
Abstract
Glutamate dehydrogenase (GDH) catalyzes the reversible deamination of l-glutamate to α-ketoglutarate and ammonia. In mammals, GDH contributes to important processes such as amino acid and carbohydrate metabolism, energy production, ammonia management, neurotransmitter recycling and insulin secretion. In humans, two isoforms of GDH are found, namely hGDH1 and hGDH2, with the former being ubiquitously expressed and the latter found mainly in brain, testis and kidney. These two iso-enzymes display highly divergent allosteric properties, especially concerning their basal activity, ADP activation and GTP inhibition. On the other hand, both enzymes are thought to predominantly localize in the mitochondrial matrix, even though alternative localizations have been proposed. To further study the subcellular localization of the two human iso-enzymes, we created HEK293 cell lines stably over-expressing hGDH1 and hGDH2. In these cell lines, immunofluorescence and enzymatic analyses verified the overexpression of both hGDH1 and hGDH2 iso-enzymes, whereas subcellular fractionation followed by immunoblotting showed their predominantly mitochondrial localization. Given that previous studies have only indirectly compared the subcellular localization of the two iso-enzymes, we co-expressed them tagged with different fluorescent dyes (green and red fluorescent protein for hGDH1 and hGDH2, respectively) and found them to co-localize. Despite the wealth of information related to the functional properties of hGDH1 and hGDH2 and the availability of the hGDH1 structure, there is still an ongoing debate concerning their metabolic role and their involvement in disease processes. Data on the localization of hGDHs, as the ones presented here, could contribute to better understanding of the function of these important human enzymes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introductory Remarks
Glutamate dehydrogenase (GDH) catalyzes the reversible deamination of l-glutamate to α-ketoglutarate and ammonia using NAD+ and NADP+ as cofactors [1]. As shown by its ubiquitous existence in almost all living species, GDH is a key enzyme for metabolism and therefore considered to be essential for cell survival [2]. In mammals, inside mitochondria, its main metabolic function is to fuel aerobic metabolism through the Krebs cycle for energy production, interconnecting in parallel amino acid and carbohydrate metabolism [3, 4]. In addition, it contributes to other important tissue and non-tissue specific processes, including autophagy, neurotransmitter recycling, ammonia management and insulin secretion [5,6,7,8,9,10,11]. To perform these processes, humans possess two GDH isoforms, hGDH1 and hGDH2, respectively, with the latter emerging from the former through retroposition in the common ancestor of modern primates more than 23 million years ago [12,13,14,15,16]. Being endowed by favorable allosteric regulatory mutations that diversified its function from the parental hGDH1 gene, hGDH2 persisted during evolution and adapted to the changing needs of the primate nervous and other systems [13, 17, 18]. Among the properties that distinguish hGDH2 from its parental hGDH1 enzyme are low basal activity that is nonetheless highly amenable to ADP and l-leucine activation, increased GTP resistance and decreased heat stability [19, 20]. In this work, we will expand further on the localization of the two human iso-enzymes aiming to probe their role in cellular metabolism and human disease processes.
The hGDH Reaction and its Regulation
Despite decades of extensive research, the direction of the highly-regulated GDH reaction in mammals (Fig. 1), including humans, is a matter of active debate [21,22,23]. Even though this reaction is reversible, and its rate influenced by multiple metabolic mediators, there seems to be a preference in humans for the GDH reaction to proceed in the direction of α-ketoglutarate production [24, 25]. The main argument for this is that the high Km of the enzyme for ammonia (in the mmolar range) corresponds to ammonia levels that would otherwise be toxic to mammalian cells [10, 26, 27]. As a counterargument, even though under physiological conditions ammonia levels in human tissues are low, they can reach mmolar levels during hypeammonemia states [28]. In addition, inside mitochondria, the ammonia levels can locally increase to levels that would allow the GDH reaction to operate in the reductive amination reaction [22, 29]. Finally, as ammonia accumulates in the tumor micro-environment, due to poor blood supply and high production rates by glutamine catabolism, it is recycled back via GDH to support amino acid synthesis for the rapidly proliferating breast cancer cells, accelerating tumor growth [30].

Reversible deamination of l-glutamate to α-ketoglutarate and ammonia by (A) hGDH1 and (B) hGDH2. NAD+ and NADP+ act as cofactors of the reaction. In the absence of activators, basal activity of hGDH1 and hGDH2 are 3–8% and 30–35% of their maximal activity, respectively. hGDH1 is allosterically activated by ADP and l-leucine and inhibited by GTP, whereas hGDH2 is insensitive to GTP inhibition but amenable to full activation by ADP and l-leucine
Irrespectively of the preferred direction of the reaction, it seems that the enzyme displays high in vivo activity with the flux through its pathway being constantly regulated [31]. This regulation is of special importance for the hGDH2 enzyme, which, in contrast to hGDH1 that displays a significant (35–40%) basal activity in the absence of allosteric effectors, has a negligible (3–5%) activity in the absence of ADP and/or l-leucine [32, 33]. This low basal activity of hGDH2 is though amenable to full activation by ADP and/or l-leucine, permitting the enzyme to act as a molecular switch in metabolic processes accompanied by significant fluctuations of these two metabolic intermediates [20]. This remarkable property was conferred to the hGDH2 enzyme by the Arg443Ser evolutionary amino-acid substitution [17]. Concerning the inhibitory regulation of these human iso-enzymes, hGDH1 is very sensitive to inhibition by GTP, and this inhibition displays a significantly co-operative behavior (Fig. 1) [34]. This property is thought to be of importance for not overloading the Krebs cycle when the cell has been saturated in energy, as GTP produced from the function of the Krebs cycle just downstream of the point of entrance of α-ketoglutarate produced by the hGDH1 reaction diffuses back to inhibit hGDH1 (negative feedback regulation) [5]. In contrast, hGDH2 shows a marked resistance to GTP inhibition, owing to the Gly456Ala substitution (Fig. 1) [18]. We should note at this point that even though ADP, l-leucine and GTP are the main allosteric regulators of the two human GDH iso-enzymes, there are numerous metabolic mediators and exogenous compounds that exert an effect on hGDH1 and hGDH2, most of the time with differing potencies [20].
Structure–Function Relationships of hGDHs
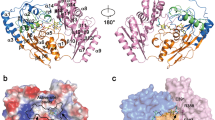
Despite their significantly diverging functional properties, hGDH1 and hGDH2 show very high amino-acid sequence homology, differing in their mature form in only 15 of their 505 amino acids (97% sequence homology), with these amino acid substitutions accumulating during evolution of the primate lineage [5, 14]. Considering their functional divergence, it is expected that these few differences in the primary structure of the two iso-enzymes translate to significant alterations in their tertiary conformation. Indeed, crystallographic studies in the bovine GDH, as well as in hGDH1 [35,36,37,38], have indicated that the Arg443Ser and Gly456Ala evolutionary amino acid substitutions (crucial for the molecular evolution of hGDH2) form part of critical regulatory regions of the mammalian enzyme, termed antenna and pivot helix, respectively (Fig. 2) [13, 39]. The antenna protrudes from the main core of the mammalian enzyme, formed by the NAD+ binding domain and the glutamate binding domain surrounding the active cleft, whereas the pivot helix forms part of the NAD+ binding domain (Fig. 2). We have previously shown that of great importance for the allosteric regulation of the enzyme is the interaction of Arg443 in the descending α-helix of the antenna of one subunit with Ser409 on the ascending α-helix on a neighboring subunit [40].

Structure of hGDH1 as (A) hexamer, (B) trimer, (C) dimer and (D) monomer. Each subunit (monomer) of the hGDH1 hexamer (composed essentially from a dimer of trimers) is represented in different color. In sections A, B and C, the red cycle indicates the interaction of the antennas of adjacent subunits. In section D (monomer of hGDH1), the antenna, pivot helix, active cleft, and the NAD+ and glutamate (GLU) binding domains are pointed out. Also in section D, the Arg443 and Gly456 residues, two of the amino acids differing between hGDH1 and hGDH2 and sites of the two most important evolutionary amino-acid substitutions (Arg443Ser and Gly456Ala, located in the antenna and pivot helix, respectively) are shown in red. The cartoon diagram was created from the coordinates of the hGDH1 hexamer (apo-form, PDB code 1 LIF) using the PyMOL Molecular Graphics System, Version 1.4, Schrodinger, LLC
In a quaternary level, the mature hGDH1 enzyme forms hexamers of identical subunits, that essentially consist of a dimer of trimers with the subunits of each trimer interacting at their antennas (Fig. 2) [35]. It is also possible that the hexamers form homopolymers, depending on the prevailing conditions [41]. As a crystallographic structure of hGDH2 has not been published yet, we cannot be certain which are the structural consequences of the evolutionary amino acid substitutions that gave hGDH2 its unique functional properties. Also, it is currently unknown if, in the tissues that both hGDH enzymes are expressed, they form hetero-hexamers, which are expected to differ markedly in their functional properties from the homo-hexamers that have been extensively studied thus far.
Expression of hGDH1 and hGDH2 in Human Tissues
Despite current lack of knowledge on the in vivo existence of hGDH1/hGDH2 hetero-hexamers, there has been considerable progress in characterizing the distribution of these iso-enzymes in human tissues [5, 42,43,44,45]. As already mentioned, hGDH1 is a housekeeping enzyme, being present in virtually all human tissues, with the highest levels found in the liver. On the other hand, hGDH2 has a significantly different pattern of expression in human tissues, as shown by immunocytochemistry and western blot studies, being expressed in brain (more intensely in astrocytes than in neurons), testis (Sertoli cells), steroid producing tissues and kidney [5, 42, 43].
Concerning the nervous system, where hGDH2, but not hGDH1, has been found in neurons, it is of special interest that both human iso-enzymes are present in astrocytes given the special role these cells have for nervous system function [44,45,46]. This role includes providing neurons with nutrients or compounds necessary for optimal function and removing excess glutamate from the synaptic cleft to be recycled back to neurons through the consecutive action of astrocytic glutamine synthetase and neuronal phosphate-activated glutaminase [5]. It is possible that the coexistence of both iso-enzymes in astrocytes enables use of hGDH1 for metabolic (housekeeping) purposes, while liberating hGDH2 for use in glutamatergic neurotransmission. The functional properties of hGDH2 (low basal activity amenable to ADP activation and GTP resistance) seem to best serve a role in glutamatergic neurotransmission. Specifically, given that energy-consuming glutamatergic neurotransmission is associated with decrease in ADP levels, this will activate hGDH2, which will continue to function despite Krebs cycle activation replenishing the energy stores of the astrocyte and thus increasing GTP levels [3]. This has recently been shown in transgenic mice overexpressing hGDH2, where under glucose deprived conditions, astrocytes were able to use alternative energy sources [47] .
Subcellular Localization of hGDHs
As the tissue distribution of the hGDH1 and hGDH2 iso-enzymes provides clues to their biological role, the same holds true for their subcellular localization. Extensive studies on the mammalian GDHs, have shown that the enzyme is predominantly localized in the mitochondrial matrix [48,49,50], even though membrane-bound [51,52,53] and nuclear forms have been described [54,55,56].
Despite the numerous studies on the mammalian non-human GDH, hGDH1 and hGDH2 subcellular localization has been less well studied. Concerning the human brain, it has been recently shown using immunohistochemistry that there is hGDH1 and hGDH2 immunostaining in the nuclear membrane of glial and neuronal cells [46, 57]. Using the GFP system, we have in the past few years expressed either hGDH1 or hGDH2 tagged with this fluorescent marker and compared its subcellular distribution with that of appropriate cellular markers. In this way, we have been able to show that both hGDH1 and hGDH2 localize in the mitochondria and the endoplasmic reticulum [32, 58]. Using the same technique, Rosso et al. [59] have concluded that while hGDH2 is specifically targeted to mitochondria, hGDH1 localizes both in mitochondria and the cytoplasm. This enhanced mitochondrial targeting of hGDH2 compared to hGDH1 was attributed to the Glu7Lys evolutionary amino acid substitution in the long cleavable presequence [59]. The importance of this Glu7Lys substitution, even though this time as part of a triple substitution, as well as the slightly more efficient targeting of hGDH2 into mitochondria, were verified in our recent study using isolated yeast mitochondria and human cell lines [60]. In this work, we also showed the importance of the two α-helices of the 53-amino acid long presequence for the highly efficient import into mitochondria [60].
The studies mentioned above rely on the indirect comparison of tagged hGDH1 and hGDH2 distribution through the use of another subcellular marker and as such they do not perform a direct comparison of the two iso-enzymes. To further characterize the localization of the two human iso-enzymes, we studied the endogenous hGDH localization in HEK293 (Human Embryonic Kidney) cells using anti-hGDH1 and anti-hGDH2 specific antibodies, and then overexpressed hGDH1 and hGDH2 in the same cell line (as well as in HeLa cells; human cervical adenocarcinoma), to verify our results with the endogenous proteins. In addition, to perform a direct comparison of the localization of the two human iso-enzymes, we have concurrently tagged hGDH1 and hGDH2 with different fluorescent dyes (GFP and RFP, respectively) and co-expressed them in mammalian cell lines (HEK293 and HeLa cells). Results showed that both human iso-enzymes colocalize in cellular structures corresponding to mitochondria.
Methods
Creation of HEK293 Cell Lines Stably Overexpressing hGDH1 and hGDH2
HEK293 cells stably over-expressing hGDH1 or hGDH2 were created according to the vector (pcDNA™3.1) manufacturer’s protocol (Invitrogen, CA, USA). The construct used for this purpose was pcDNA™3.1 (Invitrogen, CA, USA) containing either GLUD1 or GLUD2 cDNA. The cell cultures were maintained in 100 mm dishes treated with Dulbecco’s Modified Eagles Medium enriched with 10% fetal bovine serum at 37 °C in 5% CO2. We transfected HEK293 cells using Lipofectamine 2000 and 24 h post transfection, a medium containing selective antibiotic (Geneticin, Life Technologies) in final concentration of 600 µg/ml was introduced to the cell culture. The cell culture was incubated for another 2–3 weeks until single colonies were visible, while fresh medium with geneticin was introduced every 48 h. Single colonies were transferred in 24 well plates for further expansion and positive colonies were selected based on Western blot, immunocytochemistry and enzymatic activity analyses. Two colonies (col2 and col6, respectively) were selected from both the hGDH1 and hGDH2 overexpressing cells for use in the experiments described below. Each of these experiments was performed at least in triplicate (≥ three biological replicates used).
Western Blot Analyses
HEK293 cells were harvested from cultures and lysed using ice-cold lysis buffer (10 mM Tris–HCl, 0.5M NaCl, 0.5 mM EDTA, pH 7.4, 1% triton, protease inhibitor mixture). A Bradford kit (Bio-Rad Laboratories, Hercules, CA) was used for total protein quantification. Whole cell lysates were loaded on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) and transferred on nitrocellulose membrane at 350 mA for 1 h. The nitrocellulose membranes were incubated overnight with anti-GDH1 (1:5000) (Aviva Systems Biology, CA, USA) and anti-hGDH2 (1:5000) [5] primary specific polyclonal antibodies. Next, the samples were incubated with horseradish peroxidase-coupled secondary antibodies (goat anti-rabbit, 1:1000) and visualization of the proteins was done using ECL kit (Thermo Fisher scientific, Life Technologies).
Subcellular Fractionation Analyses
For the cell compartmentation assay, cells were harvested from 100 mm dishes and centrifuged at 850 g for 2 min. After the cell pellet was obtained, Mitochondria Isolation Kit for cultured cells (Thermo Fisher Scientific, Life Technologies) was used according to the reagent-based method and following the manufacturer’s instructions. In brief, cells were resuspended in Reagent A buffer, supplemented with protease inhibitor, and incubated for 2 min on ice. Reagent B was then added, and samples were vortexed and incubated on ice for 5 min, followed by centrifugation at 700×g for 10 min at 4 °C with Reagent C. The resulting supernatant was collected and subjected to centrifugation at 12,000×g for 15 min at 4 °C, to pellet mitochondria [61]. Total protein quantification of the samples was performed also using the Bradford kit (Bio-Rad Laboratories, Hercules, CA) and samples were loaded on 12% SDS–PAGE gel for electrophoresis. An antibody against MnSOD (also known as SOD2), which is an integral mitochondrial matrix protein [62, 63], was used as a mitochondrial marker evaluating the proper mitochondrial fractions preparation.
Enzymatic Activity Measurements
Whole cell lysates from HEK293 cells, obtained as described above, were used for enzymatic activity measurements. Total protein quantification of the samples was performed using Bradford kit (Bio-Rad Laboratories, Hercules, CA). Samples was stored at − 80 °C and handling for the experiments was always performed on ice. Enzymatic activity measurements were performed spectrophotometrically at 340 nm in the reductive amination direction measuring the concentration reduction of NADPH because of the reaction. The reaction mixture was 1 ml of final volume containing 50 mM triethanolamine-HCl buffer pH 8, 100 mM ammonium acetate, 100 µM NADPH and 2.6 mM EDTA. A final concentration of 10 mM α-ketoglutarate and 1 mM ADP was also added simulating maximal activating conditions for both enzymes. Specific activity was calculated in µmol/mg total protein of the samples/min.
Immunocytochemistry
Immunocytochemistry was performed in HEK293 cells cultured in 24-well plates with bottom coverslips. When cells reached ~ 70% confluency, they were fixed with 5% paraformaldehyde (PFA) at room temperature (RT) for 15 min. After fixation, cells were washed twice with phosphate-buffered saline (PBS) (pH 7.5). Blocking was performed using 0.5% triton and 2% normal goat serum (NGS) in PBS for 1 h at RT. Cells were incubated over night at 4 °C with primary antibodies against hGDH1 or hGDH2 diluted 1:5000 in blocking solution. Cells were washed in PBS and incubated with fluorescence-conjugated secondary antibodies (goat anti-rabbit, 1:200) and TO-PRO 3 for nuclear staining (1:1000, Thermo Fisher Scientific, Life Technologies) in PBS. Coverslips were mounted in Dako (Agilent Technologies) and samples were visualized using laser scanning SP2 confocal fluorescence microscopy (Leica TCS-NT Laser Scanning). Image acquiring was performed using lenses × 10, × 40 or × 63 with oil objective. The results were processed in Adobe Photoshop CS6.
Construct Creation, Transfection and Visualization of Co-expressed Human Iso-enzymes
The constructs used to study the co-localization of the two iso-enzymes, hGDH1 and hGDH2, were the hGDH1/green fluorescent protein (GFP) and the hGDH2/red fluorescence protein (RFP), respectively. To obtain the hGDH1/GFP construct, the cDNA sequence of the GLUD1 gene was firstly amplified via PCR and fused to the GFP sequence, as previously described [58]. The hGDH2/RFP construct was designed to chimerically express hGDH2 conjugated with RFP at its C-terminus. In more detail, the plasmid vector used was pmCherry-N1 (Clontech Laboratories, Inc., CA, USA) and the entire GLUD2 sequence was fused into the vector. The forward primer was designed to create an EcoRI restriction site (5′-CCGGAATTCATGTACCGCTACCTGGCC-3′). The reverse primer (5′-ATTCGCGGATCCCCCTTTGTGAAGGTCACACCA-3′) creates a BamHI restriction site and converts the stop codon TAG to AAG. The amplified product was then digested using EcoRI and BamHI restriction enzymes and introduced in to the pmCherry vector. Ligation was performed overnight at 16 °C using the T4 DNA ligase kit (New England biolabs), according to the manufacturer’s instructions. Bi-directional Sanger sequencing was performed for verification of the proper orientation and sequence of the constructs. Moreover, for fluorescent labeling of mitochondria, the mammalian expression vector pDsRed2-Mito was used (BD Biosciences Clontech, Palo Alto, CA, USA).
The two constructs, hGDH1/GFP and hGDH2/RFP, respectively, were co-transfected, in final concentration of 3 µg/well, in HEK293 and HeLa cell lines using the transfection reagent Lipofectamine 2000 (Invitrogen, CA, USA). Also, 2 μg of plasmid pDsRed2-Mito were co-transfected with 2 μg of hGDH1/GFP in HEK293 cells, to label the mitochondrial compartment with red fluorescence. The cells were maintained in six well plates with bottom coverslip and treated with Dulbecco’s Modified Eagles Medium enriched with 10% fetal bovine serum at 37 °C in 5% CO2 conditions. Transfected cells were incubated for 48 h at 37 °C, and when the expression of the enzymes was maximized, cells were fixed with 5% PFA in slides and visualized using laser scanning SP2 confocal fluorescence microscopy (Leica TCS-NT laser scanning). The lenses used for image acquiring were × 10, × 40 or × 63 with oil objective. The results were exported as TIFF files and processed in Adobe Photoshop CS6.
Statistical Analyses
For the statistical analyses, one-way ANOVA was performed using Graphpad prism 6.
Results
Overexpression of Both hGDH1 and hGDH2 Several Times Over the Endogenous Proteins
To verify the stable transformation of the HEK293 cells and study the expression levels of hGDH1 and hGDH2 in these cell lines, we initially performed western blot analyses in whole cell lysates from two different colonies for each iso-enzyme (Fig. 3). For hGDH2, comparing the signal intensity between stable cell lines overexpressing hGDH2 with the signal of the native hGDH2 enzyme showed that the exogenous protein was highly over-expressed (Fig. 3). For hGDH1, under the conditions the experiment was performed, no signal for the endogenous protein was detected.

Overexpression of the hGDH1 and hGDH2 in stably transfected HEK293 cell lines. Shown are western blot analyses of whole cell lysates from wild-type (WT) and cell lines stably overexpressing hGDH1 and hGDH2 (two different colonies for each enzyme). In the upper panel the anti-hGDH1 antibody was used, while in the lower panel the membrane was blotted for hGDH2 with the specific anti-hGDH2 antibody. Equal amounts of total protein were loaded in each of the first five lanes. In the last two lanes, purified hGDH1 and hGDH2 were used as a control
To functionally quantify the overexpression of hGDHs in stably transfected HEK 293 cells, we measured the enzymatic specific activity in whole cell lysates. As shown in Fig. 4, the hGDH specific activity in the stably over-expressing cell lines was several times over the endogenous hGDH activity. The highest activity was found in the hGDH2 overexpressing col6 cell line (Fig. 4), an activity accounting for an over-expression level of 48 times over the endogenous GDH activity.

GDH specific activity (µmol/mg protein/min) in whole HEK293 cell lysates. The samples used were wild-type (WT) HEK293 cells, stable cell lines overexpressing hGDH1 (hGDH1 col2 and col6, respectively) and stable cell lines overexpressing hGDH2 (hGDH2 col2 and col6, respectively). The GDH specific activity was significantly enhanced in stable cell lines compared to the WT cells (one-way ANOVA, p < 0.0001). The highest expression levels were found in the hGDH2 col6 cell line (0.03867 ± 0.003 µmol/mg protein/min), an activity 48 times more than the endogenous GDH activity found in the WT HEK293 cells (0.0008047 ± 0.0001 µmol/mg protein/min). The activities recorded for the rest of the stable cell lines were 0.02290 ± 0.0007, 0.01329 ± 0.0002, and 0.01683 ± 0.0015 µmol/mg protein/min for the hGDH1 col2, hGDH1 col6 and hGDH2 col2 cell lines, respectively (28.5, 16.5 and 21 times the endogenous GDH activity). ***p<0.0001 compared to the WT HEK293 cell GDH specific activity
Mitochondrial Localization of Both Overexpressed hGDH1 and hGDH2
Next, we studied the HEK293 stable cell lines by immunocytochemistry using specific anti-hGDH1 and anti-hGDH2 antibodies. In both cases, we were able to verify the overexpression of the exogenous iso-enzyme over the endogenous, as well as its presence in coarse cytoplasmic structures reminiscent of mitochondria (Figs. 5 and 6). Interestingly, in both cases, a similar immunofluorescence pattern, even though of considerably less intensity, was found in the wild-type HEK293 cells, indicative of a comparable distribution of the endogenous hGDH1/2 iso-enzyme (Figs. 5 and 6).

Exogenously introduced hGDH1 localizes in coarse cellular structures reminiscent of mitochondria. Immunocytochemistry was performed using the anti-hGDH1 specific antibody in WT HEK293 and two colonies of HEK293 cell lines stably overexpressing hGDH1 (hGDH1 col2 and col6, respectively). TO-PRO3 was used for nuclei staining and is depicted in blue. The same laser intensity was applied in all cases during confocal microscopy

Exogenously introduced hGDH2 localizes in coarse cellular structures reminiscent of mitochondria. Immunocytochemistry was performed using the anti-hGDH2 specific antibody in WT HEK293 cells and two colonies of cell lines stably overexpressing hGDH2 (hGDH2 col2 and col6, respectively). TOPRO3 was used for nuclei staining and is depicted in blue. The same laser intensity was applied in all cases during confocal microscopy
To confirm the mitochondrial localization of the enzyme, we performed subcellular fractionation experiments followed by western blot analyses. In all four stable cell line clones studied (two for each iso-enzyme), we detected the presence of hGDH1 and hGDH2 in the mitochondrial fraction (Fig. 7). For the cytosolic fraction, there was absence of hGDH1, and only small amounts of hGDH2 (Fig. 7). This could represent alternative localization of the overexpressed hGDH2 iso-enzyme or be due to small amount of cross-contamination, as shown by experiments using MnSOD as a mitochondrial marker (Fig. 8).

Mitochondrial localization of over-expressed hGDH1 and hGDH2. Western blot analyses after subcellular fragmentation (into mitochondrial and cytosolic fractions) of cell lysates of WT and stable cell line colonies (col2 for both hGDH1 and hGDH2 stable cell line, respectively). In the upper panel the anti-GDH1 antibody was used, while the lower membrane was blotted for hGDH2. These results indicate that the overexpressed enzyme is driven into mitochondria, given that under these conditions the endogenous iso-enzyme(s) are not detected. The small cytosolic band in hGDH2 over-expressing cell lines could represent alternative localization of the enzyme or be the result of negligible contamination due to excess of the enzyme (see below). The last two lanes (controls; hGDH1 C+ and hGDH2 C+) were loaded with purified recombinant hGDH1 and hGDH2, respectively

Co-localisation of hGDH2 with the mitochondrial marker MnSOD. Western blot analyses after subcellular fragmentation (into mitochondrial-Mito and cytosolic-Cyto fractions) of cell lysates of WT and stable cell line colonies (col2 for both hGDH1 and hGDH2 stable cell line). The anti-hGDH2 antibody was used, while reprobing was performed using an anti-MnSOD antibody. Results show that hGDH2 was predominantly localized in the mitochondrial fraction, with only a small amount present in the cytosolic fraction, a pattern comparable to that of MnSOD. The first two lanes (controls; hGDH1 C+ and hGDH2 C+) were loaded with purified recombinant hGDH1 and hGDH2
Co-expressed hGDH1/GFP and hGDH2/RFP Co-localize in Human Cell Lines
When we co-expressed hGDH1/GFP and hGDH2/RFP constructs in HEK293 cells, we observed that both displayed a similar punctate distribution, reminiscent of mitochondria (Fig. 9). The immunofluorescence of both chimeric proteins was overlapping (Fig. 9). The same results were obtained when we co-transfected hGDH1/GFP and hGDH2/RFP in HeLa cells (Fig. 10). The co-transfection experiment using hGDH1/GFP and pDsRed2-Mito in HEK293 cells, produced a yellow fluorescent signal on dual-color imaging, corroborating the hGDH1/GFP mitochondrial localization (Fig. 11).

Colocalization of hGDH1/GFP and hGDH2/RFP in HEK293 cells. Here we performed confocal imaging of the co-transfection of hGDH1/GFP and hGDH2/RFP constructs in HEK293 cells. The signal originating from hGDH1/GFP is shown in A, D, G and J, whereas in B, E, H and K the signal from hGDH2/RFP is detected. Finally, in C, F,I and L, the merged signal of the two fluorescent chimeric proteins is shown. Both chimeric iso-enzymes co-localize in structures reminiscent of mitochondria

Colocalization of hGDH1/GFP and hGDH2/RFP in HeLa cells. Here we performed co-transfection of the hGDH1/GFP and hGDH2/RFP constructs in HeLa cells followed by confocal imaging. A, D Signal originating from hGDH1/GFP. B, E Signal from hGDH2/RFP. C, F The merged signal of the two fluorescent chimeric proteins. As in similar experiments performed in HEK293 cells, both chimeric iso-enzymes co-localize in structures probably representing of mitochondria

Colocalization of hGDH1/GFP and pDsRed2-Mito in HEK293 cells. Here we performed co-transfection of the hGDH1/GFP (green) and pDsRed2-Mito (red, a mitochondrial marker) constructs in HEK293 cells followed by confocal imaging. A, D, G Signal originating from hGDH1/GFP. B, E, H Signal from pDsRed2-Mito. C, F, I The merged signal of the two fluorescent chimeric proteins. Both chimeric proteins co-localize in mitochondria
Discussion
The results of the studies presented above point towards a similar behavior of both iso-enzymes when over-expressed in human cell lines (either separately, or, for the first time, concurrently) as far as their localization is concerned, consistent with predominantly mitochondrial localization. This is of importance, given that these iso-enzymes co-exist in several tissues and cell types, where they are thought to play different roles due to their different regulatory properties [3, 5, 43]. Thus, it is probable that during primate evolution the new hGDH2 iso-enzyme adopted a novel role mainly due to its unique enzymatic properties rather than by being differentially targeted inside cells. This is consistent with the experiments we have recently performed in the yeast S. cerevisiae as well as in human cell lines to elucidate the properties of the targeting pre-sequence [60].
One could argue that the current results could be due to the over-expression of the two hGDH iso-enzymes. However, we got similar results in western blot studies using two colonies of hGDH1 overexpressing cells (col2 and col6, respectively) and two colonies of hGDH2 overexpressing cells (col2 and col6, respectively), with each of the four colonies showing different levels of overexpression. Also, in cells with transient hGDH1/GFP and hGDH2/RFP overexpression, we observed similar results in immunofluorescence experiments across several levels of overexpression. Finally, we noted similar immunofluorescence patterns using the anti-hGDH1 and anti-hGDH2 antibodies in wild-type HEK293 cells and hGDH1 and hGDH2 overexpressing cells, even though, as expected, the signal was less intense in the WT cells (Figs. 5, 6). These results are compatible to similar experiments performed in different cell systems using different methods [32, 58,59,60]. Also, there have been several reports in the literature of models of hGDH overexpression that were used to study the metabolic role of mammalian GDH without being compromised by the overexpression of the exogenous enzyme. For example, Carobbio et al. overexpressed GDH using a retrovirus expression system in rat and mouse pancreatic islets, as well as in a beta-cell line, for studying the glucose-induced insulin secretion profile [64]. Also, Tanizawa et al. [65, 66] used both the pcDNA3.1 vector used here and a retroviral vector to overexpress hGDH1 in COS-7 and MIN6 cells, respectively, again to study insulin secretion.
There are, also, several papers focusing on over-expression of mammalian GDH in neural tissue. These studies used transgenic mice modified to overexpress mouse mGDH1 or human hGDH2 [67, 68]. One of the pioneers in this field of research is the group of Eli Michaelis, to whom this issue is dedicated [67, 69,70,71,72,73]. Based on an over-expression model of mouse Glud1 enzyme, they were able to show age-associated changes due to excess glutamate release in the hippocampus and other areas, a phenotype similar to progressive neurodegeneration (see below) [67, 73]. Establishing models of stable overexpression of hGDH1 and hGDH2, as done here, are needed to study these important enzymes, given their significance for human disorders, as described below.
hGDH and Human Disease
Given the importance of hGDHs for cell metabolism and several tissue-specific functions, such as insulin secretion and neurotransmission, it is not unexpected that several human disorders, especially relating to carbohydrate metabolism and nervous system function, are associated to hGDH1 and hGDH1 deregulation [57, 74, 75]. Inversely, several studies have focused on the pathogenetic role of hGDHs in these human diseases aiming to develop GDH modulating strategies as a therapeutic means.
The first human disease found to be associated with mutations in a hGDH gene was the hyperinsulinism–hyperammonemia (HI/HA) syndrome, a rare autosomal dominant disorder, characterized by hypoglycemic episodes provoked by fasting or high-protein meals [76]. In a breakthrough research effort, HI/HA was found to be caused by activating mutations in the GLUD1 gene, encoding for hGDH1 [77]. Since then, many more disease-causing mutations have been identified in exons 6, 7, 11 and 12 in the GLUD1 gene [78,79,80,81,82,83,84,85,86,87,88,89,90]. Most of these missense variants affect the GTP-binding site of the enzyme, abrogating its sensitivity to GTP inhibition, with even the mutations that are remote from the GTP binding site indirectly interfering with the allosteric regulation by GTP. The resulting hyperactive hGDH1 enzyme produces increased ATP levels (increased ATP/ADP ratio) in pancreatic β-cells. Since the ATP/ADP ratio regulates insulin secretion from β-cells, hGDH1 hyperactivity leads to excess insulin release that is inappropriate to the serum glucose levels [91]. It is also possible that these activating mutations in hGDH1 lead to impaired glucagon release from pancreatic α-cells and possibly increased glucagon-like peptide 1 secretion from L-cells [92, 93]. The evidence above renders hGDHs an attractive target for diabetes treatment [94, 95].
It is interesting that there is another form of congenital hyperinsulinism caused by deregulation of hGDH1 function brought about by deficiency of the short-chain 3-hydroxy-acyl-CoA dehydrogenase enzyme (SCHAD, encoded by the HADH1 gene) [96, 97]. It has been shown that SCHAD strongly interacts with hGDH1 and inhibits its function [98], possibly in the context of a multiprotein metabolic complex (metabolon) [99]. Interestingly, hGDH1 forms part of other metabolons, as in the case of the BCAT metabolizing enzymes [4, 9].
In cases of the HI/HA syndrome caused by activating mutations of hGDH1, it has been shown that these patients develop neurological manifestations that extend beyond these expected by the hypoglycemic episodes and the coexisting hyperammonemia [100, 101]. Specifically, these patients develop psychomotor retardation, behavioral disorders and a high rate of epilepsy unrelated to hypoglycemia or hyperammonemia [85, 100, 102, 103]. It is possible that the hyperactivity of hGDH1 in brain can lead to depletion of glutamate and consequently depletion of GABA, which is synthesized in neurons by the action of glutamate decarboxylase on glutamate. Reduction of GABA, which is the main inhibitory neurotransmitter in the central nervous system, could lead to epilepsy and dystonia [104,105,106]. Alternatively, it is possible that the high ammonia levels in the context of the HI/HA syndrome could shift the balance of the hGDH1 reaction towards glutamate formation, creating excess glutamate locally that could create epileptic predisposition and excitotoxicity, leading to mental retardation. In any case, this syndrome adds evidence for the existing evidence for a role of hGDH1 deregulation in epilepsy [107, 108], paving the way for use of pharmacologic manipulation of the enzyme as an antiepileptic strategy [109].
There are also reports for a possible role of mammalian GDH in attenuating glutamate induced injury following brain ischemia. In the mouse model of Michaelis group with mouse Glud1 overexpression in neurons, the transgenic mice showed increased tolerance to brain ischemia, produced by middle cerebral artery occlusion, an effect attributed to glutamate preconditioning [110]. In this context, it is possible that hGDH modulators could serve as neuroprotective agents in brain ischemia [111]. For example, it has been shown that in a mouse model of middle cerebral artery occlusion, GDH activators provided potent neuroprotection by increasing α-ketoglutarate levels and, through the TCA cycle, replenishing ATP stores [112]. This could indicate that mouse Glud1 (the orthologous enzyme of hGDH1) mediated glutamate oxidation serves as an energy source in ischemic or other energy or nutrient deprived conditions [113].
Beyond the effects of hGDH1 and mouse Glud1 hyperactivity in the nervous system described above, there is ample evidence that hGDH1/2 deregulation could affect neurodegenerative processes. A recent study from our group [114] showed that a mutation in the GLUD2 gene (encoding for hGDH2) accelerates Parkinson’s disease onset by 6–13 years in men. This GLUD2 variant (c.T1492G) leads to the Ser445Ala change in hGDH2 and causes a gain of function by increasing the enzyme’s basal activity [114]. We can only speculate about the consequences on human brain of the hGDH2Ser445Ala substitution, but it is likely that increased glutamate oxidation could accelerate an ongoing degenerative process [114]. In other words, it could be that glutamate needs to retain appropriate levels in brain tissue, and both glutamate excess and glutamate deficiency could be harmful. On the other hand, it is possible that hGDH2 hyperactivity leads to excess synaptic glutamate, as shown for mouse Glud1 overexpression in the Michaelis transgenic model (see below). Regardless of that and even though the role of hGDH2 in the pathophysiology of Parkinson’s disease remains to be determined, these findings build upon existing data of an important contribution of hGDH1/2 deregulation on two other neurodegenerative disorders, namely olivopontocerebellar atrophy and amyotrophic lateral sclerosis [115,116,117,118,119,120]. This contribution has been known since the 1980s, when the work of Andreas Plaitakis group has shown deregulation of glutamate metabolism and GDH deficiency in patients with various cerebellar, brainstem and anterior horn disorders [115, 117,118,119,120].
Increased interest has been shown in recent years in the role of deregulation of glutamate metabolism in Alzheimer’s disease, another, far more common, age-related neurodegenerative disorder [121]. In this respect, there are several observations that point towards involvement of hGDH1/2 mediated glutamate metabolism pathways in the neurodegeneration that accompanies Alzheimer’s disease or other age-related disease phenotypes. As discussed above, the group of Elias Michaelis [67] created a transgenic (Tg) mouse model overexpressing mouse Glud1 that showed constant excess synaptic glutamate release in specific brain regions such as the striatum and the CA1 region of the hippocampus. This was associated with progressive neurodegeneration in many brain areas, but especially in the CA1 hippocampal region where a significant reduction in the number of dendritic spines and axonal nerve terminals was observed. Notably, the degeneration process became more intense and extended to other brain regions as the animals were growing older. Based on these findings, Elias Michaelis and his group quite insightfully suggested that GDH1 overexpression can accelerate age-related neuronal loss and dendritic dysfunction in a manner analogous to that previously observed in Alzheimer’s disease [67, 73]. This suggestion, when examined under the light of the above described hGDH1/2 alterations in other degenerative disorders, possibly opens a new path for the elucidation of the pathophysiology of Alzheimer’s disease, a disease that poses an overwhelming burden in modern societies with aging populations.
Overall for the role of hGDH1 and hGDH2 in human neurodegenerative disorders, there is an apparent discrepancy among various published results on the effect of hGDH1/2 overactivity and of hGDH1/2 loss of function, both in animal models and in human studies. However, given the heterogeneity of the experimental models used and the complexity of the neurodegenerative processes involved, it could be possible that hyperactivity or loss of function of hGDH1 and/or hGDH2 could be protective in one clinical context and deleterious in another.
Finally, there is an emerging role for hGDH1 and hGDH2 in tumorigenesis [122]. As described above, human breast cancer cells use the GDH reaction to incorporate ammonia into amino acid biosynthesis and promote their growth [30]. This is of special importance under conditions of glutamine shortage. Specifically, under glutamine depletion, these breast cancer cell lines exploit the GDH reaction for biosynthetic purposes, and the main iso-enzyme contributing to this processes is hGDH2 [123]. Concerning the role of the GDH reaction in the metastatic potential of cancer cells, there is evidence that upregulation of hGDH1 activity could mediate lung cancer metastasis through metabolic reprogramming of glutaminolysis [124]. Back to the brain, there is increasing literature on the role of hGDH1 and hGDH2 in human gliomas [125, 126]. Specifically, for hGDH2, it has been recently shown that it promoted glioma growth by compensating for mutant isocitrate dehydrogenase (IDHR132H) metabolic derangements and that this effect was mediated through the adaptively evolved allosteric domain [127, 128]. These observations open the way for possibly using hGDH1 and hGDH2 inhibitors [129, 130] in cancer therapeutics.
In summary, hGDH1 and hGDH2 have important roles in metabolism and are also thought to be involved in the pathogenesis of various neurological and non-neurological syndromes, including HI/HA, diabetes, epilepsy, Parkinson’s disease, Alzheimer’s disease, amyotrophic lateral sclerosis, and various tumors, including gliomas. It is expected that in the near future pharmacological manipulation of the two human GDH iso-enzymes could be of therapeutic potential in at least some of these disease processes.
References
Strecker HJ (1953) Glutamic dehydrogenase. Arch Biochem Biophys 46(1):128–140. https://doi.org/10.1016/0003-9861(53)90176-3
Hudson R, Daniel R (1993) L-glutamate dehydrogenases: distribution, properties and mechanism. Comp Biochem Physiol B 106(4):767–792
Plaitakis A, Zaganas I (2001) Regulation of human glutamate dehydrogenases: implications for glutamate, ammonia and energy metabolism in brain. J Neurosci Res 66(5):899–908. https://doi.org/10.1002/jnr.10054
Hutson SM, Islam MM, Zaganas I (2011) Interaction between glutamate dehydrogenase (GDH) and L-leucine catabolic enzymes: intersecting metabolic pathways. Neurochem Int 59(4):518–524. https://doi.org/10.1016/j.neuint.2011.05.001
Zaganas I, Spanaki C, Plaitakis A (2012) Expression of human GLUD2 glutamate dehydrogenase in human tissues: functional implications. Neurochem Int 61(4):455–462. https://doi.org/10.1016/j.neuint.2012.06.007
Li M, Li C, Allen A, Stanley CA, Smith TJ (2014) Glutamate dehydrogenase: structure, allosteric regulation, and role in insulin homeostasis. Neurochem Res 39(3):433–445. https://doi.org/10.1007/s11064-013-1173-2
Fahien LA, MacDonald MJ (2011) The complex mechanism of glutamate dehydrogenase in insulin secretion. Diabetes 60(10):2450–2454. https://doi.org/10.2337/db10-1150
McKenna M, Stridh M, McNair L, Sonnewald U, Waagepetersen H, Schousboe A (2016) Glutamate oxidation in astrocytes: roles of glutamate dehydrogenase and aminotransferases. J Neurosci Res 94(12):1561–1571. https://doi.org/10.1002/jnr.23908
McKenna MC (2011) Glutamate dehydrogenase in brain mitochondria: do lipid modifications and transient metabolon formation influence enzyme activity? Neurochem Int 59(4):525–533. https://doi.org/10.1016/j.neuint.2011.07.003
Spanaki C, Plaitakis A (2012) The role of glutamate dehydrogenase in mammalian ammonia metabolism. Neurotox Res 21(1):117–127. https://doi.org/10.1007/s12640-011-9285-4
Lorin S, Tol MJ, Bauvy C, Strijland A, Poüs C, Verhoeven AJ, Codogno P, Meijer AJ (2013) Glutamate dehydrogenase contributes to leucine sensing in the regulation of autophagy. Autophagy 9(6):850–860. https://doi.org/10.4161/auto.24083
Shashidharan P, Michaelidis TM, Robakis NK, Kresovali A, Papamatheakis J, Plaitakis A (1994) Novel human glutamate dehydrogenase expressed in neural and testicular tissues and encoded by an X-linked intronless gene. J Biol Chem 269(24):16971–16976
Zaganas IV, Kanavouras K, Borompokas N, Arianoglou G, Dimovasili C, Latsoudis H, Vlassi M, Mastorodemos V (2014) The odyssey of a young gene: structure–function studies in human glutamate dehydrogenases reveal evolutionary-acquired complex allosteric regulation mechanisms. Neurochem Res 39(3):471–486. https://doi.org/10.1007/s11064-014-1251-0
Burki F, Kaessmann H (2004) Birth and adaptive evolution of a hominoid gene that supports high neurotransmitter flux. Nat Genet 36:1061. https://doi.org/10.1038/ng1431 https://www.nature.com/articles/ng1431#supplementary-information
Varki A (2004) How to make an ape brain. Nat Genet 36:1034. https://doi.org/10.1038/ng1004-1034
Shashidharan P, Plaitakis A (2014) The discovery of human of GLUD2 glutamate dehydrogenase and its implications for cell function in health and disease. Neurochem Res 39(3):460–470. https://doi.org/10.1007/s11064-013-1227-5
Zaganas I, Spanaki C, Karpusas M, Plaitakis A (2002) Substitution of Ser for Arg-443 in the regulatory domain of human housekeeping (GLUD1) glutamate dehydrogenase virtually abolishes basal activity and markedly alters the activation of the enzyme by adp and l-Leucine. J Biol Chem 277(48):46552–46558. https://doi.org/10.1074/jbc.M208596200
Zaganas I, Plaitakis A (2002) Single amino acid substitution (G456A) in the vicinity of the GTP binding domain of human housekeeping glutamate dehydrogenase markedly attenuates GTP inhibition and abolishes the cooperative behavior of the enzyme. J Biol Chem 277(29):26422–26428. https://doi.org/10.1074/jbc.M200022200
Shashidharan P, Clarke DD, Ahmed N, Moschonas N, Plaitakis A (1997) Nerve tissue-specific human glutamate dehydrogenase that is thermolabile and highly regulated by ADP. J Neurochem 68(5):1804–1811. https://doi.org/10.1046/j.1471-4159.1997.68051804.x
Zaganas I, Kanavouras K, Mastorodemos V, Latsoudis H, Spanaki C, Plaitakis A (2009) The human GLUD2 glutamate dehydrogenase: localization and functional aspects. Neurochem Int 55(1):52–63. https://doi.org/10.1016/j.neuint.2009.03.001
Engel PC (2014) Glutamate dehydrogenases: the why and how of coenzyme specificity. Neurochem Res 39(3):426–432. https://doi.org/10.1007/s11064-013-1089-x
Bailey J, Bell ET, Bell JE (1982) Regulation of bovine glutamate dehydrogenase. The effects of pH and ADP. J Biol Chem 257(10):5579–5583
Yudkoff M (1997) Brain metabolism of branched-chain amino acids. Glia 21 (1):92–98. https://doi.org/10.1002/(SICI)1098-1136(199709)21:1%3C92::AID-GLIA10%3E3.0.CO;2-W
Cooper AJL (2012) The role of glutamine synthetase and glutamate dehydrogenase in cerebral ammonia homeostasis. Neurochem Res 37(11):2439–2455. https://doi.org/10.1007/s11064-012-0803-4
Smith HQ, Li C, Stanley CA, Smith TJ (2017) Glutamate dehydrogenase, a complex enzyme at a crucial metabolic branch point. Neurochem Res. https://doi.org/10.1007/s11064-017-2428-0
Marcaggi P, Coles JA (2001) Ammonium in nervous tissue: transport across cell membranes, fluxes from neurons to glial cells, and role in signalling. Prog Neurobiol 64(2):157–183. https://doi.org/10.1016/S0301-0082(00)00043-5
Adeva MM, Souto G, Blanco N, Donapetry C (2012) Ammonium metabolism in humans. Metab Clin Exp 61(11):1495–1511. https://doi.org/10.1016/j.metabol.2012.07.007
Zaganas I, Pajęcka K, Wendel Nielsen C, Schousboe A, Waagepetersen HS, Plaitakis A (2013) The effect of pH and ADP on ammonia affinity for human glutamate dehydrogenases. Metab Brain Dis 28(2):127–131. https://doi.org/10.1007/s11011-013-9382-6
Tsacopoulos M, Poitry-Yamate CL, Poitry S (1997) Ammonium and glutamate released by neurons are signals regulating the nutritive function of a glial cell. J Neurosci 17(7):2383–2390
Spinelli JB, Yoon H, Ringel AE, Jeanfavre S, Clish CB, Haigis MC (2017) Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science 358(6365):941–946. https://doi.org/10.1126/science.aam9305
Li M, Li C, Allen A, Stanley CA, Smith TJ (2012) The structure and allosteric regulation of mammalian glutamate dehydrogenase. Arch Biochem Biophys 519(2):69–80. https://doi.org/10.1016/j.abb.2011.10.015
Mastorodemos V, Zaganas I, Spanaki C, Bessa M, Plaitakis A (2005) Molecular basis of human glutamate dehydrogenase regulation under changing energy demands. J Neurosci Res 79(1–2):65–73. https://doi.org/10.1002/jnr.20353
Kanavouras K, Mastorodemos V, Borompokas N, Spanaki C, Plaitakis A (2007) Properties and molecular evolution of human GLUD2 (neural and testicular tissue-specific) glutamate dehydrogenase. J Neurosci Res 85(5):1101–1109. https://doi.org/10.1002/jnr.21197
Plaitakis A, Metaxari M, Shashidharan P (2000) Nerve tissue-specific (GLUD2) and housekeeping (GLUD1) human glutamate dehydrogenases are regulated by distinct allosteric mechanisms. J Neurochem 75(5):1862–1869. https://doi.org/10.1046/j.1471-4159.2000.0751862.x
Smith TJ, Schmidt T, Fang J, Wu J, Siuzdak G, Stanley CA (2002) The structure of apo human glutamate dehydrogenase details subunit communication and allostery. J Mol Biol 318(3):765–777. https://doi.org/10.1016/S0022-2836(02)00161-4
Peterson PE, Pierce J, Smith TJ (1997) Crystallization and characterization of bovine liver glutamate dehydrogenase. J Struct Biol 120(1):73–77. https://doi.org/10.1006/jsbi.1997.3899
Peterson PE, Smith TJ (1999) The structure of bovine glutamate dehydrogenase provides insights into the mechanism of allostery. Structure 7(7):769–782. https://doi.org/10.1016/S0969-2126(99)80101-4
Smith TJ, Peterson PE, Schmidt T, Fang J, Stanley CA (2001) Structures of bovine glutamate dehydrogenase complexes elucidate the mechanism of purine regulation 1 Edited by I. A. Wilson. J Mol Biol 307(2):707–720. https://doi.org/10.1006/jmbi.2001.4499
Li M, Li C, Allen A, Stanley CA, Smith TJ (2011) The structure and allosteric regulation of glutamate dehydrogenase. Neurochem Int 59(4):445–455. https://doi.org/10.1016/j.neuint.2010.10.017
Mastorodemos V, Kanavouras K, Sundaram S, Providaki M, Petraki Z, Kokkinidis M, Zaganas I, Logothetis DE, Plaitakis A (2015) Side-chain interactions in the regulatory domain of human glutamate dehydrogenase determine basal activity and regulation. J Neurochem 133(1):73–82. https://doi.org/10.1111/jnc.13019
Banerjee S, Schmidt T, Fang J, Stanley CA, Smith TJ (2003) Structural studies on ADP activation of mammalian glutamate dehydrogenase and the evolution of regulation. Biochemistry 42(12):3446–3456. https://doi.org/10.1021/bi0206917
Spanaki C, Kotzamani D, Petraki Z, Drakos E, Plaitakis A (2015) Expression of human GLUD1 and GLUD2 glutamate dehydrogenases in steroid producing tissues. Mol Cell Endocrinol 415:1–11. https://doi.org/10.1016/j.mce.2015.07.020
Spanaki C, Zaganas I, Kleopa KA, Plaitakis A (2010) Human GLUD2 glutamate dehydrogenase is expressed in neural and testicular supporting cells. J Biol Chem 285(22):16748–16756. https://doi.org/10.1074/jbc.M109.092999
Spanaki C, Kotzamani D, Petraki Z, Drakos E, Plaitakis A (2014) Heterogeneous cellular distribution of glutamate dehydrogenase in brain and in non-neural tissues. Neurochem Res 39(3):500–515. https://doi.org/10.1007/s11064-013-1235-5
Hull J, Usmari Moraes M, Brookes E, Love S, Conway ME (2018) Distribution of the branched-chain α-ketoacid dehydrogenase complex E1α subunit and glutamate dehydrogenase in the human brain and their role in neuro-metabolism. Neurochem Int 112:49–58. https://doi.org/10.1016/j.neuint.2017.10.014
Spanaki C, Kotzamani D, Kleopa K, Plaitakis A (2016) Evolution of GLUD2 glutamate dehydrogenase allows expression in human cortical neurons. Mol Neurobiol 53(8):5140–5148. https://doi.org/10.1007/s12035-015-9429-2
Nissen JD, Lykke K, Bryk J, Stridh MH, Zaganas I, Skytt DM, Schousboe A, Bak LK, Enard W, Pääbo S, Waagepetersen HS (2017) Expression of the human isoform of glutamate dehydrogenase, hGDH2, augments TCA cycle capacity and oxidative metabolism of glutamate during glucose deprivation in astrocytes. Glia 65(3):474–488. https://doi.org/10.1002/glia.23105
Salganicoff L, Robertis ED (1965) Subcellular distribution of the enzymes of the glutamic acid, glutamine and γ-aminobutyric acid cycles in rat brain*. J Neurochem 12(4):287–309. https://doi.org/10.1111/j.1471-4159.1965.tb06766.x
Aoki C, Milner TA, Berger SB, Sheu KFR, Blass JP, Pickel VM (1987) Glial glutamate dehydrogenase: ultrastructural localization and regional distribution in relation to the mitochondrial enzyme, cytochrome oxidase. J Neurosci Res 18(2):305–318. https://doi.org/10.1002/jnr.490180207
Rothe F, Brosz M, Storm-Mathisen J (1994) Quantitative ultrastructural localization of glutamate dehydrogenase in the rat cerebellar cortex. Neuroscience 62(4):1133–1146. https://doi.org/10.1016/0306-4522(94)90349-2
Colon AD, Plaitakis A, Perakis A, Berl S, Clarke DD (1986) Purification and characterization of a soluble and a particulate glutamate dehydrogenase from rat brain. J Neurochem 46(6):1811–1819. https://doi.org/10.1111/j.1471-4159.1986.tb08500.x
Rajas F, Rousset B (1993) A membrane-bound form of glutamate dehydrogenase possesses an ATP-dependent high-affinity microtubule-binding activity. Biochem J 295(2):447–455. https://doi.org/10.1042/bj2950447
Lee W, Shin S, Cho S, Park J (1999) Purification and characterization of glutamate dehydrogenase as another isoprotein binding to the membrane of rough endoplasmic reticulum. J Cell Biochem 76(2):244–253
Pragnya P, Mrutyunjay S, Divya S, Shashibhal Matacharan P, Madan Mohan C, Jogeswar Satchidananda P (2011) Characterization of nuclear glutamate dehydrogenase of chicken liver and brain. Protein Pept Lett 18(12):1194–1203. https://doi.org/10.2174/092986611797642698
di Prisco G, Banay-Schwartz M, Strecker HJ (1968) Glutamate dehydrogenase in nuclear and mitochondrial fractions of rat liver. Biochem Biophys Res Commun 33(4):606–612. https://doi.org/10.1016/0006-291X(68)90339-2
Lai J, Sheu K, Kim Y, Clarke D, Blass J (1986) The subcellular localization of glutamate dehydrogenase (GDH): is GDH a marker for mitochondria in brain? Neurochem Res 11(5):733–744
Plaitakis A, Kalef-Ezra E, Kotzamani D, Zaganas I, Spanaki C (2017) The glutamate dehydrogenase pathway and its roles in cell and tissue biology in health and disease. Biology 6(1):11
Mastorodemos V, Kotzamani D, Zaganas I, Arianoglou G, Latsoudis H, Plaitakis A (2009) Human GLUD1 and GLUD2 glutamate dehydrogenase localize to mitochondria and endoplasmic reticulum. Biochem Cell Biol 87(3):505–516. https://doi.org/10.1139/O09-008
Rosso L, Marques AC, Reichert AS, Kaessmann H (2008) Mitochondrial targeting adaptation of the hominoid-specific glutamate dehydrogenase driven by positive darwinian selection. PLOS Genet 4(8):e1000150. https://doi.org/10.1371/journal.pgen.1000150
Kalef-Ezra E, Kotzamani D, Zaganas I, Katrakili N, Plaitakis A, Tokatlidis K (2016) Import of a major mitochondrial enzyme depends on synergy between two distinct helices of its presequence. Biochem J 473(18):2813–2829. https://doi.org/10.1042/bcj20160535
Ebrahimi-Fakhari D, Saffari A, Wahlster L, Di Nardo A, Turner D, Lewis Tommy L, Conrad C, Rothberg Jonathan M, Lipton Jonathan O, Kölker S, Hoffmann Georg F, Han M-J, Polleux F, Sahin M (2016) Impaired mitochondrial dynamics and mitophagy in neuronal models of tuberous sclerosis complex. Cell Rep 17(4):1053–1070. https://doi.org/10.1016/j.celrep.2016.09.054
Karnati S, Lüers G, Pfreimer S, Baumgart-Vogt E (2013) Mammalian SOD2 is exclusively located in mitochondria and not present in peroxisomes. Histochem Cell Biol 140(2):105–117. https://doi.org/10.1007/s00418-013-1099-4
Lin C-C, Kurashige M, Liu Y, Terabayashi T, Ishimoto Y, Wang T, Choudhary V, Hobbs R, Liu L-K, Lee P-H, Outeda P, Zhou F, Restifo NP, Watnick T, Kawano H, Horie S, Prinz W, Xu H, Menezes LF, Germino GG (2018) A cleavage product of Polycystin-1 is a mitochondrial matrix protein that affects mitochondria morphology and function when heterologously expressed. Sci Rep 8(1):2743. https://doi.org/10.1038/s41598-018-20856-6
Carobbio S, Ishihara H, Fernandez-Pascual S, Bartley C, Martin-Del-Rio R, Maechler P (2004) Insulin secretion profiles are modified by overexpression of glutamate dehydrogenase in pancreatic islets. Diabetologia 47(2):266–276. https://doi.org/10.1007/s00125-003-1306-2
Tanizawa Y, Nakai K, Sasaki T, Anno T, Ohta Y, Inoue H, Matsuo K, Koga M, Furukawa S, Oka Y (2002) Unregulated elevation of glutamate dehydrogenase activity induces glutamine-stimulated insulin secretion. Identification and characterization of a GLUD1 gene mutation and insulin secretion studies with MIN6 cells overexpressing the mutant glutamate dehydrogenase. Diabetes 51(3):712–717. https://doi.org/10.2337/diabetes.51.3.712
Anno T, Uehara S, Katagiri H, Ohta Y, Ueda K, Mizuguchi H, Moriyama Y, Oka Y, Tanizawa Y (2004) Overexpression of constitutively activated glutamate dehydrogenase induces insulin secretion through enhanced glutamate oxidation. Am J Physiol-Endocrinol Metab 286(2):E280–E285. https://doi.org/10.1152/ajpendo.00380.2003
Bao X, Pal R, Hascup KN, Wang Y, Wang W-T, Xu W, Hui D, Agbas A, Wang X, Michaelis ML, Choi I-Y, Belousov AB, Gerhardt GA, Michaelis EK (2009) Transgenic expression of Glud1 (glutamate dehydrogenase 1) in neurons: in vivo model of enhanced glutamate release, altered synaptic plasticity, and selective neuronal vulnerability. J Neurosci 29(44):13929–13944. https://doi.org/10.1523/jneurosci.4413-09.2009
Li Q, Guo S, Jiang X, Bryk J, Naumann R, Enard W, Tomita M, Sugimoto M, Khaitovich P, Pääbo S (2016) Mice carrying a human GLUD2 gene recapitulate aspects of human transcriptome and metabolome development. Proc Natl Acad Sci 113(19):5358–5363. https://doi.org/10.1073/pnas.1519261113
Wang X, Bao X, Pal R, Agbas A, Michaelis EK (2010) Transcriptomic responses in mouse brain exposed to chronic excess of the neurotransmitter glutamate. BMC Genom 11(1):360. https://doi.org/10.1186/1471-2164-11-360
Michaelis EK, Wang X, Pal R, Bao X, Hascup KN, Wang Y, Wang WT, Hui D, Agbas A, Choi IY, Belousov A, Gerhardt GA (2011) Neuronal Glud1 (glutamate dehydrogenase 1) over-expressing mice: increased glutamate formation and synaptic release, loss of synaptic activity, and adaptive changes in genomic expression. Neurochem Int 59(4):473–481. https://doi.org/10.1016/j.neuint.2011.03.003
Hascup KN, Bao X, Hascup ER, Hui D, Xu W, Pomerleau F, Huettl P, Michaelis ML, Michaelis EK, Gerhardt GA (2011) Differential levels of glutamate dehydrogenase 1 (GLUD1) in Balb/c and C57BL/6 mice and the effects of overexpression of the Glud1 gene on glutamate release in striatum. ASN Neuro 3(2):AN20110005. https://doi.org/10.1042/an20110005
Choi I-Y, Lee P, Wang W-T, Hui D, Wang X, Brooks WM, Michaelis EK (2014) Metabolism changes during aging in the hippocampus and striatum of Glud1 (glutamate dehydrogenase 1) transgenic mice. Neurochem Res 39(3):446–455. https://doi.org/10.1007/s11064-014-1239-9
Wang X, Patel ND, Hui D, Pal R, Hafez MM, Sayed-Ahmed MM, Al-Yahya AA, Michaelis EK (2014) Gene expression patterns in the hippocampus during the development and aging of Glud1(glutamate dehydrogenase 1) transgenic and wild type mice. BMC Neurosci 15(1):37. https://doi.org/10.1186/1471-2202-15-37
Plaitakis A, Zaganas I, Spanaki C (2013) Deregulation of glutamate dehydrogenase in human neurologic disorders. J Neurosci Res 91(8):1007–1017. https://doi.org/10.1002/jnr.23176
Kelly A, Stanley C (2001) Disorders of glutamate metabolism. Dev Disabil Res Rev 7(4):287–295
Palladino AA, Stanley CA (2010) The hyperinsulinism/hyperammonemia syndrome. Rev Endocr Metab Disord 11(3):171–178. https://doi.org/10.1007/s11154-010-9146-0
Stanley CA, Lieu YK, Hsu BYL, Burlina AB, Greenberg CR, Hopwood NJ, Perlman K, Rich BH, Zammarchi E, Poncz M (1998) Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene. N Engl J Med 338(19):1352–1357. https://doi.org/10.1056/nejm199805073381904
Tran C, Konstantopoulou V, Mecjia M, Perlman K, Mercimek-Mahmutoglu S, Kronick Jonathan B (2015) Hyperinsulinemic hypoglycemia: think of hyperinsulinism/hyperammonemia (HI/HA) syndrome caused by mutations in the GLUD1 gene. J Pediatr Endocrinol Metab. https://doi.org/10.1515/jpem-2014-0441
Aka S, Alanay Y, Boodhansingh K, Stanley C, Semiz S (2016) Seizures and diagnostic difficulties in hyperinsulinism-hyperammonemia syndrome. Turk J Pediatr 58(5):541–544. https://doi.org/10.24953/turkjped.2016.05.014
Ninković D, Sarnavka V, Bašnec A, Ćuk M, Ramadža Danijela P, Fumić K, Kušec V, Santer R, Barić I (2016) Hyperinsulinism-hyperammonemia syndrome: a de novo mutation of the GLUD1 gene in twins and a review of the literature. J Pediatr Endocrinol Metab. https://doi.org/10.1515/jpem-2016-0086
Kapoor RR, Flanagan SE, Arya VB, Shield JP, Ellard S, Hussain K (2013) Clinical and molecular characterisation of 300 patients with congenital hyperinsulinism. Eur J Endocrinol 168(4):557–564. https://doi.org/10.1530/eje-12-0673
Corrêa-Giannella ML, Freire DS, Cavaleiro AM, Fortes MAZ, Giorgi RR, Pereira MAA (2012) Hyperinsulinism/hyperammonemia (HI/HA) syndrome due to a mutation in the glutamate dehydrogenase gene. Arq Bras Endocrinol Metabol 56:485–489
Aso K, Okano Y, Takeda T, Sakamoto O, Ban K, Iida K, Yamano T, Shintaku H (2011) Spectrum of glutamate dehydrogenase mutations in Japanese patients with congenital hyperinsulinism and hyperammonemia syndrome. Osaka City Med J 57(1):1–9
Diao C, Chen S, Xiao X, Wang T, Sun X, Wang O, Song H, Zhang Y, Yu M, Zhang Q, Wang H (2010) Two unrelated chinese patients with hyperinsulinism/hyperammonemia (HI/HA) syndrome due to mutations in glutamate dehydrogenase gene. J Pediatr Endocrinol Metab. https://doi.org/10.1515/JPEM.2010.23.7.733
Kapoor RR, Flanagan SE, Fulton P, Chakrapani A, Chadefaux B, Ben-Omran T, Banerjee I, Shield JP, Ellard S, Hussain K (2009) Hyperinsulinism–hyperammonaemia syndrome: novel mutations in the GLUD1 gene and genotype–phenotype correlations. Eur J Endocrinol 161(5):731–735. https://doi.org/10.1530/eje-09-0615
de Lonlay P, Benelli C, Fouque F, Ganguly A, Aral B, Dionisi-Vici C, Touati G, Heinrichs C, Rabier D, Kamoun P, Robert J-J, Stanley C, Saudubray J-M (2001) Hyperinsulinism and hyperammonemia syndrome: report of twelve unrelated patients. Pediatr Res 50:353. https://doi.org/10.1203/00006450-200109000-00010
Santer R, Kinner M, Passarge M, Superti-Furga A, Mayatepek E, Meissner T, Schneppenheim R, Schaub J (2001) Novel missense mutations outside the allosteric domain of glutamate dehydrogenase are prevalent in European patients with the congenital hyperinsulinism-hyperammonemia syndrome. Hum Genet 108(1):66–71. https://doi.org/10.1007/s004390000432
Yasuda K, Koda N, Kadowaki H, Ogawa Y, Kimura S, Kadowaki T, Akanuma Y (2001) A Japanese case of congenital hyperinsulinism with hyperammonemia due to a mutation in glutamate dehydrogenase (GLUD1) gene. Intern Med 40(1):32–37. https://doi.org/10.2169/internalmedicine.40.32
Barrosse-Antle M, Su C, Chen P, Boodhansingh KE, Smith TJ, Stanley CA, De León DD, Li C (2017) A severe case of hyperinsulinism due to hemizygous activating mutation of glutamate dehydrogenase. Pediatr Diabetes 18(8):911–916. https://doi.org/10.1111/pedi.12507
Snider KE, Becker S, Boyajian L, Shyng SL, MacMullen C, Hughes N, Ganapathy K, Bhatti T, Stanley CA, Ganguly A (2013) Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J Clin Endocrinol Metab 98(2):E355–E363. https://doi.org/10.1210/jc.2012-2169
Stanley CA (2009) Regulation of glutamate metabolism and insulin secretion by glutamate dehydrogenase in hypoglycemic children1–4. Am J Clin Nutr 90(3):862S–866S. https://doi.org/10.3945/ajcn.2009.27462AA
Kibbey RG, Choi CS, Lee H-Y, Cabrera O, Pongratz RL, Zhao X, Birkenfeld AL, Li C, Berggren P-O, Stanley C, Shulman GI (2014) Mitochondrial GTP insensitivity contributes to hypoglycemia in hyperinsulinemia hyperammonemia by inhibiting glucagon release. Diabetes 63(12):4218–4229. https://doi.org/10.2337/db14-0783
Andersson LE, Shcherbina L, Al-Majdoub M, Vishnu N, Arroyo CB, Carrara JA, Wollheim CB, Fex M, Mulder H, Wierup N, Spégel P (2017) Glutamine-elicited secretion of glucagon-like peptide 1 (GLP-1) is governed by an activated glutamate dehydrogenase. Diabetes. https://doi.org/10.2337/db16-1441
Göhring I, Mulder H (2012) Glutamate dehydrogenase, insulin secretion, and type 2 diabetes: a new means to protect the pancreatic β-cell? J Endocrinol 212(3):239–242. https://doi.org/10.1530/joe-11-0481
Pournourmohammadi S, Grimaldi M, Stridh MH, Lavallard V, Waagepetersen HS, Wollheim CB, Maechler P (2017) Epigallocatechin-3-gallate (EGCG) activates AMPK through the inhibition of glutamate dehydrogenase in muscle and pancreatic ß-cells: a potential beneficial effect in the pre-diabetic state? Int J Biochem Cell Biol 88:220–225. https://doi.org/10.1016/j.biocel.2017.01.012
Stanley CA (2011) Two genetic forms of hyperinsulinemic hypoglycemia caused by dysregulation of glutamate dehydrogenase. Neurochem Int 59(4):465–472. https://doi.org/10.1016/j.neuint.2010.11.017
De Lonlay P, Giurgea I, Sempoux C, Touati G, Jaubert F, Rahier J, Ribeiro M, Brunelle F, Nihoul-Fékété C, Robert J-J, Saudubray J-M, Stanley C, Bellanné-Chantelot C (2005) Dominantly inherited hyperinsulinaemic hypoglycaemia. J Inherit Metab Dis 28(3):267–276. https://doi.org/10.1007/s10545-005-7057-0
Li C, Chen P, Palladino A, Narayan S, Russell LK, Sayed S, Xiong G, Chen J, Stokes D, Butt YM, Jones PM, Collins HW, Cohen NA, Cohen AS, Nissim I, Smith TJ, Strauss AW, Matschinsky FM, Bennett MJ, Stanley CA (2010) Mechanism of hyperinsulinism in short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency involves activation of glutamate dehydrogenase. J Biol Chem 285(41):31806–31818. https://doi.org/10.1074/jbc.M110.123638
Narayan SB, Master SR, Sireci AN, Bierl C, Stanley PE, Li C, Stanley CA, Bennett MJ (2012) Short-chain 3-hydroxyacyl-coenzyme a dehydrogenase associates with a protein super-complex integrating multiple metabolic pathways. PLoS ONE 7(4):e35048. https://doi.org/10.1371/journal.pone.0035048
Bahi-Buisson N, Roze E, Dionisi C, Escande F, Valayannopoulos V, Feillet F, Heinrichs C (2008) Neurological aspects of hyperinsulinism–hyperammonaemia syndrome. Dev Med Child Neurol 50(12):945–949. https://doi.org/10.1111/j.1469-8749.2008.03114.x
Kelly A, Stanley CA (2008) Neurological aspects in hyperinsulinism-hyperammonaemia syndrome. Dev Med Child Neurol 50(12):888–888. https://doi.org/10.1111/j.1469-8749.2008.03149.x
Nakano K, Kobayashi K, Okano Y, Aso K, Ohtsuka Y (2012) Intractable absence seizures in hyperinsulinism-hyperammonemia syndrome. Pediatr Neurol 47(2):119–122. https://doi.org/10.1016/j.pediatrneurol.2012.04.019
Balasubramaniam S, Kapoor R, Yeow J, Lim P, Flanagan S, Ellard S, Hussain K (2011) Biochemical evaluation of an infant with hypoglycemia resulting from a novel de novo mutation of the GLUD1 gene and hyperinsulinism-hyperammonemia syndrome. J Pediatr Endocrinol Metab 24(7–8):573–577
Treiman DM (2001) GABAergic mechanisms in epilepsy. Epilepsia 42(Suppl 3):8–12
Shetty AK, Upadhya D (2016) GABA-ergic cell therapy for epilepsy: advances, limitations and challenges. Neurosci Biobehav Rev 62:35–47. https://doi.org/10.1016/j.neubiorev.2015.12.014
Levy LM, Hallett M (2002) Impaired brain GABA in focal dystonia. Ann Neurol 51(1):93–101
Malthankar-Phatak GH, De Lanerolle N, Eid T, Spencer DD, Behar KL, Spencer SS, Kim JH, Lai JCK (2006) Differential glutamate dehydrogenase (GDH) activity profile in patients with temporal lobe epilepsy. Epilepsia 47(8):1292–1299. https://doi.org/10.1111/j.1528-1167.2006.00543.x
Sherwin A, Quesney F, Gauthier S, Olivier A, Robitaille Y, McQuaid P, Harvey C, van Gelder N (1984) Enzyme changes in actively spiking areas of human epileptic cerebral cortex. Neurology 34(7):927–933
Rasgado Lourdes AV, Reyes Guillermo C, Díaz Fernando V (2015) Modulation of brain glutamate dehydrogenase as a tool for controlling seizures. Acta Pharm. https://doi.org/10.1515/acph-2015-0033
Badawi Y, Pal R, Hui D, Michaelis EK, Shi H (2015) Ischemic tolerance in an in vivo model of glutamate preconditioning. J Neurosci Res 93(4):623–632. https://doi.org/10.1002/jnr.23517
Kim AY, Baik EJ (2018) Glutamate dehydrogenase as a neuroprotective target against neurodegeneration. Neurochem Res. https://doi.org/10.1007/s11064-018-2467-1
Kim AY, Jeong K-H, Lee JH, Kang Y, Lee SH, Baik EJ (2017) Glutamate dehydrogenase as a neuroprotective target against brain ischemia and reperfusion. Neuroscience 340:487–500. https://doi.org/10.1016/j.neuroscience.2016.11.007
Hohnholt MC, Andersen VH, Andersen JV, Christensen SK, Karaca M, Maechler P, Waagepetersen HS (2017) Glutamate dehydrogenase is essential to sustain neuronal oxidative energy metabolism during stimulation. J Cereb Blood Flow Metab. https://doi.org/10.1177/0271678x17714680
Plaitakis A, Latsoudis H, Kanavouras K, Ritz B, Bronstein JM, Skoula I, Mastorodemos V, Papapetropoulos S, Borompokas N, Zaganas I, Xiromerisiou G, Hadjigeorgiou GM, Spanaki C (2009) Gain-of-function variant in GLUD2 glutamate dehydrogenase modifies Parkinson’s disease onset. Eur J Hum Genet 18:336. https://doi.org/10.1038/ejhg.2009.179 https://www.nature.com/articles/ejhg2009179#supplementary-information
Plaitakis A, Caroscio JT (1987) Abnormal glutamate metabolism in amyotrophic lateral sclerosis. Ann Neurol 22(5):575–579. https://doi.org/10.1002/ana.410220503
Sorbi S, Tonini S, Giannini E, Piacentini S, Marini P, Amaducci L (1986) Abnormal platelet glutamate dehydrogenase activity and activation in dominant and nondominant olivopontocerebellar atrophy. Ann Neurol 19(3):239–245. https://doi.org/10.1002/ana.410190304
Plaitakis A (1990) Glutamate dysfunction and selective motor neuron degeneration inamyotrophic lateral sclerosis: a hypothesis. Ann Neurol 28(1):3–8. https://doi.org/10.1002/ana.410280103
Plaitakis A, Berl S, Yahr MD (1984) Neurological disorders associated with deficiency of glutamate dehydrogenase. Ann Neurol 15(2):144–153. https://doi.org/10.1002/ana.410150206
Plaitakis A, Nicklas WJ, Desnick RJ (1980) Glutamate dehydrogenase deficiency in three patients with spinocerebellar syndrome. Ann Neurol 7(4):297–303. https://doi.org/10.1002/ana.410070403
Plaitakis A, Flessas P, Natsiou A, Shashidharan P (1993) Glutamate dehydrogenase deficiency in cerebellar degenerations: clinical, biochemical and molecular genetic aspects. Can J Neurol Sci 20(Suppl 3):S109–S116
Burbaeva GS, Boksha IS, Tereshkina EB, Savushkina OK, Starodubtseva LI, Turishcheva MS (2005) Glutamate metabolizing enzymes in prefrontal cortex of Alzheimer’s disease patients. Neurochem Res 30(11):1443–1451. https://doi.org/10.1007/s11064-005-8654-x
Jin L, Li D, Alesi Gina N, Fan J, Kang H-B, Lu Z, Boggon Titus J, Jin P, Yi H, Wright Elizabeth R, Duong D, Seyfried Nicholas T, Egnatchik R, DeBerardinis Ralph J, Magliocca Kelly R, He C, Arellano Martha L, Khoury Hanna J, Shin Dong M, Khuri Fadlo R, Kang S (2015) Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer Cell 27(2):257–270. https://doi.org/10.1016/j.ccell.2014.12.006
Takeuchi Y, Nakayama Y, Fukusaki E, Irino Y (2018) Glutamate production from ammonia via glutamate dehydrogenase 2 activity supports cancer cell proliferation under glutamine depletion. Biochem Biophys Res Commun 495(1):761–767. https://doi.org/10.1016/j.bbrc.2017.11.088
Jin L, Chun J, Pan C, Kumar A, Zhang G, Ha Y, Li D, Alesi GN, Kang Y, Zhou L, Yu W-M, Magliocca KR, Khuri FR, Qu C-K, Metallo C, Owonikoko TK, Kang S (2018) The PLAG1-GDH1 axis promotes anoikis resistance and tumor metastasis through CamKK2-AMPK signaling in LKB1-deficient lung cancer. Mol Cell 69(1):87–99.e87. https://doi.org/10.1016/j.molcel.2017.11.025
Zhang J, Wang G, Mao Q, Li S, Xiong W, Lin Y, Ge J (2016) Glutamate dehydrogenase (GDH) regulates bioenergetics and redox homeostasis in human glioma. Oncotarget 295:799–800
Yang C, Sudderth J, Dang T, Bachoo RG, McDonald JG, DeBerardinis RJ (2009) Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Can Res 69(20):7986–7993. https://doi.org/10.1158/0008-5472.Can-09-2266
Waitkus MS, Pirozzi CJ, Moure CJ, Diplas BH, Hansen LJ, Carpenter AB, Yang R, Wang Z, Ingram BO, Karoly ED, Mohney RP, Spasojevic I, McLendon RE, Friedman HS, He Y, Bigner DD, Yan H (2018) Adaptive evolution of the GDH2 allosteric domain promotes gliomagenesis by resolving IDH1R132H-induced metabolic liabilities. Cancer Res 78(1):36–50. https://doi.org/10.1158/0008-5472.Can-17-1352
Chen R, Nishimura MC, Kharbanda S, Peale F, Deng Y, Daemen A, Forrest WF, Kwong M, Hedehus M, Hatzivassiliou G, Friedman LS, Phillips HS (2014) Hominoid-specific enzyme GLUD2 promotes growth of IDH1R132H glioma. Proc Natl Acad Sci 111(39):14217–14222. https://doi.org/10.1073/pnas.1409653111
Li M, Allen A, Smith TJ (2007) High throughput screening reveals several new classes of glutamate dehydrogenase inhibitors. Biochemistry 46(51):15089–15102. https://doi.org/10.1021/bi7018783
Zhu M, Fang J, Zhang J, Zhang Z, Xie J, Yu Y, Ruan JJ, Chen Z, Hou W, Yang G, Su W, Ruan BH (2017) Biomolecular interaction assays identified dual inhibitors of glutaminase and glutamate dehydrogenase that disrupt mitochondrial function and prevent growth of cancer cells. Anal Chem 89(3):1689–1696. https://doi.org/10.1021/acs.analchem.6b03849
Acknowledgements
This work would not have been completed without the valuable assistance of Dr. Christina Dimovasili. We would also like to thank Dr. Dimitra Kotzamani and Dr. Maria Savaki for their help with confocal microscopy, Cynthia Manassaki for coordinating the overall project and Rena Skoula for her continuous aid with technical issues. Finally, we cordially acknowledge the vision and inspiration to work in this exciting field of research provided to us by Professor Andreas Plaitakis.
Funding
This work has been supported by intramural grants of the University of Crete, Heraklion, Crete, Greece, through the Special Account for Research - Research Funding Programs: (1) “Structural studies on human glutamate dehydrogenase iso-enzymes” (Grant Code: KA4374), (2) “Knock-out of GLUD1 and GLUD2 genes from human cell lines, using the CRISPR/Cas9 system” (Grant Code: KA4435).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declared that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Mathioudakis, L., Bourbouli, M., Daklada, E. et al. Localization of Human Glutamate Dehydrogenases Provides Insights into Their Metabolic Role and Their Involvement in Disease Processes. Neurochem Res 44, 170–187 (2019). https://doi.org/10.1007/s11064-018-2575-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-018-2575-y