma to diffuse through a sheath region. As a typical sheath thickness at atmospheric pressure is of the order of 100 µm [37], this requires a diffusion time of the order of 10 to 100 µs for H and CH3 at 400 K, suggesting that radicals with a lifetime of ~ 1 µs might even in the case of plasma in direct contact with the catalyst not dominantly contribute to plasma-catalyst interactions. We showed in this work that plasma-catalyst synergy is possible due to long-lived radical species such as alkylperoxy radicals with lifetimes comparable or larger than these diffusive timescales. While these species might be less reactive in the gas phase they can still play an important role in plasma catalysis as they are less impacted by transport limitations on the typical length scales between pellets of hundreds of micrometers in pack-bed reactors.
Conclusion
In this work, the mechanisms underpinning the interaction of plasma with Fe/γ-Al2O3 catalyst in the context of direct conversion of methane to CO, CO2 and oxygenates at room temperature are investigated by incorporating the catalyst reactor downstream of the plasma jet. An enhancement of CH3OH in the presence of a catalyst compared to the plasma only case is observed for the shortest distance between the plasma and catalyst studied in this work. Correlations between MBMS measurements and estimates of species lifetimes suggest that this synergistic effect is caused by radical species most likely CH3O2. It was possible to exclude dominant contributions of plasma-enabled heating on methanol formation for the synergistic effect observed in this work while it was shown that gas heating even as low as 30–40 °C was significantly impacting desorption rates of CH3OH on alumina particles. While most studies in plasma catalysis seem to focus on Elay-Rideal reactions by primary radicals such as H and CH3, this study shows that surface reactions induced by secondary more long-lived radicals such as alkylperoxy radicals might be less impacted by transport limitations and their role in surface reactions might deserve more attention.
References
Ravi M, Ranocchiari M, van Bokhoven JA (2017) The direct catalytic oxidation of methane to methanol-a critical assessment. Angew Chemie Int Ed 56(52):16464–16483
Joghee P, Malik JN, Pylypenko S, O’Hayre R (2015) A review on direct methanol fuel cells–in the perspective of energy and sustainability. MRS Energy Sustain 2(1):3
Larkin DW, Lobban LL, Mallinson RG (2001) The direct partial oxidation of methane to organic oxygenates using a dielectric barrier discharge reactor as a catalytic reactor analog. Catal Today 71(1–2):199–210
Bogaerts A, Tu X, Whitehead JC, Centi G, Lefferts L, Guaitella O, Azzolina-Jury F, Kim H-H, Murphy AB, Schneider WF et al (2020) The 2020 plasma catalysis roadmap. J Phys D Appl Phys 53(44):443001
Nozaki T, Okazaki K (2013) Non-thermal plasma catalysis of methane: principles, energy efficiency, and applications. Catal Today 211:29–38
Kim J, Go DB, Hicks JC (2017) Synergistic effects of plasma-catalyst interactions for CH 4 activation. Phys Chem Chem Phys 19(20):13010–13021
Kim J, Abbott MS, Go DB, Hicks JC (2016) Enhancing C-H bond activation of methane via temperature-controlled. Catalyst-Plasma Interactions ACS Energy Lett 1(1):94–99
Astafan A, Batiot-Dupeyrat C, Pinard L (2019) Mechanism and kinetic of coke oxidation by nonthermal plasma in fixed-bed dielectric barrier reactor. J Phys Chem C 123(14):9168–9175
Chawdhury P, Wang Y, Ray D, Mathieu S, Wang N, Harding J, Bin F, Tu X, Subrahmanyam C (2021) A promising plasma-catalytic approach towards single-step methane conversion to oxygenates at room temperature. Appl Catal B Environ 284:119735
De Bie C, Van Dijk J, Bogaerts A (2015) The dominant pathways for the conversion of methane into oxygenates and syngas in an atmospheric pressure dielectric barrier discharge. J Phys Chem C 119(39):22331–22350
Yi Y, Li S, Cui Z, Hao Y, Zhang Y, Wang L, Liu P, Tu X, Xu X, Guo H et al (2021) Selective oxidation of CH4 to CH3OH through plasma catalysis: insights from catalyst characterization and chemical kinetics modelling. Appl Catal B Environ 296:120384
Lustemberg PG, Palomino RM, Gutiérrez RA, Grinter DC, Vorokhta M, Liu Z, Ramírez PJ, Matolín V, Ganduglia-Pirovano MV, Senanayake SD et al (2018) Direct conversion of methane to methanol on ni-ceria surfaces: metal-support interactions and water-enabled catalytic conversion by site blocking. J Am Chem Soc 140(24):7681–7687
Li Y, Jiang J, Hinshelwood M, Zhang S, Bruggeman P, Oehrlein GS (2021) Characterization of Plasma Catalytic Decomposition of Methane: Role of Atomic O and Reaction Mechanism. J Phys D Appl Phys. https://doi.org/10.1088/1361-6463/ac4728
Hofmann S, van Gessel AFH, Verreycken T, Bruggeman P (2011) Power dissipation, gas temperatures and electron densities of cold atmospheric pressure helium and argon RF plasma Jets. Plasma Sources Sci Technol 20(6):065010
Jiang J, Luo Y, Moldgy A, ArandaGonzalvo Y, Bruggeman PJ (2020) Absolute spatially and time-resolved O, O3, and air densities in the effluent of a modulated RF-driven atmospheric pressure plasma jet obtained by molecular beam mass spectrometry. Plasma Process Polym 17(6):1900163
Baiocchi FA, Wetzel RC, Freund RS (1984) Electron-impact ionization and dissociative ionization of the CD3 and CD2 free radicals. Phys Rev Lett 53(8):771–774
Itikawa Y, Ichimura A, Onda K, Sakimoto K, Takayanagi K, Hatano Y, Hayashi M, Nishimura H, Tsurubuchi S (1989) Cross sections for collisions of electrons and photons with oxygen molecules. J Phys Chem Ref Data 18(1):23–42
Fu HB, Hu YJ, Bernstein ER (2006) Generation and detection of alkyl peroxy radicals in a supersonic jet expansion. J Chem Phys 125(1):014310
Meloni G, Zou P, Klippenstein SJ, Ahmed M, Leone SR, Taatjes CA, Osborn DL (2006) Energy-resolved photoionization of alkylperoxy radicals and the stability of their cations. J Am Chem Soc 128(41):13559–13567
Nixon KL, Pires WAD, Neves RFC, Duque HV, Jones DB, Brunger MJ, Lopes MCA (2016) Electron impact ionisation and fragmentation of methanol and ethanol. Int J Mass Spectrom 404:48–59
Zawadzki M (2018) Electron-impact ionization cross section of formic acid. Eur Phys J D 72(1):12
Vacher JR, Jorand F, Blin-Simiand N, Pasquiers S (2008) Partial ionization cross-sections of acetone and 2-butanone. Int J Mass Spectrom 273(3):117–125
Możejko P (2007) Calculations of electron impact ionization cross section for simple biomolecules: formic and acetic acids. Eur Phys J Spec Top 144(1):233–237
Große-Kreul S, Hübner S, Schneider S, Ellerweg D, von Keudell A, Matejčík S, Benedikt J (2015) Mass Spectrometry of Atmospheric Pressure Plasmas. Plasma Sources Sci Technol 24(4):044008
NIST (2010) Standard Reference Database 69: NIST Chemistry WebBook; National Institute of Standards and Technology
Loenders B, Engelmann Y, Bogaerts A (2021) Plasma-catalytic partial oxidation of methane on Pt(111): a microkinetic study on the role of different plasma species. J Phys Chem C 125(5):2966–2983
Eckert Z, Tsolas N, Togai K, Chernukho A, Yetter RA, Adamovich IV (2018) Kinetics of plasma-assisted oxidation of highly diluted hydrocarbon mixtures excited by a repetitive nanosecond pulse discharge. J Phys D Appl Phys 51(37):374002
Golda J, Biskup B, Layes V, Winzer T, Benedikt J (2020) Vacuum ultraviolet spectroscopy of cold atmospheric pressure plasma jets. Plasma Process Polym 17(6):1900216
Greenler RG (1962) Infrared study of the adsorption of methanol and ethanol on aluminum oxide. J Chem Phys 37(9):2094–2100
Korhonen ST, Bañares MA, Fierro JLG, Krause AOI (2007) Adsorption of methanol as a probe for surface characteristics of zirconia-, alumina-, and zirconia/alumina-supported chromia catalysts. Catal Today 126(1–2):235–247
Schauermann S, Hoffmann J, Johánek V, Hartmann J, Libuda J (2002) Adsorption, decomposition and oxidation of methanol on alumina supported palladium particles. Phys Chem Chem Phys 4(15):3909–3918
Plessis P, Marmet P, Dutil R (1983) Ionisation and appearance potentials of ch4 by electron impact. J Phys B At Mol Phys 16(7):1283–1294
Nozaki T, Muto N, Kado S, Okazaki K (2004) Dissociation of vibrationally excited methane on Ni catalyst: Part 1. Application to methane steam reforming. Catal Today 89:57–65
Nozière B, Hanson DR (2017) Speciated monitoring of gas-phase organic peroxy radicals by chemical ionization mass spectrometry: cross-reactions between CH3O2, CH3(CO)O2, (CH3)3CO2, and c-C6H11O2. J Phys Chem A 121(44):8453–8464
Jiang J, Kondeti SKVS, Nayak G, Bruggeman PJ (2021) Experimental and modeling studies of the plasma chemistry in a humid ar radiofrequency atmospheric pressure plasma jet. J. Phys. D. Appl. Phys. 55(22):225206
Agarwal N, Freakley SJ, McVicker RU, Althahban SM, Dimitratos N, He Q, Morgan DJ, Jenkins RL, Willock DJ, Taylor SH et al (2017) Aqueous Au-Pd colloids catalyze selective CH4 oxidation to CH3OH with O2 under mild conditions. Science 358(6360):223–227
Benedikt J, Hecimovic A, Ellerweg D, von Keudell A (2012) Quadrupole mass spectrometry of reactive plasmas. J Phys D Appl Phys 45(40):403001
Acknowledgements
This material is based upon work supported by the National Science Foundation (CBET 1703439). The work has significantly benefited from methods and techniques developed in the framework of work supported by the US Department of Energy, Office of Science, Office of Fusion Energy Sciences General Plasma Science program under Award Number DE-SC0020232 and DE-SC0001939.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Jiang, J., Bruggeman, P.J. Investigation of the Mechanisms Underpinning Plasma-Catalyst Interaction for the Conversion of Methane to Oxygenates. Plasma Chem Plasma Process 42, 689–707 (2022). https://doi.org/10.1007/s11090-022-10251-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11090-022-10251-5