
Abstract
Mammalian glutaminases catalyze the stoichiometric conversion of l-glutamine to l-glutamate and ammonium ions. In brain, glutaminase is considered the prevailing pathway for synthesis of the neurotransmitter pool of glutamate. Besides neurotransmission, the products of glutaminase reaction also fulfill crucial roles in energy and metabolic homeostasis in mammalian brain. In the last years, new functional roles for brain glutaminases are being uncovered by using functional genomic and proteomic approaches. Glutaminases may act as multifunctional proteins able to perform different tasks: the discovery of multiple transcript variants in neurons and glial cells, novel extramitochondrial localizations, and isoform-specific proteininteracting partners strongly support possible moonlighting functions for these proteins. In this chapter, we present a critical account of essential works on brain glutaminase 80 years after its discovery. We will highlight the impact of recent findings and thoughts in the context of the glutamate/glutamine brain homeostasis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
6.1 Introduction
Glutamate (Glu) is the most important neurotransmitter in normal brain function : about 80–90 % of all brain synapses release this nonessential amino acid (Braitenberg and Schüz 1998; Attwell and Laughlin 2001). Glu does not cross the blood-brain barrier (BBB) (Hawkins 2009) and therefore must be synthesized in neurons from local precursors. The enzyme phosphate-activated glutaminase (GA) catalyzes the hydrolytic deamidation of glutamine (Gln) to Glu and ammonium ions and appears as the major Gln metabolizing enzyme in brain (Hertz 1979; Kvamme 1984). This enzyme has been considered as the principal pathway for neurotransmitter Glu production (Nicklas et al. 1987); accordingly, the bulk of Glu released at the synapses derives from precursor Gln (Bradford et al. 1978; Hamberger et al. 1979; Thanki et al. 1983; Laake et al. 1995; Hertz and Zielke 2004), while inhibition of GA depletes the stores of Glu (Conti and Minelli 1994). Nevertheless, presynaptic Glu is not always necessarily derived from Gln: synthesis from tricarboxylic acid cycle (TCA) intermediates also contributes to generation of excitatory Glu (Waagepetersen et al. 2005; Takeda et al. 2012), although the exact contribution of each source is a matter of debate and remains to be established (Kam and Nicoll 2007).
Enzymes that break the amide bond of Gln and transfer the ammonia to other specific substrates in biosynthetic pathways are known as glutamine amidotransferases (EC 2.4.2). However, these enzymes should not be confused with GA, which is a glutamine amidohydrolase (EC 3.5.1.2), a genuine hydrolytic enzyme releasing the γ-glutamyl group and ammonium into the media without transfer to any other acceptor molecule. The catalytic reaction requires no energy and is essentially irreversible: the K′eq is 322 at pH 7.0 and 25 °C (Benzinger et al. 1959), which indicates that synthesis of Glu is strongly favored. Mammalian glutaminases only show reactivity toward free Gln and cannot deamidate internal Gln residues in proteins, although such activity has been reported for microbial protein glutaminases (Yamaguchi et al. 2001). On the other hand, prokaryotic asparaginases may also deamidate free Gln and this has been used as a chemotherapeutic treatment to deplete plasma Gln levels in children suffering leukemia (Holcenberg et al. 1979). However, mammalian asparaginases (l-asparaginase-amidohydrolase, EC 3.5.1.1) do not utilize Gln as substrates (Schalk et al. 2014).
Besides the transmitter pool of Glu in excitatory synapses, there is a metabolic pool with great relevance because this amino acid is a primary metabolic fuel for brain cells (Erecinska and Silver 1990) and a precursor of many other important metabolites (Fonnum 1984). In fact, the first step in the mitochondrial breakdown of glutamine to pyruvate/lactate, a pathway known as glutaminolysis, is initiated by GA and has been largely recognized as a fundamental process for energy supply in the bioenergetics of many normal and transformed cell types (Kovacevic and McGivan 1983; Márquez et al. 2015). Furthermore, Gln can be used through glutaminolysis as an anaplerotic substrate to replenish TCA intermediates (DeBerardinis et al. 2007).
6.2 Historical Account: Pioneer Works
Krebs, in his pioneer studies on amino acids metabolism, discovered GA while studying the enzymatic hydrolysis of Gln in animal tissues (Krebs 1935). Krebs first noticed the existence of two different types of GA, classifying them as “brain type” (also present in kidney) and “hepatic type.” Lately, Errera and Greenstein (1949) coined the term “phosphate-activated” GA to describe the enzymatic activity which was strongly stimulated in tissue extracts after addition of inorganic phosphate (Pi). The enzyme is widely distributed in mammalian tissues where it fulfills essential tasks related to tissue-specific function (Curthoys and Watford 1995).
The traditional terms, kidney- and liver-type isoenzymes , denoted marked differences in molecular structures, kinetic, regulatory, and immunological properties that were early detected and justified their study separately (Kovacevic and McGivan 1983). Historically, kidney-type enzymes were the first GAs isolated and characterized in mammalian tissues. Kvamme’s group leads the way in this area by isolating to apparent homogeneity pig renal and pig brain GAs (Kvamme et al. 1970; Svenneby et al. 1973). Later rat kidney (Curthoys et al. 1976), cow brain (Chiu and Boeker 1979), pig brain (Nimmo and Tipton 1980), and rat brain (Haser et al. 1985; Kaneko et al. 1987) GAs were also purified. Kidney-type enzymes were isolated in highly purified states from cancer cells indeed (Quesada et al. 1988; Segura et al. 1995).
Most kidney-type enzymes undergo specific and extensive polymerization in the presence of phosphate and phosphate–borate buffers. The reversible association–dissociation equilibrium has been advantageously used to purify this GA isoform. In sharp contrast, the liver-type isoenzymes do not show aggregation and self-assembly in high molecular mass polymers. This fact, along with a remarkable susceptibility to proteolytic degradation, has hampered their purification; thus, liver-type isozymes have been considerably more refractory to purification than their kidney-type counterparts. Actually, the isolation of pure rat liver isoform was achieved almost two decades after the first kidney-type enzyme was isolated (Heini et al. 1987; Smith and Watford 1988), although partial purifications were also previously reported (Huang and Knox 1976; Patel and McGivan 1984). Finally, it is worthy to mention that the classical terms kidney(K)-type and liver(L)-type are currently obsolete and should be avoided for the sake of coherence: the new expression data for mammalian GA do not support any more such kind of terminology (see Matés et al. 2013 for an update on GA nomenclature).
6.3 Mammalian Glutaminase Genes
The mammalian GA family members are encoded by two paralogous genes, Gls and Gls2, presumably derived from a common ancestral gene by duplication and divergent evolution (Porter et al. 2002; Pérez-Gómez et al. 2003). In humans, the GLS gene is located on chromosome 2 and encodes GA isozymes classically referred to as kidney-type (K-type), while GLS2 is located on chromosome 12 and codes for liver-type (L-type) isozymes (Aledo et al. 2000). Orthologous genes have been described in rat (Chung-Bok et al. 1997) and mouse (Mock et al. 1989). The human GLS gene (cited in many publications with the incorrect term GLS1: see HUGO Gene nomenclature, http://www.genenames.org/) spans 82 kb and splits into 19 exons (Porter et al. 2002). Two different transcripts arise from this gene (Fig. 6.1): the KGA transcript, originally found in kidney, composed by 18 exons and formed by joining exons 1–14 and 16–19, and the GAC transcript which appears by alternative splicing and uses only the first 15 exons (Elgadi et al. 1999; Porter et al. 2002) (Fig. 6.1).

Human glutaminase genes and mRNA transcripts . (Top panel) Human glutaminase GLS gene and alternative transcripts KGA and GAC. (Bottom panel) Human glutaminase GLS2 gene and alternative transcripts GAB and LGA. Each gene is shown with introns depicted as solid light blue lines and exons as numbered dark blue boxes. The promoter regions are also indicated on the 5′-end of each gene, including the alternative promoter of the GLS2 gene on intron 1. Dashed red lines indicate the exons forming KGA and GAB mRNA transcripts, while dotted dark green lines comprise exons involved in the generation of transcripts GAC and LGA. The transcription start site is marked by an arrow and numbered as +1
The human GLS2 gene has a length of approximately 18 kb and splits into 18 exons (Pérez-Gómez et al. 2003) (Fig. 6.1). Two transcripts have been identified from the mammalian Gls2 gene: the canonical long transcript termed GAB, formed by joining all 18 exons of the gene (Pérez-Gómez et al. 2003), and the short transcript LGA that lacks exon 1 and was originally identified in rat liver (Smith and Watford 1990) (Fig. 6.1). Human GAB transcript was isolated as a cDNA clone from ZR-75 breast cancer cells encoding a protein of 602 amino acids, which is 67 amino acids longer than rat liver LGA protein (Gómez-Fabre et al. 2000) (Fig. 6.1).
Although located in different chromosomes, the human GLS and GLS2 genes share a considerable degree of sequence similarity. Apart from the additional exon present in the human GLS gene, the main differences in the coding sequences of both genes are located at exons 1 and 18. Exon 1 shares 62.5 % similarity, but it codes for 129 amino acids in KGA and only for 61 amino acids in GAB, accounting for most of the 67 extra amino acids of KGA protein at the N-terminal. The sequences encoded by exon 1 contain the signals involved in mitochondrial targeting and translocation processes (Shapiro et al. 1991; Gómez-Fabre et al. 2000) (Fig. 6.2). Likewise, exon 18, which codes for the C-terminal region of both proteins, shows the lowest sequence similarity (29.4 %). This region of human GLS2 proteins has been demonstrated to be involved in the recognition of PDZ (PSD95\Dlg\ZO1 domains) interaction modules (Olalla et al. 2001) (Fig. 6.2). Therefore, the most significant differences between human GLS and GLS2 exons are located in regions involved with organelle targeting and protein–protein interactions, which may help to explain their differential function and regulation. Conversely, exons 3–17 of GLS and GLS2 mRNA transcripts have the same length and show a high sequence similarity.

Schematic illustration of the four human GA isoforms . The main signature sequences, motifs, and domains characterized or identified by sequence analysis are highlighted. Starting from the N-terminal end, the following domains are shown: mitochondrial import presequence, nuclear receptor interaction motif (LXXLL), glutaminase domain, ankyrin repeats, APC/C-Cdh1 (anaphase-promoting complex/cyclosome and activator protein Cdh1)-specific recognition motif (KEN) or KEN box, and PDZ protein recognition module (ESMV). The sequence coded by unique exon 15 (GAC isoform) and alternative first exon (LGA isoform) is also highlighted in yellow (For further information see text.)
The GLS isoform KGA is found in all tissues with GA activity with the exception of postnatal liver (Curthoys and Watford 1995; Aledo et al. 2000), although expression in liver endothelial cells has also been reported (Lohmann et al. 1999). The GAC splice variant, first isolated from a human HT-29 colon carcinoma cDNA library, is expressed predominantly in human cardiac muscle and pancreas, appreciably in placenta, kidney, and lung, but not in brain and liver (Elgadi et al. 1999). Other authors also found GAC expression in rat kidney and pig renal cells (Porter et al. 2002). On the other hand, GA transcripts derived from the Gls2 gene were originally thought to be present in adult liver tissue and absent in extrahepatic tissues (Smith and Watford 1990; Curthoys and Watford 1995). This restricted pattern of expression was changed after new findings demonstrated the expression of GLS2 transcripts in extrahepatic tissues like brain, pancreas, and breast cancer cells (Gómez-Fabre et al. 2000). Moreover, simultaneous expression of both GLS and GLS2 isoenzymes is a more frequent event than previously thought, as has been demonstrated in human tissues (Aledo et al. 2000) and cancer cells (Turner and McGivan 2003; Pérez-Gómez et al. 2005). Therefore, the molecular portrait of GA isoform expression in mammalian tissues involves multiple transcripts even in a single cell type. The abundance of a particular GA mRNA species may significantly change depending upon the tissue type or the developmental or metabolic state of the tissue . The physiological meaning of the existence of different species of mRNA coding for GA is not fully understood. However, it is tempting to speculate that each transcript may represent a specific target for different stimuli, the overall GA expression being the balance between these stimuli.
6.4 Glutaminase Expression in Brain
6.4.1 GA Transcripts and Proteins Expressed in Brain Cells
As mentioned before, the Gls-encoded KGA isoform was the first GA enzyme isolated and characterized. Furthermore, very similar molecular and kinetic properties (see next section) were found for brain and kidney KGA (Haser et al. 1985; Kvamme 1998) while, in parallel, brain KGA cDNA sequences were also available after their cloning from rat and human brain (Banner et al. 1988; Nagase et al. 1998). These early studies established the classical pattern of GA expression in mammals, where KGA was originally thought as the only glutaminase present in brain (Haser et al. 1985; Curthoys and Watford 1995). Nowadays, the pattern of GA expression in mammalian tissues has been shown to be considerably more complex. Concretely, we first reported that human brain expresses GLS2 transcripts, in addition to the KGA isoform; these transcripts were ubiquitously expressed in brain regions with the strongest signal appearing in cerebral cortex (Aledo et al. 2000; Gómez-Fabre et al. 2000). Shortly after, Northern analysis and immunocytochemistry in brain of diverse mammalian species (human, monkey, rat, cow, mouse and rabbit) confirmed simultaneous expression of GLS (KGA) and GLS2 (GAB and/or LGA) isoenzymes; interestingly, both isoforms colocalize in numerous cells throughout the brain (Olalla et al. 2002).
The existence of alternative transcripts of the Gls2 gene was recently demonstrated in brain and liver of three mammalian species: human, rat, and mouse (Martín-Rufián et al. 2012). Two GLS2 transcript variants showing alternative first exons were amplified: the long GAB transcript, previously cloned from ZR-75 breast cancer cells (Gómez-Fabre et al. 2000), and the short LGA transcript first characterized in rat liver (Smith and Watford 1990; Chung-Bok et al. 1997). Cloning and primer extension analysis of the human brain LGA transcript, to map its transcription start site (TSS) , unambiguously demonstrated that this short GLS2 isoform arises by a combination of two mechanisms of transcriptional regulation: alternative transcription initiation and alternative promoter. The LGA variant contains both the TSS and the alternative promoter in the first intron of the Gls2 gene, which is located 7 kb away from the canonical promoter of the GAB isoform (Martín-Rufián et al. 2012) (Fig. 6.1). Therefore, three main conclusions can be drawn from these studies: (1) two alternative GLS2 transcripts, GAB and LGA, are expressed in mammalian brain (and liver); (2) GAB is a new GA isozyme and not the human orthologous of the classical rat liver LGA isozyme; and (3) the short transcript variant LGA is not liver specific and is also expressed in brain.
Quantitative measurements of GLS2 transcript variants by real-time qRT-PCR demonstrated that the ratio of the two products varied very widely between tissues and species (Martín-Rufián et al. 2012). Thus, while LGA was slightly more abundant than GAB in mouse brain, the opposite was found in rat brain where GAB mRNA level was fourfold the amount of LGA. Despite this, GLS isoforms (KGA + GAC) showed the greatest abundance in mouse and rat brain, accounting for more than 90 % of total GA transcripts and becoming the predominant GA in mammalian brain (Martín-Rufián et al. 2012). In human brain tissue and cultured neurons and astrocytes, KGA and GAC transcripts showed similar expression levels (Szeliga et al. 2008). In liver, there was also a dramatic change in the relative abundance of GLS2 transcripts, which showed to be absolutely dependent upon the species: GAB was the predominant isoform in mouse, while the short LGA transcript was the most prominent in rat. These strikingly different expression patterns observed for GLS2 transcript variants imply selective transcriptional regulatory mechanisms being operative in distinct tissues and mammalian species.
Alternative mRNA isoforms not always translate into different protein isoforms. In fact, they can be involved in the regulation of translation initiation, RNA editing, and other processes. Therefore, additional experimental evidence at the protein level is required to assess the functionality of GLS and GLS2 mRNA variants. To our knowledge, evidence supporting expression, at the protein level, of the four different GA mRNAs detected in brain tissue (KGA, GAC, GAB, and LGA) have been obtained for all of them except for GAC isozyme. Isoform-specific antibodies against whole proteins, truncated proteins, and peptide sequences were widely used to reveal KGA and GAB isoforms in mammalian brain (Kaneko et al. 1987; Aoki et al. 1991; Laake et al. 1999; Olalla et al. 2002, 2008). The human brain LGA cDNA was in vitro transcribed and translated in a rabbit reticulocyte lysate system and GA activity was consistently detected for the whole LGA translated product. Moreover, immunoblot analysis of rat brain tissue, isolated rat brain mitochondria, and SHSY-5Y human neuroblastoma cells were consistent with co-expression of both GAB and LGA proteins (Martín-Rufián et al. 2012).
6.4.2 Molecular and Kinetic Properties
The subunit composition of rat brain (and kidney) KGA has been characterized : a heterotetrameric enzyme having two different subunits of 66 kDa and 68 kDa with a 3:1 stoichiometry (Haser et al. 1985; Shapiro et al. 1987; Perera et al. 1991). These two GA polypeptides are produced in vivo from a common precursor of 74 kDa (Shapiro et al. 1991; Holcomb et al. 2000) (Table 6.1). On the other hand, the KGA protomer of pig brain has been reported to contain identical subunits of 64 kDa (Svenneby et al. 1973) or 73 kDa (Nimmo and Tipton 1980). The full length human variant GAC cDNA codes for a protein of 598 amino acids with a molecular mass of about 65 kDa (Elgadi et al. 1999) (Table 6.1). Both KGA and GAC isoforms undergo specific and extensive polymerization in the presence of phosphate and phosphate–borate buffers. Thus, the purified native enzyme may appear in three different states : (1) a protomeric and inactive state in Tris–HCl buffer formed by two subunits (dimeric form); (2) an active form by combination of two protomers in the presence of Pi (four subunits, tetrameric form); and (3) a polymeric state with molecular masses higher than 2,000,000 in the presence of phosphate–borate buffer for KGA (Godfrey et al. 1977) or with Pi concentrations as low as 20 mM for GAC (Ferreira et al. 2013). These enzymes require a polyvalent anion for activity. In vitro, inorganic phosphate (Pi) is the most prominent stimulator of GA. Furthermore, there is a correlation between tetramer formation induced by Pi and enzyme activation (Svenneby 1971, 1972; Godfrey et al. 1977; Morehouse and Curthoys 1981). The tetramer–dimer equilibrium is concentration dependent; concretely, for the GAC isoform, it has been shown to occur also in the absence of Pi just by raising the enzyme concentration, which argues in favor of GAC having a greater tendency to oligomerize than KGA: the unique C-terminal sequence of GAC seems to be an important determinant for oligomerization (Cassago et al. 2012).
Purified brain KGA is an allosteric enzyme highly sensitive to changes in the level of Pi (Haser et al. 1985). For the purified enzyme, the activity was completely dependent on added Pi; the phosphate activation curve was sigmoidal with half-maximal activation at concentrations of 13.5 mM (Svenneby 1971) to 25 mM (Haser et al. 1985) and a Hill index of 1.4–1.5. In contrast, rat and pig brain KGA showed hyperbolic kinetics with regard to their dependence on Gln (Haser et al. 1985; Nimmo and Tipton 1981), although other authors have pointed out that this Michaelis–Menten behavior only occurs at pH 8, whereas at higher pH values the plots become concave upward indicating positive cooperativity (Kvamme 1984). Anyway, the K M for Gln was dependent on the Pi concentration : increasing the Pi concentration from 10 mM to 150 mM decreased the K M for Gln from 14 mM to 5 mM (Haser et al. 1985).
GLS isoforms are very sensitive to changes in the levels of Pi, but whether Pi is the true physiological stimulator of GA in brain remains to be determined. However, considering its brain concentration and the fact that it can be rapidly altered during neuronal activity, its candidature as an important physiological regulator of brain GA in vivo was postulated (Erecinska and Silver 1990). For example, in synaptosomes , the Pi activation curve of KGA was very steep and hyperbolic (Bradford and Ward 1976), instead of the sigmoidal curve seen in homogenates and purified enzyme preparations. This distinct kinetic behavior with Pi has been explained by the different intrasynaptosomal Pi concentration compared to the medium (Erecinska and Silver 1990). In addition, synaptosomes treated with KCl and veratridine, to simulate a state of increased neuronal activity in vivo, showed enhanced GA activity concomitantly with the rise in Pi, thereby authors postulated that increase in Pi was the main factor responsible for GA activation under depolarizing conditions (Erecinska et al. 1990).
The concentration of Pi in brain is 1–2 mM (Siesjö 1978; Erecinska and Silver 1989) but a sharp rise might be expected during periods of intense neuronal activation, due to the lowering of the cell energy charge after hydrolysis of high-energy compounds including nucleotide triphosphates . Interestingly, intramitochondrial levels of Pi can significantly increase in a rapid way in cellular models, like cancer cells, characterized by a strong induction of KGA/GAC activity: we found that tumor cell’s mitochondria incubated with 0.5 mM Gln double their Pi concentration one minute after Gln exposure (Medina et al. 1988). Additional support for the candidature of Pi as a relevant in vivo effector of Gls-encoded GAs has recently come from structural data, obtained by X-ray analysis of truncated GAC proteins containing the GA domain (Cassago et al. 2012) (Fig. 6.2). They showed that Pi binds inside the catalytic pocket, resulting in allosteric stabilization of tetramers and facilitating substrate entry by outcompeting with the product, Glu, to guarantee enzyme cycling. This competence of Pi versus Glu for binding to the active site nicely explains the decreased inhibitory effect of Glu in the presence of increasing Pi concentrations (Shapiro et al. 1982). This competitive behavior between Pi and Glu was early postulated from kinetic analysis of KGA in human brain synaptosomes (Svenneby et al. 1986) and rat cortex homogenates (Bradford et al. 1984).
Main effectors of brain GA activity are, in fact, compounds that alter the Pi activation. For example, calcium activates the hydrolysis of Gln in brain synaptosomes (Kvamme et al. 1983), but it seems to affect GA indirectly by modulating the level of free intramitochondrial Pi (Erecinska et al. 1990). In fact, Ca2+ fails to stimulate purified brain KGA (Kvamme et al. 1983), reinforcing the notion that activation is rather an indirect effect. Another effector of possible physiological relevance is H+. The enzyme’s optimum pH is between 8 and 9; therefore, brain metabolism of Gln increases at alkaline pH (Erecinska et al. 1990), while strong reduction in GA activity was noted after lowering the pH of the incubation medium of synaptosomes (Kvamme and Olsen 1981). Of note, during increased neuronal activity the intracellular pH decreases and, hence, pH microdomains are generated in active regions, like dendrites, where these acid shifts are particularly larger (Willoughby and Schwiening 2002). Thus, it is tempting to speculate a decrease on neuronal GA function during periods of intense electrical activity. In contrast, an intracellular alkalinization is observed in mammalian astrocytes in response to repetitive neuronal activity (Chesler and Kraig 1989). Most important, in cultured astrocytes alkalinization induces Glu uptake and promotes a threefold increase in Gln content (Brookes 1997). Therefore, concrete populations of glial cells containing functional mitochondrial GA (see next section) might be activated through astrocytic alkalinization to enhance the production of endogenous Gln-derived Glu. Therefore, pH fluctuations during synaptic activity may also influence GA activity and cerebral function, being a physiologically important process during periods of intense neuronal electrical activity.
In brain, a wide variety of endogenous and exogenous effectors can modulate GA activity in the range of mM concentrations : sulphate, chloride, carboxylic acids, nucleotide triphosphates, riboflavin phosphate, acyl-CoA derivatives and the dye bromothymol blue (Erecinska and Silver 1990; Kvamme et al. 2000). The main inhibitor of brain KGA (and all GLS isoforms) was the first to be discovered: Glu, one product of the reaction (Krebs 1935). The inhibition is competitive (Chiu and Boeker 1979; Haser et al. 1985) and strongly dependent on relative Pi levels : the inhibitory effect of Glu sharply decreases at higher Pi concentration (Svenneby 1971; Shapiro et al. 1982; Campos et al. 1998), in agreement with the active site’s mechanism explained before. The relative concentrations of Gln and Glu in glutamatergic terminals suggest that GA can be strongly inhibited in nerve cells as in their terminals (Kvamme et al. 2000). Furthermore, in vitro and in vivo measurements of GA activity in brain support this notion. Assuming an extracellular Gln concentration of 0.5 mM, the activity of pig brain GA in vivo with 5–8 mM Pi was calculated to be at most 5–10 % of that obtained at saturating concentrations of Gln and Pi (Kvamme 1998). Accordingly, in synaptosomal homogenates the GA rate was found to be 124 nmol/mg per min at 37 °C, whereas in intact synaptosomes it was only 7 nmol/mg per min (Ward and Bradford 1979). This dramatic decrease can be explained by Glu inhibition, since Glu concentration is at least 10 mM in intact nerve endings (Erecinska and Silver 1990) and 10 mM Glu inhibits GA by over 90 % in broken synaptosomes (Bradford et al. 1978). Moreover, a low rate of GA activity (4–5 nmol/mg per min at 28 °C) was calculated from the flux of 15N from 0.5 mM [15N]-Gln in nerve endings incubated at 2 mM Pi in the medium (Yudkoff et al. 1989). Finally, the in vivo GA activity in intact brain of hyperammonemic rats measured by 15N nuclear magnetic resonance (NMR) is about 1 % of the reported in vitro activity shown by rat brain homogenates (Kanamori and Ross 1995). Taking together, these results suggest that GA activity in intact brain is maintained at a low level by a short-term regulation of its key effectors, the most likely ones being Pi, Glu and H+. In conclusion, the situation is considerably more complex in vivo: GA activity will largely depend on the interplay of Gln, Glu, and Pi true concentrations in neurons and glial cells, as well as transient pH fluctuations induced by neural activity.
Conflicting data were reported for ammonium ions , the second end product of GA reaction, as effectors of brain GLS isoforms. In partially purified KGA protein isolated from pig brain (Nimmo and Tipton 1981) and cow brain (Chiu and Boeker 1979) ammonium showed no effect, as well as in cultured astrocytes and neurons (Kvamme et al. 1982; Hogstad et al. 1988). Nonetheless, inhibition of GA was reported in particulate preparations (Bradford and Ward 1976; Wallace and Dawson 1992) and homogenates (Benjamin 1981), while an activation effect was found for the purified pig brain enzyme (Svenneby 1971). In any case, the low concentration of ammonia in the CNS (<0.2 mM) makes unlikely a relevant effect of ammonia on GA activity, at least under normal physiological conditions. Other nonspecific inhibitors are compounds that react with thiol groups as well as Gln analogs like 6-diazo-5-oxonorleucine (DON) (Curthoys and Watford 1995; Campos et al. 1998).
A recently discovered GLS-specific inhibitor is BPTES [bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide] , a small molecule which prevents the formation of large phosphate-induced oligomers (Robinson et al. 2007). The inhibition mechanism was elucidated after structural data were recently obtained from crystallized mammalian GLS isoforms. Protein crystals were grown alone and in combination with Gln, Glu, Pi, and novel inhibitors. BPTES inhibition is achieved through an uncompetitive mechanism interfering with the Pi-induced allosteric activation: the GA domain (Fig. 6.2) of human KGA forms a highly symmetrical tetramer containing two molecules of BPTES that are positioned at the dimer/dimer interfaces; binding of BPTES triggers a major conformational change of the active site residues and also freezes the enzyme in a stable inactive tetramer (Thangavelu et al. 2012). Further, X-ray diffraction data of crystallized human and mouse GAC proteins demonstrated that BPTES inhibits this isoenzyme indeed, locking the GAC tetramer in a non-catalytic state and avoiding formation of larger active polymers (DeLaBarre et al. 2011; Ferreira et al. 2013). Other small molecules which are potent inhibitors of GLS isoenzymes are dibenzophenanthridines; in concrete, the compound 968, a bromo-benzophenanthridinone, is a potent specific inhibitor of the GAC isoform now receiving much attention as anticancer drug (Wang et al. 2010). It is also an allosteric inhibitor, but with a different mechanism to that of BPTES : it preferentially binds to an inactive monomeric state of GAC and prevents it from undergoing the required conformational changes to achieve activation (Stalnecker et al. 2015).
Much less is known with regard to the regulation of GLS2 isoforms in mammalian brain. The classical rat liver LGA isozyme has been characterized at the molecular and kinetic levels. The distinct kinetic behavior of mammalian GLS and GLS2 isozymes has been a hallmark frequently used to distinguish between GA isoforms. The main kinetic differences have been observed in the dependence of the activator Pi, low for GLS2, high for GLS; the relative affinity for the substrate Gln, higher in GLS than in GLS2 isoforms; and the inhibitory effect of Glu, a unique characteristic only reported for GLS isozymes (Kovacevic and McGivan 1983; Curthoys and Watford 1995). Also, rat liver LGA was completely dependent on the presence of ammonia, which is an obligatory activator at physiological concentrations (Verhoeven et al. 1983; Patel and McGivan 1984; Snodgrass and Lund 1984). This activation effect was largely dependent on the pH and ammonia (NH3), rather than ammonium ions, as the activating species (McGivan and Bradford 1983). Rat liver LGA has a subunit molecular mass of 58 kDa (Heini et al. 1987; Smith and Watford 1988) (Table 6.1) and a flat pH curve with an optimum between 7.8 and 8.2.
The only GLS2 isoenzyme expressed in brain that has been characterized in a purified form is the GAB isoform . The recombinant human GAB protein was expressed in Sf9 insect cells (infected with recombinant baculovirus) and affinity purified (Campos-Sandoval et al. 2007). GAB is expressed as a precursor protein of 66 kDa and a mature, processed form, of about 63 kDa, after cleavage of the first 38-39 amino acids at the N-terminal sequence (Table 6.1). Its kinetic characterization demonstrated an allosteric behavior (Hill index of 2.7) with low affinity for Gln (S0.5 values of 32 and 64 mM for high (150 mM) and low (5 mM) Pi, respectively), and low dependence for Pi as expected for a GLS2 isoenzyme. Surprisingly, GAB was inhibited by Glu, a characteristic only shown by GLS isoforms (Table 6.1). The inhibition has an IC50 value of 50 mM at low Pi concentrations (5 mM) and suboptimal Gln concentration (20 mM). These data are in the range of Ki values reported for Glu competitive inhibition of GLS isoenzymes at high Pi concentrations (Shapiro et al. 1982). The Glu inhibition of GAB disappears at high Pi concentrations (up to 100 mM Glu). This was the first report of inhibition by Glu of a purified Gls2-encoded GA isoform; previous studies using partially purified (Patel and McGivan 1984) or pure homogeneous (Smith and Watford 1988) LGA preparations from rat liver, as well as mitochondria isolated from human liver (Snodgrass and Lund 1984), always confirmed that liver LGA is not inhibited by Glu (up to 50 mM).
Another remarkable difference for human GAB was seen in relation with its main activators : it is scarcely activated by ammonia, unlike the liver LGA enzyme which is strictly dependent on ammonia. Partially purified LGA from rat and human liver, as well as LGA in intact and disrupted liver mitochondria, have an absolute requirement for ammonia as an obligatory activator (Häussinger and Sies 1979; Patel and McGivan 1984 and references therein). In fact, the ammonia activation has been considered the particular distinguishing feature of LGA isoforms . In liver, the physiological significance of this effect was considered in the context of feed-forward activation of hepatic GA by ammonia produced in the intestine (Häussinger and Sies 1979); thus, ammonia levels are amplified inside the mitochondria to increase flux through urea cycle via carbamoyl phosphate synthetase I (EC 6.3.4.16) activation (Häussinger 1983). However, as mentioned before, the ammonia levels in brain are kept low under normal circumstances, whereby activation of cerebral GA isozymes by this effector does not seem to play a relevant physiological role.
The existence of GLS2 isoforms with distinctive kinetic properties different from classical K- and L-type isozymes were also noted by other authors, although using homogenates or crude cell extracts instead of purified enzymes. For example, McGivan’s group found mRNAs species identical to human GAB in several colorectal tumors, but without any evidence for the presence of an enzyme with LGA-type kinetics (Turner and McGivan 2003). In conclusion, more studies are needed to further characterize GLS2 protein isoforms in mammalian brain. Tissues like brain, which express both GLS and GLS2 isoforms, are endowed with a powerful system to guarantee biosynthesis of transmitter Glu and glutaminolysis under many different conditions, including a wide range of substrate, activator, and inhibitor concentrations.
Unlike GLS isoforms, relevant structural information from X-ray diffraction data and/or NMR studies at atomic resolution has not yet been published for GLS2 isoforms; hence, the search for specific compounds that block their activity has been hampered. Nonetheless, some selective inhibitors have been recently found for GLS2 isoforms : apomorphine showed greater affinity than BPTES for GA enzymes but with similar activities toward GLS and GLS2 isoforms (Thomas et al. 2013). More recently, a drug discovery screening with natural products discovered a series of alkyl benzoquinones that preferentially inhibit GLS2 isoenzymes and display antitumor activity (Lee et al. 2014).
6.4.3 Regional, Cellular, and Subcellular Locations
One of the main histochemical markers used for identification of excitatory glutamatergic neurons has been the enzyme GA (reviewed by Kaneko 2000), as expected from its role as a main source of transmitter Glu. Nonetheless, Glu also plays important roles in brain general metabolism and it is also a precursor of inhibitory neurotransmitter γ-aminobutyric acid (GABA); for these reasons, immunoreactivity against the amino acid Glu cannot be considered specific for identification of glutamatergic neurons. Traditionally, it was thought that the only cerebral GA isoform was KGA, whereby antibodies against this isoform were the unique tools employed in most immunocytochemical studies dealing with regional, cellular, and subcellular localization of brain GA.
A survey on relevant studies dealing with the immunocytochemical localization of brain KGA (most studies done in rat) indicates expression in cerebral cortex, glutamatergic pyramidal neurons, and GABAergic interneurons (Donoghue et al. 1985; Akiyama et al. 1990; Aoki et al. 1991; Kaneko et al. 1992, 1995; Kaneko and Mizuno 1994, 1996); hippocampus, glutamatergic pyramidal and granular neurons (Altschuler et al. 1985); basal telencephalon, striatum, nucleus accumbens, cholinergic and GABAergic neurons (Aoki et al. 1991; Kaneko and Mizuno 1992; Manns et al. 2001); cerebellum, glutamatergic granular cells (Wenthold et al. 1986; Kaneko et al. 1989; Laake et al. 1999); thalamus, glutamatergic neurons (Kaneko and Mizuno 1996); mesencephalon and rhombencephalon, catecholaminergic and serotonergic neurons, cochlear nucleus (Kaneko et al. 1990; Godfrey et al. 1994); and brainstem, spinal cord, respiratory, and motor neurons (Magnusson et al. 1986; Senba et al. 1991; Yezierski et al. 1993; Turman and Chandler 1994; Pilowsky et al. 1997).
The first reports demonstrating expression of GLS2 isoforms in mammalian brain were published 15 years ago: Northern analysis of GA transcripts in human brain indicated simultaneous expression of GLS and GLS2 mRNAs (Aledo et al. 2000; Gómez-Fabre et al. 2000). Shortly after, co-expression was also demonstrated in brain of other mammalian species such as cow, mouse, rabbit, and rat, while chicken only displayed GLS transcript (Olalla et al. 2002). This pattern of expression was in conflict with that previously reported for the rat (Smith and Watford 1990). The co-expression of both types of GA isoforms was confirmed at the protein level by biochemical and immunological approaches in rat and monkey brain (Olalla et al. 2002) (Fig. 6.3). Both isozymes , KGA and GAB/LGA (GAB antibodies do not discriminate between GLS2 isoforms and would recognize both GAB and LGA proteins), are ubiquitously expressed in brain regions with the strongest signal appearing in cerebral cortex (Olalla et al. 2002). Since Gln serves as an oxidative substrate in most cells, the wide distribution of GA throughout the mammalian brain was not unexpected. Nevertheless, GA was highly concentrated in the cerebral cortex, which is consistent with Glu as a major excitatory neurotransmitter of projection neurons in this area. An excellent correlation between the KGA and GLS2 expression patterns was observed at the regional and cellular level.

Summary of confocal and immuno-EM results for KGA and GLS2 in neurons from mammalian brain. (a) Rat cortical neuronal nuclei (layer II) immunolabeled with anti-GAB isoform-specific antibody; (b) monkey cortical neurons displaying cytoplasmic particulate immunoreaction deposits with anti-KGA antibody; (c) double-labeled neuron showing granular cytoplasmic localization for KGA protein (blue) and nuclear localization for GLS2 protein (brown); (d–f) double-labeling immunofluorescence with anti-mitochondria (red, d), and anti-KGA (green, e) reveals a mitochondrial localization for this isoform (yellow, f); (g–i) double labeling with propidium iodide (red, g) and anti-GAB (green, h) shows that many cells, but not all, express nuclear GLS2 protein (i, white arrows show double-labeled cells). From (d–i), confocal laser scanning microscopy of rat brain sections. BV blood vessel. Scale bars = 20 μm (a), 25 μm (b), 10 μm (c), 40 μm (d–i). Mit mitochondria (Adapted from research originally published by Olalla et al. (2002) in Journal of Biological Chemistry, © the American Society for Biochemistry and Molecular Biology)
Most of the total brain GA activity is found in the crude P2 fraction, which consists of both free mitochondria and synaptosomes (Bradford and Ward 1976; Svenneby et al. 1986) and 40–60 % is retained in purified synaptosomes (Ward and Bradford 1979). GAs have been traditionally considered as mitochondrial enzymes: it was first proposed by Errera and Greenstein (1949) in liver and later shown experimentally (Guha 1961). In brain, the isoenzyme KGA has been convincingly demonstrated to be expressed in mitochondria by immunocytochemistry (Aoki et al. 1991; Laake et al. 1999; Olalla et al. 2002) (Fig. 6.3).
However, the submitochondrial localization of GA has been a very controversial matter and has not been determined conclusively. This issue has profound physiological implications in cerebral metabolism and energy homeostasis . To illustrate the discrepancies, we can say that almost all possible submitochondrial localizations have been proposed for GA, including matrix soluble (Kalra and Brosnan 1974), matrix side of the inner mitochondrial membrane (IMM) (Shapiro et al. 1985), and simultaneous presence in both halves of the IMM existing two populations of GA: one oriented in the intermembrane space (c-side) and the other in the matrix space (m-side) (Kvamme and Olsen 1979). The last authors observed an incomplete blockade of GA activity in rat brain mitochondria by Glu, ammonia and the SH-group reagent N-ethylmaleimide (NEM) (Kvamme and Olsen 1981; Kvamme et al. 1983) and used this evidence as the basis to propose that GA exists in two forms located in separate sites: the NEM-sensitive form was located at the outer surface of the IMM (c-side), whereas the insensitive form was situated at the inner surface (m-side) of the IMM. Pig brain mitochondria was also shown to contain two major forms of GA: a readily extractable soluble enzyme located in the matrix and a membrane-bound enzyme located in the IMM; only this latter enzyme was associated with mitochondria in nerve endings (Nimmo and Tipton 1979).
Nonetheless, other authors have questioned these results (Erecinska and Silver 1990), arguing that almost complete (98 %) inhibition of GA by Glu has been demonstrated in particulate preparations of brain (Krebs 1935; Bradford et al. 1978), as also happens in isolated mitochondria from kidney and tumor cells (Shapiro et al. 1982; Campos et al. 1998). In addition, NEM crosses membranes easily and, hence, it is also hard to explain how this sulfhydryl reagent would not inhibit GA completely wherever located. For example, the inhibition pattern of Ehrlich tumor cell KGA with SH-reagents of different permeability clearly indicated that the IMM becomes a barrier to access to essential KGA residues (Aledo et al. 1997): in particulate fractions, mitochondria and mitoplasts, the membrane-permeant reagent NEM strongly inhibited KGA activity, but almost no effect was detected with membrane-impermeant reagents such as mersalyl and p-chloromercuriphenylsulphonic acid (PCMPS) . However, at the same concentrations, mersalyl and PCMPS, as well as NEM, were very effective inhibitors of the soluble KGA (Aledo et al. 1997).
Later on, Kvamme and associates have suggested predominant c-side localization for the GA isoenzymes from kidney and brain, based on the inactivation by non-permeant SH-reagents and the lack of mixing between the Gln-derived Glu and the endogenous matrix Glu (Kvamme et al. 1991; Roberg et al. 1995). This location would not necessarily require a mitochondrial Gln transport while, at the same time, GA would be under the influence of the cytoplasmic Glu, Pi and Gln concentrations. Most notably, Gln-derived Glu should be imported through the IMM to be engaged in TCA reactions. In sharp contrast, all the KGA activity in kidney and tumor cell mitochondria was found to be localized on the inner surface of the IMM , and most of their functionally sensitive domain was located on the m-side of the IMM, as inferred from convergent enzymatic, immunological and chemical modification studies (Curthoys and Weiss 1974; Shapiro et al. 1985; Aledo et al. 1997).
Although submitochondrial localization of GA might differ from organism to organism, recent data support association with the inner surface of the IMM and, most notably, that the active site of GA must be facing the matrix side. Metabolic studies employing [U-13C]Gln and NMR spectroscopy in cultured neurons have established that almost 50 % of the Gln metabolism involves TCA reactions (Waagepetersen et al. 2005). This result means that part of the transmitter Glu pool could be ascribed to transamination pathways but, at the same time, it is not compatible with most of the GA-derived Glu being generated outside the matrix. Similar findings were obtained using isolated rat brain mitochondria (Bak et al. 2008). In addition, an interesting observation also emerged from this last study: a 50 % decrease in the mitochondrial Glu level was detected in the presence of histidine, an inhibitor of Gln uptake in cerebral mitochondria (Albrecht et al. 2000). The authors interpreted these results as evidence of GA acting from the mitochondrial matrix; actually, Gln transport was a prerequisite for Glu formation and further metabolism in the TCA cycle (Bak et al. 2008). In fact, the location of GA’s active site in the c-side of the IMM, being accessible to the cytosol, poses several questions difficult to ascertain, such as the necessity for very active mitochondrial Gln carriers, as those reported for rat kidney and tumor cells (see Matés et al. 2009 and references therein). In contrast, the generation of Glu into the matrix would facilitate their further metabolism for bioenergetics or anaplerotic purposes .
There was another noteworthy and unexpected finding concerning the subcellular location of GLS2 isoforms in brain: immunocytochemistry in monkey and rat brain revealed that GLS2-type GA immunoreactivity was mostly concentrated in neuronal nuclei (Olalla et al. 2002) (Fig. 6.3). For the first time, an extramitochondrial localization for a mammalian GA was reported, because GA enzymes were considered to be exclusively mitochondrial enzymes (Kovacevic and McGivan 1983; Curthoys and Watford 1995). Nevertheless, GLS2 was not the first neurotransmitter-synthesizing enzyme being located in the cell nucleus. In 1999, the group of Jane Rylett reported the nuclear localization of the 82 kDa form of human choline acetyltransferase [EC 2.3.1.6] (Resendes et al. 1999). We found clear isoform segregation in rat and monkey brain: KGA protein was detected in mitochondria, whereas GLS2-type GA protein was mostly localized in neuronal nuclei, albeit a minor cytoplasmic immunolabeling was also detected (Olalla et al. 2002) (Fig. 6.3). Of note, both isoforms colocalize in numerous cells throughout the brain and many neuronal cells, but not all, expressed nuclear GLS2-type GA protein (Fig. 6.3). Furthermore, the nuclear GLS2 was catalytically active, although showing kinetic characteristics atypical for classical L-type isozymes. As discussed above, the novel GAB isozyme in purified form shows mixed kinetic characteristics of GLS and GLS2-type isoforms (Campos-Sandoval et al. 2007) and seems the most plausible candidate for brain nuclear GA (de la Rosa et al. 2009). It is noteworthy that human functional GAB is targeted to both mitochondria and nucleus in Sf9 cells, and in both locations the protein was catalytically active (Campos-Sandoval et al. 2007), reinforcing the view that this isoform may be targeted to different subcellular locations, including the cell nucleus.
The expression in vivo of functional GA enzymes in astrocytes is also controversial and has been a matter of debate fueled by puzzled results. Primary cultures of astrocytes displayed efficient Gln uptake, strong GA activity (Schousboe et al. 1979; Kvamme et al. 1982), and expression of GA mRNA transcripts (Szeliga et al. 2008). However, these in vitro results were questioned arguing that GA may somehow be induced by the Gln present in the growth media and by the length of culturing (Erecinska and Silver 1990; Borg et al. 1985). On the other hand, contradictory results were obtained for the in situ expression of glial GA in brain tissue: immunohistochemical studies showed expression of KGA protein and GA activity in rat brain astrocytes (Aoki et al. 1991; Würdig and Kugler 1991), but others failed to detect immunoreactivity in non-neuronal elements such as glia and blood vessels (Kaneko et al. 1987; Akiyama et al. 1990; Laake et al. 1999). Therefore, the prevalent and currently accepted view in mammalian brain states that GA is almost exclusively confined in neuronal cells and shows a limited or no expression in glial cells, with only a few exceptions: in vitro cultured astrocytes and activated microglia cells that produce Glu in an autocrine manner through upregulation of GA expression (Takeuchi et al. 2006). Interestingly, previous studies suggested the possible existence of two GA enzyme forms in cultured astrocytes and neurons by the differences observed in kinetic properties of their main effectors (Hogstad et al. 1988).
The occurrence of GA in astrocytes in vivo has not been conclusively proved. Hence, a comprehensive study was devised to elucidate expression of GA in neuroglia and, more concretely, in astrocytes (Cardona et al. 2015) (Fig. 6.4). We demonstrated by in vivo and in vitro approaches that astrocytes express at least two GA isoforms: KGA and GLS2 (GAB/LGA), essentially considered as exclusive neuronal enzymes. Immunocytochemistry in rat and human brain tissues employing isoform-specific antibodies revealed expression of both GLS (KGA) and GLS2 (GAB/LGA) isozymes in glutamatergic and GABAergic neuronal populations as well as in astrocytes. Nevertheless, there was a different and clear subcellular distribution: KGA isoform was always present in mitochondria while GAB/LGA was preferentially detected in both nucleus and mitochondria (Fig. 6.4). Immunocytochemistry in cultured astrocytes confirmed the same pattern previously seen in brain tissue samples (Fig. 6.4). Astrocytic GA expression was also assessed at the mRNA level by real-time qRT-PCR: transcripts of four GA isozymes were detected, but with marked differences on their absolute copy number. The predominance of GLS isoforms over GLS2 transcripts was remarkable (ratio of 144:1). Finally, we proved that astrocytic GA proteins possess enzymatic activity by in situ activity staining: discrete populations of astrocytes were labeled in the cortex, cerebellum, and hippocampus of rat brain, demonstrating functional catalytic activity which can be ascribed to mitochondrial GA. In conclusion, subpopulations of astroglial cells possess GA activity in vivo; they are capable of generating Glu through GA pathway by utilizing Gln either internally generated or produced in neighboring cells or taken from the extracellular fluid (ECF, Fig. 6.5). Nonetheless, available experimental data predict an in vivo GA activity considerably lower in astrocytes as compared with that shown by neurons.

Summary of confocal and immuno-EM microscopy results for KGA and GLS2 in astrocytes from mammalian brain. (a) Double immunofluorescence for KGA (red) and glial fibrillary acidic protein (GFAP) (green) in human brain temporal lobe sections. Punctate KGA immunostaining is clearly seen in the cytoplasm of neurons (open arrows) and astrocytes (white arrows). Asterisks (*) indicate the nuclei of neurons and astrocytes. (b) Immuno-EM detection of KGA in the mitochondria of rat brain astrocytes. KGA labeling was restricted to the mitochondria in astrocytes (immunoreactive mitochondria indicated with white arrows). (c, d) GLS2 in rat brain astrocytes: confocal microscopy and double immunofluorescence for GFAP (green) and GLS2 (red) in rat hippocampus. Merge images show the location of GAB in the nuclei (c, d) and specialized foot processes of perivascular astrocytes (d). Asterisk indicates a blood vessel. (e, f) GA activity in rat brain astrocytes. Double labeling in rat cerebellum sections for GFAP (green, e) and GA activity staining (blue formazan precipitate, f). In vivo GA activity appears as a granular or punctate staining (black arrows) in the cytoplasm of astrocytes (f). Scale bars = 10 μm (a), 0,5 μm (b), 20 μm (c–f) (Adapted from Cardona et al. (2015), GLIA 63:365–82)

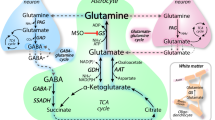
Schematic illustration of the Glu/Gln cycle between neurones and astrocytes at the tripartite synapsis . This is a simplified model highlighting the main functions of GA isozymes, even if not enough evidence exists for a particular task. In the latter case, a question mark means “not enough evidence.” For example, in neuronal nuclei the presence of GLS2 has been demonstrated; however, the nuclear function of this GA has not been fully ascertained. Two alternative nuclear functions appear: regulation of the Gln/Glu levels or transcriptional regulation (arrow pointing toward DNA). The relative contribution of GLS and GLS2 isoforms in the synthesis of the neurotransmitter Glu pool remains to be clarified. In neuronal body, Gln is converted to Glu by mitochondrial KGA and GLS2, whereas the existence of a cytosolic GA, which may contribute to the Glu transmitter pool, has not yet been confirmed. Synaptic Glu is primarily taken up by astrocytes, mostly converted to Gln by GS in the cytosol, and then cycled back to neurones (through N and A carriers) where GA regenerates the transmitter Glu. Exogenous Glu may be transported to glial mitochondria (dashed arrow) and converted to α-KG by GDH or transamination, followed by oxidation into the TCA cycle. Astroglial Gln may be exchanged with blood. Some Gln can be oxidatively degraded by astrocytic GA and TCA cycle after being transported into mitochondria through the uncharacterized MGC; the ammonium generated might be channeled to cytosolic Gln synthesis. Experimental evidence support expression of both KGA and GLS2 isoforms in astrocyte. GLS2 and GIP may interact in vivo and astrocytes are a likely anatomic substrate for their coupling in brain. The colocalization of GIP and GLS2 in perivascular end feet surrounding capillaries might be related to the regulation of the vascular tone through Glu receptors shown in endothelial cells. Caytaxin interacts with KGA and relocalizes it from mitochondria to neurites, while Bmcc1s blocks the targeting of precursor KGA (pKGA) to the mitochondria. The detailed anatomy of synapses, astrocytes, and blood vessels is not portrayed. MGC, Mitochondrial Gln Carrier; ECF, extracellular fluid; GLS2, GAB and/or LGA isoforms; KGA, Gls-encoded long GA isoform; α-KG, α-ketoglutarate
The presence of a non-mitochondrial brain GA in the neuronal cytoplasm has also been suggested by immunocytochemistry studies (Aoki et al. 1991). Nevertheless, the relevance of extramitochondrial GA to Gln/Glu metabolism deserves further investigations and is presently unknown, although interaction of GA with newly identified protein-interacting partners might give rise to transient sporadic appearance of GA in cytosol of neurons and astrocytes (see Fig. 6.5 and Sect. 6.6). On the other hand, it should be emphasized that the existence of a cytosolic GA in astrocytes seems unlikely taking into account the strong inhibition that synaptic Glu may exert. Most important, the simultaneous operation of GA and GS in the cytosol will give rise to a futile cycle with high energy expenditure in the form of ATP.
6.5 Role of GA in Glutamatergic Neurotransmission
The importance of GA in glutamatergic synaptic function has been largely recognized. Physiological, biochemical, immunological, and NMR spectroscopic data indicate that neurotransmitter Glu is mainly generated through GA reaction (Hertz 2004), although the relative contribution of each GA isoform to the transmitter pool is presently unknown. Quantitation of absolute mRNA levels of GA isoforms in total brain reveals a clear predominance of GLS isoforms versus GLS2 transcripts (Martín-Rufián et al. 2012). Although quantitative assessment of individual protein levels of neuronal GA isozymes is unknown, immunocytochemical results seem to support a greater abundance of KGA versus GLS2 isoforms (Olalla et al. 2002; Cardona et al. 2015). A knockout (KO) mice model for the Gls gene has been generated (Masson et al. 2006). Mice lacking the Gls gene die shortly after birth due to altered functioning of key glutamatergic neural networks, stressing the importance of GLS isozymes (mainly KGA) in glutamatergic transmission. Nevertheless, there was persistence of glutamatergic activity in null mutant mice and the quantal size was not reduced in cultured Gls −/− cortical neurons, suggesting that there were adequate levels of intrasynaptic Glu under conditions of basal activity; in fact, synaptic transmission was only impaired under intense stimulation (Masson et al. 2006). These results strongly suggest that alternative sources of vesicular Glu were activated in Gls null mutants: Gls2-encoded GA isoenzymes are the most suitable candidates to maintain releasable Glu stores after induction through a compensatory mechanism. On the other hand, the persistence of glutamatergic transmission in these mice would also be accounted for upregulation of other Glu-synthetic pathways , such as transamination reactions or by direct neuronal Glu reuptake.
The homeostasis of Glu and Gln in brain should be carefully regulated due to the toxic effects elicited by an excess of Glu. The Glu/Gln cycle between neurons and astrocytes is a central pathway for neurotransmitter recycling and to deal with Glu toxicity (Hertz 1979; Berl and Clarke 1983) (Fig. 6.5). The Glu released by neurons is taken up by nearby astrocytes at the synaptic cleft through efficient Glu transport systems and then converted to Gln by glutamine synthetase (GS; EC 6.3.1.2), an enzyme exclusively located in astrocytes (Norenberg and Martínez-Hernández 1979). The Glu-derived Gln is finally exported back to neurons where GA generates neurotransmitter Glu. In vivo 13C-NMR kinetic studies, metabolic modeling and flux analysis to distinguish the Glu/Gln cycle from other sources of isotopic Gln labeling (particularly Gln synthesis by glial anaplerosis), have demonstrated that the Glu/Gln cycle between astrocytes and neurones is a major pathway for neuronal Glu repletion in rat and human cerebral cortex and hippocampus (Shen et al. 1999; Lebon et al. 2002). Furthermore, these studies concluded that the rate of the Glu/Gln cycle is very high and similar in magnitude to the rate of glucose oxidation , supporting the model proposed by Magistretti and coworkers which couples neuronal activity to glucose utilization and where glial glycolytic ATP represents the major source of energy for neurotransmission (Magistretti et al. 1999; Magistretti and Pellerin 1999).
The existence of GA isoforms in astrocytes is of great importance to fully understand the bioenergetics of glial cells and, particularly, to shed light into transmitter recycling and short-term regulation of the Glu/Gln cycle. The mitochondrial location of KGA and GLS2 isoforms in astrocytes may be a control mechanism allowing broad and fine tuning of Glu production depending on their energetic needs and/or synaptic activity. In this context, it is noteworthy the significant increase in mitochondrial GLS2 detected in cultured astrocytes, using a standard culture medium containing Gln, compared with the scarce mitochondrial label observed in rat brain tissue (Cardona et al. 2015). This fact easily illustrates how astrocytes can modulate expression of GA isoforms, depending on the environmental conditions to which they are exposed, to maintain their energy and metabolic homeostasis. Mammalian KGA and GLS2 proteins possess different kinetic and regulatory properties (Table 6.1); therefore, they reach full catalytic activity at different cellular concentrations of Gln, Glu, Pi, and other effectors.
At first sight, recent data demonstrating functional expression of two GA isoforms in astrocytes are in apparent conflict with the compartmentation of the key enzymes in the Glu/Gln cycle (Norenberg and Martínez-Hernández 1979; Donoghue et al. 1985; Erecinska and Silver 1990; Kaneko et al. 1995) and seem hard to reconcile with the constant supply of synaptic Glu being received by these glial cells, which may render superfluous the existence of astrocytic GA. Nonetheless, a GA activity in astrocytes could fit well with the Glu/Gln cycle, because it would endorse astrocytes with an endogenous mitochondrial source of Glu (and ammonium) whose synthesis from Gln does not require energy. This endogenous, non-synaptic, Gln-derived Glu could be converted through glutamate dehydrogenase (GDH) or transamination to α-ketoglutarate and then proceeds via TCA cycle to satisfy astrocytic energetic needs (Fig. 6.5). Catabolism of this endogenous Gln-derived Glu may cooperate with the strong energy and biosynthetic needs required by astrocytes, thus preserving most of the exogenous synaptic Glu to be transformed back to Gln and redirected toward neurons. In this way, astrocytes will not significantly deplete synaptic Glu stores saving most of it for Gln synthesis in the cytosol; such mechanism might be particularly relevant in periods of great synaptic activity. In addition, the ammonium generated in mitochondria by this glutaminolytic process could be channeled to Gln synthesis in cytosol, providing an additional source of nitrogen needed to recycle Glu in Gln through GS (Fig. 6.5).
The Glu/Gln cycle is a key mechanism for homeostatic control of Glu, Gln, and GABA concentrations; however, it is not always a stoichiometric pathway (McKenna 2007) because part of the exogenous Glu taken by astrocytes does not end as Gln but used to fulfill other energy and metabolic purposes instead (Sonnewald et al. 1993). Actually, it has been estimated that 10–30 % of synaptic-derived Glu is oxidized by astrocytes and must be replenished to keep the carbon balance of the Glu/Gln cycle (McKenna et al. 1996; Gamberino et al. 1997; Hutson et al. 1998). In vivo studies, using 13C-NMR spectroscopy , indicated that glial anaplerosis from glucose, through the astrocyte-specific enzyme pyruvate carboxylase (PC; EC 6.4.1.1), is the main contributor for replacing the oxidatively degraded Glu (Rothman et al. 1999; Lebon et al. 2002). In this sense, a GA activity will increase the metabolic versatility of astrocytes in such way that they do not have to exclusively rely on PC for de novo synthesis of Glu. The existence of a functional GA in astrocytes will diminish their anaplerotic pressure , which is important because Glu synthesis by anaplerosis would be detrimental if it is not matched by degradation through cataplerotic routes (Sonnewald 2014). Furthermore, the fast induction of GS protein in active synapse, which rapidly converts the captured synaptic Glu into Gln, means that the flux of Glu to the TCA cycle became negligible as compared with the flux through GS (Fonseca et al. 2005). Actually, these authors conclude that astrocytes adapted to a constant supply of Glu by increasing Glu uptake and GS activity in such a way that Glu was converted almost exclusively through GS. Under these conditions, the supply of endogenous Glu by GA would be a valuable resource for astrocyte bioenergetics.
An additional advantage of the existence of astrocyte GA can be related to the mitochondrial import of Glu. Generation of mitochondrial Glu in situ avoids another problem faced by synaptic cytosolic Glu: its import into astrocytic mitochondria. It has been shown that brain astrocytes do not express the main aspartate–glutamate carrier (Aralar/AGC1) (Ramos et al. 2003; Berckich et al. 2007); therefore, exogenous Glu must enter into astrocytes uniquely by the Glu/hydroxyl carrier. Studies with Aralar KO mice underscored the importance of this carrier for Glu transport in brain and skeletal muscle mitochondria (Jalil et al. 2005), while further evidence has shown that no other Glu carrier can substitute for Aralar in those tissues (Satrústegui et al. 2007 and references therein). Notwithstanding, it should be stated that evidence for anatomic and physical linkages between the astroglial Na+-dependent Glu transporters (GLT-1/EAAT2, GLAST/EAAT1) and mitochondria has been recently reported (Whitelaw and Robinson 2013 and references therein). On the other hand, Gln can be actively concentrated into astrocyte mitochondria using a high-affinity mitochondrial Gln carrier (Roberg et al. 1999) and then be released into the mitochondrial matrix near the GA catalytic site. The generation of Glu into the matrix facilitates its further catabolism for bioenergetics or biosynthetic purposes. Thus, Gln-derived Glu is generated into the mitochondrial matrix without the need for an additional carrier, as required for exogenous Glu.
Finally, it is noteworthy to mention the role of neuronal GA in the synthesis of GABA . The Gln-derived Glu may also contribute to the synthesis of GABA at inhibitory synapses. We have recently found expression of both GLS and GLS2 isoforms in a scarcity of Purkinje cells and in many GABAergic neurons at the molecular layer of the cerebellum, which point toward GA as an important biosynthetic source of Glu at inhibitory synapses indeed (Cardona et al. 2015). Early studies did not detect KGA immunoreactivity in GABAergic neurons of neocortex from rodents and monkey (Altschuler et al. 1984, 1985; Donoghue et al. 1985; Kaneko et al. 1992; Kaneko and Mizuno 1994). However, more recent studies presented immunocytochemical evidence of the localization of KGA in GABAergic neurons in the cat visual cortex (Van der Gucht et al. 2003) and thalamus (Fisher 2007), thereby suggesting that Gln can be a metabolic precursor for GABA synthesis. Interestingly, co-expression of KGA and glutamic acid decarboxylase (GAD; EC 4.1.1.15) was also reported in rat hippocampus and basal forebrain neurons (Kaneko and Mizuno 1994; Manns et al. 2001). GABA is produced by GAD, and two GAD isoforms, encoded by different independently regulated genes, are also expressed in brain (Bu et al. 1992). Both forms can synthesize transmitter GABA, but have different roles in the coding of information by GABA-containing neurones and different subcellular localizations (Soghomonian and Martin 1998). Thus, each of the two main neurotransmitters in the CNS—Glu and GABA—can be synthesized by two isozymes coded by distinct genes, a unique situation different from all the other neurotransmitters. Glu and GABA production must be a process exquisitely regulated to ensure a proper Glu homeostasis. Although the need for several brain GA isoforms is not completely understood, it may represent the biochemical basis to achieve this fine modulation under different physiological circumstances.
6.6 New Functions Beyond Their Role in Glutamatergic Transmission
The mammalian Gls and Gls2 genes are presumed to have been derived by gene duplication of a common ancestral gene, followed by gradual changes in the sequences of both copies (Chung-Bok et al. 1997; Pérez-Gómez et al. 2003). As paralogous proteins, GLS and GLS2 isoforms may acquire different functions during their evolution while maintaining a strong similarity in sequence and three-dimensional structure (Fig. 6.2). Furthermore, GA has been endowed with consensus protein motifs and domains that might support its role as multifunctional proteins (Fig. 6.2). We strongly support this view for GA in mammalian tissues and, with special relevance, for the nuclear localization of GLS2 isoforms in neurons and astrocytes, whose significance remains to be fully determined. The nuclear function has been related with transcriptional regulation associated to differentiation (Olalla et al. 2002; Márquez et al. 2006). First indications about this role for GLS2 isoenzymes came from cancer studies. We postulated a completely different role for GLS and GLS2 isoforms in cancer based on their relative expression patterns in human leukemia, breast cancer cells, and hepatocellular transformation: the data suggest that upregulation of GLS isoforms correlates with increased rates of proliferation, whereas prevalence of the GLS2 isoforms seems to be related with differentiated and quiescent cell states (Pérez-Gómez et al. 2005). Therefore, we proposed that the process of malignant transformation shifts the pattern of GA expression in such way that GLS becomes upregulated while GLS2 is frequently repressed. In accordance with this, human T98G glioma cells showed almost null expression of GLS2, but overexpression of human GAB induced a marked change in the cell’s transcriptome correlated with a reversion of their transformed phenotype (Szeliga et al. 2009, see also chapter 9– in this book). Taking into account its presence in cell’s nuclei, it has been speculated that GAB overexpression may contribute in altering the transcriptional program of glioma cells, yielding a less malignant and more differentiated phenotype, but the concrete molecular mechanisms underpinning this phenotypical change are unknown.
In this regard, important insights into GLS2 function in neuronal differentiation have been recently published (Velletri et al. 2013). The authors show that GLS2 is under the control of TAp73, a p53 family member, during neuronal differentiation of neuroblastoma cells induced by retinoic acid (Table 6.1). Indeed, GLS2 overexpression increased neurite outgrowth. Furthermore, inhibition of GA activity in cultures of mouse embryonary cortical neurons resulted in impaired neuritogenesis. In order to gain insights into the role of GLS2 in vivo, they also analyzed GLS2 expression in developing cerebellum. Interestingly, GLS2 transcript significantly increased between postnatal day 0 (P0) and P8, a development stage characterized by a massive expansion of granule cell progenitors (Velletri et al. 2013). Convergent pieces of evidence were obtained from primary human keratinocytes, where it was shown that GLS2 is a target gene regulated by the transcription factor TAp63, also belonging to the p53 family (Table 6.1). GLS2 and TAp63 expression increases during the in vitro differentiation of keratinocytes, while depletion of GLS2 inhibits skin differentiation both at molecular and cellular levels (Giacobbe et al. 2013). Altogether, these data reinforce our hypothesis relating GLS2 with cell differentiation. With regard to GLS, experimental evidence supporting a key role in the proliferation of neural cells has been reported. Thus, both GLS isoforms (KGA and GAC) were upregulated during neurogenesis of human neural progenitor cells (NPCs) , and their expression pattern positively correlated with the neuronal marker MAP-2 (microtubule-associated protein 2) (Wang et al. 2014). Most important, studies of cultured human NPCs after siRNA silencing of GLS suggest a critical role of GLS isoforms for proliferation and survival of NPCs (Wang et al. 2014).
Another potential nuclear function for GA could be the regulation of Gln/Glu levels (Olalla et al. 2002), taking into account that Gln is a signal molecule involved in gene expression (Bungard and McGivan 2004; Häussinger and Schliess 2007). Therefore, the significance of its nuclear localization could be as simple as being an enzyme controlling in situ the Gln levels in the nucleoplasm and, thus, being indirectly involved in the expression of Gln-regulated genes (Fig. 6.5). Actually, GA activity was found in nuclear fractions isolated from rat brain, although with kinetic properties distinct from the classical Gls2-encoded LGA isoform (Olalla et al. 2002). Interestingly, purified human GAB isozyme showed mixed kinetic characteristics of GLS and GLS2 isoforms (Campos-Sandoval et al. 2007), in accord with the results obtained from nuclear rat brain extracts. Strikingly, no GA activity was revealed in astrocytic nuclei despite the clear immunoreactivity revealed for GLS2 isoforms; however, this result may arise because the NBT-based activity assay was not sensitive enough for low activity levels found in nucleus (Cardona et al. 2015).
In the search for discovering new physiological functions for brain GA, proteomics approaches, such as two-hybrid genetic screenings and immunoprecipitations (IP) coupled to mass spectrometry (MS) analysis, have proven to be very useful strategies for isolation of potential protein-interacting partners of GA. A yeast two-hybrid genetic assay was performed by screening a human brain cDNA library with the C-terminal region of human GAB. Two PDZ (PSD95/Dlg/ZO1) domain-containing proteins were isolated: α-1-syntrophin (SNT) and glutaminase-interacting protein (GIP), also known as tax-interacting binding protein-1 (TIP-1) (Olalla et al. 2001). One of the most common protein modules involved in scaffolding interactions are the PDZ domains, which are responsible for a wide array of protein–protein interactions in the CNS and other tissues (Kornau et al. 1995). These modules bind to short sequences at the extreme C-terminus of target proteins, thereby nucleating the formation of specific functional complexes. The last four amino acids of the C-terminal end of human GAB, ESMV, conforms a consensus sequence required for interaction with PDZ proteins (Fig. 6.2) (Olalla et al. 2001). The relevance of these residues for the human GAB–GIP binding was demonstrated by NMR and fluorescence spectroscopy studies, allowing determination of the dissociation constant (K d) for their binding: 1.66 mM, which means a moderate affinity suitable for regulatory functions (Banerjee et al. 2008).
The physiological relevance of the GLS2–GIP interaction was first assessed by immunocytochemical studies in brain, in order to ascertain whether both proteins may be interacting partners in vivo (Olalla et al. 2008). GIP colocalizes with GLS2 in cerebral cortex astrocytes from rat brain. Double-label studies and confocal microscopy confirmed colocalization in astrocytes cell bodies and processes, including their perivascular end feet (Fig. 6.5). The presence of GIP in astrocytes was also confirmed by immuno-electron microscopy (EM), which revealed immunoreactive astrocytic end feet surrounding perivascular endothelial cells. These results strengthen the view that GA and GIP may interact in vivo and point to astrocytes as a likely anatomic substrate for their coupling in brain (Fig. 6.5).
Analysis of rat brain neurones by EM also found GIP immunoreactivity in the nuclear envelope (Olalla et al. 2008). This nuclear location agrees with previous studies on GIP which described its participation in the regulation of transcription: GIP physically interacts with beta-catenin inhibiting its transcriptional activity, being a critical component of the beta-catenin regulatory network (Kanamori et al. 2003). While human GLS2 has structural determinants needed for mitochondrial targeting (Gómez-Fabre et al. 2000), it does not possess a discernible nuclear localization signal (Fig. 6.2); therefore, the mechanism by which it reaches the nucleus is unclear. The PDZ-protein recognition motif would be implicated in GLS2 specific targeting to selective cellular locations (Olalla et al. 2008; Márquez et al. 2009). Of note, PDZ proteins have also been reported to play an important role in the targeting of protein-interacting partners to concrete subcellular localizations, including cell nucleus (Kausalya et al. 2004), and several PDZ proteins are known to localize to the nucleus (Hsueh et al. 2000). Therefore, we cannot exclude that GIP may be involved in the targeting of GLS2 to neuronal nuclei.
In addition to PDZ domains, human GAB has other sequence motifs and conserved modules that may be essential for its nuclear import (Márquez et al. 2006). For example, it has a consensus motif for interaction with nuclear receptors: the sequence LGDLL on exon 2 (residues 72–76) conform to the consensus nuclear receptor box LXXLL (Fig. 6.2). This sequence motif allows for specific interaction with nuclear receptors and is primarily found in coactivators of nuclear receptors (Heery et al. 1997). Nuclear translocation of a mitochondrial enzyme containing a mitochondrial targeting sequence, but lacking a specific nuclear targeting signal, is not without precedent: mitochondrial 3-hydroxy-3-methylglutarylCoA synthase has been detected in nuclei and its nuclear translocation seems to involve interactions with nuclear hormone receptors through its LXXLL motif (Meertens et al. 1998).
Other protein–protein interaction motifs present in the primary structure of GLS2 proteins (and KGA, but not in the GAC isoform) are two ankyrin repeats in the C-terminal region (Fig. 6.2). Ankyrin repeats are about 33 amino acids long consisting of a β-turn and two antiparallel α-helices and have been found in proteins of diverse function such as transcriptional initiators, cell cycle regulators, cytoskeletal, ion transporters and signal transducers (Sedgwick and Smerdon 1999). The ankyrin-repeat proteins mediate many important protein–protein interactions in virtually all species. Of particular relevance in this context is the existence of transcription factors (GA-binding protein) and transcriptional regulators (IkB protein family, ANCO proteins) whose ankyrin repeats are essential for proper transcriptional activity (Sedgwick and Smerdon 1999; Zhang et al. 2004). Furthermore, ankyrin-repeat motifs are critically required for nuclear localization of signaling enzymes (Hozumi et al. 2003) and transcriptional cofactors (Sedgwick and Smerdon 1999), being able to functionally substitute for a classical nuclear localization signal (Sachdev et al. 1998). Therefore, the involvement of this modular motif in the nuclear import of GLS2 will deserve further attention.
A second PDZ protein isolated as protein-interacting partner of human GAB was SNT. In brain, the PDZ domain of SNT is known to bind to protein kinases, to the neural nitric oxide synthase (nNOS) , and to the water channel aquaporin-4 (AQP4) as a mechanism for selective targeting to unique subcellular sites (Adams et al. 2001). In astrocytes, it has been shown that SNT is a central organizer of the dystrophin complex, an important molecular scaffold for localization of AQP4 at the BBB (Bragg et al. 2006). SNT is the best characterized PDZ scaffold in astrocytes and is concentrated in the perivascular end feet of astrocytes anchoring the AQP4 water channel to these membranes (Neely et al. 2001). The coupling between AQP4 and SNT may be indirect and studies with KO mice for SNT have suggested that additional anchoring proteins (GIP?) may be needed for clustering AQP4 in the end feet of brain astrocytes (Amiry-Moghaddam et al. 2004). At present, the functional role of GIP in the perivascular end feet is unknown and whether GIP or SNT may act as scaffold proteins for GLS2 in astrocytes remains to be determined. Interestingly, GLS2 possesses the consensus C-terminal –SXV sequence, like the potassium channel Kir4.1, required for interaction with the dystrophin complex.
On the other hand, the localization of GIP and GLS2 in astrocytes, particularly in perivascular end feet surrounding blood vessels and capillaries, might be related to the regulation of the vascular tone (Fig. 6.5). Of note, a perivascular and pial localization of GA in rat brain was previously reported (Kaneko and Mizuno 1988). Indeed, we have found GLS2 in concrete perivascular end feet (Cardona et al. 2015), as well as colocalization of GIP and GLS2 in astrocytic processes (Olalla et al. 2008). Thus, it is tempting to speculate that GLS2 targeting to specialized foot processes of concrete perivascular astrocytes might be a plausible mechanism implicated in the regulation of the vascular function, considering the key role of astrocytes in cerebrovascular regulation (Koehler et al. 2006). The fact that Glu is a vasoactive compound (Fergus and Lee 1997) and the existence of Glu receptors in perivascular glia and vascular endothelial cells (Collard et al. 2002; Nedergaard et al. 2002) would implicate GLS2–GIP interaction in regulating the vascular tone (Fig. 6.5).
GLS isoforms can also traffic in brain and unexpected locations have been lately reported for them. The first interacting partner for a GLS protein was shown to be the brain-specific protein BNIP-H (for BNIP-2 homology) or caytaxin , a protein exclusively expressed in neural tissues and encoded by a gene associated with human cerebellar Cayman ataxia (Buschdorf et al. 2006). This protein contains a novel protein–protein interaction domain known as the BNIP-2 and Cdc42GAP homology (BCH) domain. The regional distribution of caytaxin in mouse brain broadly matched the pattern of expression previously known for KGA . In PC12 cells stimulated with nerve growth factor for 24 h to induce differentiation to neuronal phenotype, the co-transfection of KGA and caytaxin induced a specific redistribution of KGA to neurite terminals independently of, and away from, mitochondria (Fig. 6.5); therefore, caytaxin relocalized KGA from cell body to neurite terminals. Neuronal trafficking of KGA was specifically ascribed to its interaction with caytaxin, which also reduces the steady-state levels of Glu by inhibiting KGA activity (Buschdorf et al. 2006). Thus, the interaction KGA–caytaxin seems relevant for regulating the homeostasis of Glu synthesis important for proper neurotransmission and neuronal cell growth (Fig. 6.5). For example, absence of functional caytaxin would increase Glu levels in the cell bodies of neurons leading to neurotoxicity and/or abnormal neuronal growth. In agreement with these findings, a whole-brain proteomic analysis of Gls-deficient mice (Gls −/− and Gls +/−) has revealed a link between GLS isoforms and ATPases, ion channels, nucleic acid and lipid metabolism, calcium, Akt and retinol signaling, cytoskeletal components and protein synthesis machinery, among others. This protein network involves key players of the cellular assembly and organization, cell signaling and cell cycle (Bae et al. 2013). Thus, reduction of GLS expression by genetic knockdown of Gls gene in mice suggests a crucial role of these proteins in neuronal maturation and development.
A second member of the BCH domain-containing family of proteins (related to caytaxin) has been identified as direct interacting partner of KGA isoform. BMCC1 (Bcl2, the adenovirus E1B 19 kDa interacting protein 2 and the Cdc42 GAP homology BCH motif-containing molecule at the carboxy-terminal region 1) is a large polypeptide expressed in brain which encodes several isoforms; one of them, BMCC1s, is a novel short isoform of BMCC1 predominantly expressed in the mouse brain and involved in the regulation of cytoskeleton dynamics and cell survival (Arama et al. 2012). Proteomic analysis of BMCC1s-binding proteins identified KGA as an interacting partner. When primary cultures of cortical neurons were contransfected with both proteins, the subcellular distribution of KGA was drastically modified: almost half of transfected cells lost its unique KGA mitochondrial profile, being also detected in the cytoplasm (Boulay et al. 2013). The authors concluded that BMCC1s preferentially interacts with the 74 kDa cytosolic monomeric precursor of KGA (pKGA) and, thus, it cannot reach the mitochondria for being processed to the mature catalytically active form (Fig. 6.5).
The hypothesis of brain GA isoforms being regulated by protein-interacting partners is appealing because it may provide a mechanism for both control of Glu synthesis and targeting of cerebral GA to concrete cellular compartments . In fact, GIP has been shown to inhibit GLS2-type GA activity in crude extracts of rat liver (Aledo et al. 2001), whereas two binding partners recently discovered for KGA, caytaxin and BMCC1s, were also able to inhibit the catalytic activity of KGA, although by different mechanisms: direct interaction or blocking its path to the mitochondria.
6.7 Summary
Eighty years after its discovery, the knowledge accumulated on brain GA is considerable. Significant progress has been made in this field stressing the relevance of GA, but still a number of sound questions remain to be answered. Based on novel and growing pieces of evidence summarized in this chapter, the roles of GA in cerebral function are multiple and spreading out. Nowadays, the pattern of GA expression has been shown to be considerably more complex: the old concept of only one type of GA expressed in mammalian brain (KGA) was changed at the light of overwhelming evidence demonstrating that, at least, four GA isoenzymes are expressed. The concept of an exclusively mitochondrial location for GA was also demonstrated to be wrong as new subcellular locations (e.g., nucleus, cytosol) have been described for these proteins. Even more, the concept of GA as exclusive neuron-specific enzyme has been challenged by recent findings reporting in vivo expression of functional GA isoforms in astrocytes. The interactome of brain GA is starting to be uncovered. To reach their final destinations in brain cells, GA may interact with newly discovered scaffold proteins. Such arrangements could provide the molecular basis for selective and regulated targeting to concrete cellular locations and control of Glu synthesis by blocking GA activity. These unanticipated findings open a new avenue of research on how GA may affect the homeostasis of Glu/Gln in the tripartite synapsis. Besides their classical role in glutamatergic transmission, novel emerging roles for GA include transcriptional control, neuronal growth and differentiation, and cerebrovascular regulation. Thus, GA may be added to the growing list of moonlighting proteins, that is, proteins having a second (or third) function.
There are several key issues about GA function in brain that remain unresolved. Given our new knowledge about the presence of GA in astrocytes, it is expected that in the next years we can determine the function of GA in glial cells in physiological conditions and whether astrocytic GA contribute to the changes in brain function under pathological conditions. Further studies will undoubtedly reveal insights into the nuclear role of GA; particularly the role of GLS2 and/or Gln in the regulation of gene expression. The physiological relevance of the interaction between GA- and protein-binding partners, as well as their implications in neurological disorders, still remain to be elucidated. We expect that the function of KGA in neurogenesis and neuronal growth and development can be fully addressed in the next future as well. The expression of, at least, four GA in mammalian brain is so far unexplained. The source of neurotransmitter Glu and what is the relative contribution of each GA isoform to the transmitter pool of Glu are outstanding questions for future work, as well as elucidation of the factors which regulate GA activity in vivo, and hence Glu supply, during neuronal stimulation.
Abbreviations
- AGC:
-
Aspartate–glutamate carrier
- BBB:
-
Blood-brain barrier
- EM:
-
Electron microscopy
- GA:
-
Phosphate-activated glutaminase
- GAB:
-
Gls2-encoded long GA isoform
- GAC:
-
Gls-encoded short GA isoform
- GAD:
-
Glutamic acid decarboxylase
- GDH:
-
Glutamate dehydrogenase
- GFAP:
-
Glial fibrillary acidic protein
- GIP:
-
Glutaminase-interacting protein
- GS:
-
Glutamine synthetase
- IMM:
-
Inner mitochondrial membrane
- KGA:
-
Gls-encoded long GA isoform
- LGA:
-
Gls2-encoded short GA isoform
- NBT:
-
Nitroblue tetrazolium
- NMR:
-
Nuclear magnetic resonance
- NPCs:
-
Neural progenitor cells
- PDZ:
-
PSD95/Dlg/ZO1 domains
- TCA:
-
Tricarboxylic acid cycle
References
Adams ME, Mueller HA, Froehner SC (2001) In vivo requirement of the alphasyntrophin PDZ domain for the sarcolemmal localization of nNOS and aquaporin-4. J Cell Biol 155:113–122
Akiyama H, Kaneko T, Mizuno N, McGeer PL (1990) Distribution of phosphate-activated glutaminase in the human cerebral cortex. J Comp Neurol 297:239–252
Albrecht J, Dolinska M, Hilgier W, Lipkowski AW, Nowacki J (2000) Modulation of glutamine uptake and phosphate-activated glutaminase activity in rat brain mitochondria by amino acids and their synthetic analogues. Neurochem Int 36:341–347
Aledo JC, de Pedro E, Gómez-Fabre PM, Núñez de Castro I, Márquez J (1997) Submitochondrial localization and membrane topography of Ehrlich ascitic tumour cell glutaminase. Biochim Biophys Acta 1323:173–184
Aledo JC, Gómez-Fabre PM, Olalla L, Márquez J (2000) Identification of two human glutaminase loci and tissue-specific expression of the two related genes. Mamm Genome 11:1107–1110
Aledo JC, Rosado A, Olalla L, Campos JA, Márquez J (2001) Overexpression, purification, and characterization of glutaminase-interacting protein, a PDZ domain protein from human brain. Protein Expr Purif 23:411–418
Altschuler RA, Wenthold RJ, Schwartz AM, Haser WG, Curthoys NP, Parakkal MH et al (1984) Immunocytochemical localization of glutaminase-like immunoreactivity in the auditory nerve. Brain Res 291:173–178
Altschuler RA, Monaghan DT, Haser WG, Wenthold RJ, Curthoys NP, Cotman CW (1985) Immunocytochemical localization of glutaminase-like and aspartate aminotransferase-like immunoreactivities in the rat and guinea pig hippocampus. Brain Res 330:225–233
Amiry-Moghaddam M, Frydenlund DS, Ottersen OP (2004) Anchoring of aquaporin-4 in brain: molecular mechanisms and implications for the physiology and pathophysiology of water transport. Neuroscience 129:999–1010
Aoki C, Kaneko T, Starr A, Pickel VM (1991) Identification of mitochondrial and non-mitochondrial glutaminase within select neurons and glia of rat forebrain by electron microscopic immunocytochemistry. J Neurosci Res 28:531–548
Arama J, Boulay AC, Bosc C, Delphin C, Loew D, Rostaing P et al (2012) Bmcc1s, a novel brain-isoform of Bmcc1, affects cell morphology by regulating MAP6/STOP functions. PLoS One 7:e35488
Attwell D, Laughlin SB (2001) An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab 21:1133–1145
Bae N, Wang Y, Li L, Rayport S, Lubec G (2013) Network of brain protein level changes in glutaminase deficient fetal mice. J Proteomics 80:236–249
Bak LK, Zieminska E, Waagepetersen HS, Schousboe A, Albrecht J (2008) Metabolism of [U-13C]Glutamine and [U-13C]Glutamate in isolated rat brain mitochondria suggests functional phosphate-activated glutaminase activity in matrix. Neurochem Res 33:273–278
Banerjee M, Huang C, Marquez J, Mohanty S (2008) Probing the structure and function of human glutaminase-interacting protein: a possible target for drug design. Biochemistry 47:9208–9219
Banner C, Hwang JJ, Shapiro RA, Wenthold RJ, Nakatani Y, Lampel KA, Thomas JW, Huie D, Curthoys NP (1988) Isolation of a cDNA for rat brain glutaminase. Brain Res 427:247–254
Benjamin AM (1981) Control of glutaminase activity in rat brain cortex in vitro: influence of glutamate, phosphate, ammonium, calcium and hydrogen ions. Brain Res 208:363–377
Benzinger T, Kitzinger C, Hems R, Burton K (1959) Free-energy changes of the glutaminase reaction and the hydrolysis of the terminal pyrophosphate bond of adenosine triphosphate. Biochem J 71:400–407
Berckich DA, Ola MS, Cole J, Sweatt AJ, Hutson SM (2007) Mitochondrial transport proteins of the brain. J Neurosci Res 85:3367–3377
Berl S, Clarke DD (1983) The metabolic compartmentation concept. In: Hertz L, Kvamme E, McGeer EG, Schousboe A (eds) Glutamine, glutamate and GABA in the central nervous system. Liss, New York, pp 205–217
Borg J, Spitz B, Hamel G, Mark J (1985) Selective culture of neurons from rat cerebral cortex: morphological characterization, glutamate uptake and related enzymes during maturation in various culture media. Brain Res 350:37–49
Boulay AC, Burbassi S, Lorenzo HK, Loew D, Ezan P, Giaume C et al (2013) Bmcc1s interacts with the phosphate-activated glutaminase in the brain. Biochimie 95:799–807
Bradford HF, Ward HK (1976) On glutaminase activity in mammalian synaptosomes. Brain Res 110:115–125
Bradford HF, Ward HK, Thomas AJ (1978) Glutamine: a major substrate for nerve endings. J Neurochem 30:1453–1459
Bradford HF, Ward HK, Sandberg M (1984) Kinetic properties of glutaminase from cerebral cortex. Neurochem Res 9:751–757
Bragg AD, Amiry-Moghaddam M, Ottersen OP, Adams ME, Froehner SC (2006) Assembly of a perivascular astrocyte protein scaffold at the mammalian blood–brain barrier is dependent on alpha-syntrophin. Glia 53:879–890
Braitenberg V, Schüz A (1998) Cortex: statistics and geometry of neuronal connectivity, 2nd edn. Springer, Berlin
Brookes N (1997) Intracellular pH as a regulatory signal in astrocyte metabolism. Glia 21:64–73
Bu DF, Erlander MG, Hitz BC, Tillakaratne NJ, Kaufman DL, Wagner-McPherson CB, Evans GA, Tobin AJ (1992) Two human glutamate decarboxylases, 65-kDa GAD and 67-kDa GAD, are each encoded by a single gene. Proc Natl Acad Sci U S A 89:2115–2119
Bungard CI, McGivan JD (2004) Glutamine availability up-regulates expression of the amino acid transporter protein ASCT2 in HepG2 cells and stimulates the ASCT2 promoter. Biochem J 382:27–32
Buschdorf JP, Chew LL, Zhang B, Cao Q, Liang FY, Liou YC et al (2006) Brain-specific BNIP-2-homology protein Caytaxin relocalises glutaminase to neurite terminals and reduces glutamate levels. J Cell Sci 119:3337–3350
Campos JA, Aledo JC, del Castillo-Olivares A, del Valle AE, Núñez de Castro I, Márquez J (1998) Involvement of essential cysteine and histidine residues in the activity of isolated glutaminase from tumour cells. Biochim Biophys Acta 1429:275–283
Campos-Sandoval JA, López de la Oliva AR, Lobo C, Segura JA, Matés JM, Alonso FJ, Márquez J (2007) Expression of functional human glutaminase in baculovirus system: affinity purification, kinetic and molecular characterization. Int J Biochem Cell Biol 39:765–773
Cardona C, Sánchez-Mejías E, Dávila JC, Martín-Rufián M, Campos-Sandoval JA, Vitorica J et al (2015) Expression of Gls and Gls2 glutaminase isoforms in astrocytes. Glia 63:365–382
Cassago A, Ferreira AP, Ferreira IM, Fornezari C, Gomes ER, Greene KS et al (2012) Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proc Natl Acad Sci U S A 109:1092–1097
Chesler M, Kraig RP (1989) Intracellular pH transients of mammalian astrocytes. J Neurosci 9:2011–2019
Chiu JF, Boeker EA (1979) Cow brain glutaminase: partial purification and mechanism of action. Arch Biochem Biophys 196:493–500
Chung-Bok M-I, Vincent N, Jhala U, Watford M (1997) Rat hepatic glutaminase: identification of the full coding sequence and characterization of a functional promoter. Biochem J 324:193–200
Collard CD, Park KA, Montalto MC, Alapati S, Buras JA, Stahl GL et al (2002) Neutrophil-derived glutamate regulates vascular endothelial barrier function. J Biol Chem 277:14801–14811
Conti F, Minelli A (1994) Glutamate immunoreactivity in rat cerebral cortex is reversibly abolished by 6-diazo-5-oxo-L-norleucine (DON), an inhibitor of phosphate-activated glutaminase. J Histochem Cytochem 42:717–726
Curthoys NP, Watford M (1995) Regulation of glutaminase activity and glutamine metabolism. Annu Rev Nutr 15:133–159
Curthoys NP, Weiss RF (1974) Regulation of renal ammoniagenesis. Subcellular localization of rat kidney glutaminase isoenzymes. J Biol Chem 249:3261–3266
Curthoys NP, Kuhlenschmidt T, Godfrey SS (1976) Regulation of renal ammoniagenesis. Purification and characterization of phosphate-dependent glutaminase from rat kidney. Arch Biochem Biophys 174:82–89
de la Rosa V, Campos-Sandoval JA, Martín-Rufián M, Cardona C, Matés JM, Segura JA, Alonso FJ, Márquez J (2009) A novel glutaminase isoform in mammalian tissues. Neurochem Int 55:76–84
DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S et al (2007) Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A 104:19345–19350
DeLaBarre B, Gross S, Fang C, Gao Y, Jha A, Jiang F, Song JJ, Wei W, Hurov JB (2011) Full-length human glutaminase in complex with an allosteric inhibitor. Biochemistry 50:10764–10770
Donoghue JP, Wenthold RJ, Altschuler RA (1985) Localization of glutaminase-like and aspartate aminotransferase-like immunoreactivity in neurons of cerebral neocortex. J Neurosci 5:2597–2608
Elgadi KM, Meguid RA, Qian M, Souba WW, Abcouwer SF (1999) Cloning and analysis of unique human glutaminase isoforms generated by tissue-specific alternative splicing. Physiol Genomics 1:51–62
Erecinska M, Silver IA (1989) ATP and brain function. J Cereb Blood Flow Metab 9:12–19
Erecinska M, Silver IA (1990) Metabolism and role of glutamate in mammalian brain. Prog Neurobiol 35:245–296
Erecinska M, Zaleska MM, Nelson D, Nissim I, Yudkoff M (1990) Neuronal glutamine utilization: glutamine/glutamate homeostasis in synaptosomes. J Neurochem 54:2057–2069
Errera M, Greenstein JP (1949) Phosphate-activated glutaminase in kidney and other tissues. J Biol Chem 178:495–502
Fergus A, Lee KS (1997) Regulation of cerebral microvessels by glutamatergic mechanisms. Brain Res 754:35–45
Ferreira AP, Cassago A, Gonçalves Kde A, Dias MM, Adamoski D, Ascenção CF et al (2013) Active glutaminase C self-assembles into a supratetrameric oligomer that can be disrupted by an allosteric inhibitor. J Biol Chem 288:28009–28020
Fisher RS (2007) Co-localization of glutamic acid decarboxylase and phosphate-activated glutaminase in neurons of lateral reticular nucleus in feline thalamus. Neurochem Res 32:177–186
Fonnum F (1984) Glutamate: a neurotransmitter in mammalian brain. J Neurochem 42:1–11
Fonseca LL, Monteiro MAR, Alves PM, Carrondo MJT, Santos H (2005) Cultures of rat astrocytes challenged with a steady supply of glutamate: new model to study flux distribution in the glutamate-glutamine cycle. Glia 51:286–296
Gamberino WC, Berkich DA, Lynch CJ, Xu B, LaNoue KF (1997) Role of pyruvate carboxylase in facilitation of synthesis of glutamate and glutamine in cultured astrocytes. J Neurochem 69:2312–2325
Giacobbe A, Bongiorno-Borbone L, Bernassola F, Terrinoni A, Markert EK, Levine AJ et al (2013) p63 regulates glutaminase 2 expression. Cell Cycle 12:1395–1405
Godfrey S, Kuhlenschmidt T, Curthoys P (1977) Correlation between activation and dimer formation of rat renal phosphate-dependent glutaminase. J Biol Chem 252:1927–1931
Godfrey DA, Ross CD, Parli JA, Carlson L (1994) Aspartate aminotransferase and glutaminase activities in rat olfactory bulb and cochlear nucleus; comparisons with retina and with concentrations of substrate and product amino acids. Neurochem Res 19:693–703
Gómez-Fabre PM, Aledo JC, del Castillo-Olivares A, Alonso FJ, Núñez de Castro I, Campos JA, Márquez J (2000) Molecular cloning, sequencing and expression studies of the human breast cancer cell glutaminase. Biochem J 345:365–375
Guha SR (1961) Intercellular localization of glutaminase I in rat liver. Enzymologia 23:94–100
Hamberger AC, Chiang GH, Nylén ES, Scheff SW, Cotman CW (1979) Glutamate as a CNS transmitter. I. Evaluation of glucose and glutamine as precursors for the synthesis of preferentially released glutamate. Brain Res 168:513–530
Haser WG, Shapiro RA, Curthoys NP (1985) Comparison of the phosphate-dependent glutaminase obtained from rat brain and kidney. Biochem J 229:399–408
Häussinger D (1983) Hepatocyte heterogeneity in glutamine and ammonia metabolism and the role of an intercellular glutamine cycle during ureogenesis in perfused rat liver. Eur J Biochem 133:269–275
Häussinger D, Schliess F (2007) Glutamine metabolism and signaling in the liver. Front Biosci 12:371–391
Häussinger D, Sies H (1979) Hepatic glutamine metabolism under the influence of the portal ammonia concentration in the perfused rat liver. Eur J Biochem 101:179–184
Hawkins RA (2009) The blood-brain barrier and glutamate. Am J Clin Nutr 90(Suppl):867S–874S
Heery DM, Kalkhoven E, Hoare S, Parker MG (1997) A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature 387:733–736
Heini HG, Gebhardt R, Brecht A, Mecke D (1987) Purification and characterization of rat liver glutaminase. Eur J Biochem 162:541–546
Hertz L (1979) Functional interactions between neurons and astrocytes I. Turnover and metabolism of putative amino acid transmitters. Prog Neurobiol 13:277–323
Hertz L (2004) Intercellular metabolic compartmentation in the brain: past, present and future. Neurochem Int 45:285–296
Hertz L, Zielke HR (2004) Astrocytic control of glutamatergic activity: astrocytes as stars of the show. Trends Neurosci 27:735–743
Hogstad S, Svenneby G, Torgner IA, Kvamme E, Hertz L, Schousboe A (1988) Glutaminase in neurons and astrocytes cultured from mouse brain: kinetic properties and effects of phosphate, glutamate, and ammonia. Neurochem Res 13:383–388
Holcenberg JS, Borella LD, Camitta BM, Ring BJ (1979) Human pharmacology and toxicology of succinylated Acinetobacter glutaminase-asparaginase. Cancer Res 39:3145–3151
Holcomb T, Taylor L, Trohkimoinen J, Curthoys NP (2000) Isolation, characterization and expression of a human brain mitochondrial glutaminase cDNA. Brain Res Mol Brain Res 76:56–63
Hozumi Y, Ito T, Nakano T, Nakagawa T, Aoyagi M, Kondo H et al (2003) Nuclear localization of diacylglycerol kinase zeta in neurons. Eur J Neurosci 18:1448–1457
Hsueh YP, Wang TF, Yang FC, Sheng M (2000) Nuclear translocation and transcription regulation by the membrane-associated guanylate kinase CASK/LIN-2. Nature 404:298–302
Huang YZ, Knox WE (1976) A comparative study of glytaminase isozymes in rat tissues. Enzyme 21:408–426
Hutson SM, Berkich D, Drown P, Xu B, Aschner M, LaNoue KF (1998) Role of branched-chain aminotransferase isoenzymes and gabapentin in neurotransmitter metabolism. J Neurochem 71:863–874
Jalil MA, Begum L, Contreras L, Pardo B, Iijima M, Li MX et al (2005) Reduced N-acetylaspartate levels in mice lacking aralar, a brain- and muscle-type mitochondrial aspartate-glutamate carrier. J Biol Chem 280:31333–31339
Kalra J, Brosnan JT (1974) The subcellular localization of glutaminase isoenzymes in rat kidney cortex. J Biol Chem 249:3255–3260
Kam K, Nicoll R (2007) Excitatory synaptic transmission persists independently of the glutamate-glutamine cycle. J Neurosci 27:9192–9200
Kanamori K, Ross BD (1995) In vivo activity of glutaminase in the brain of hyperammonaemic rats measured by 15N nuclear magnetic resonance. Biochem J 305:329–336
Kanamori M, Sandy P, Marzinotto S, Benetti R, Kai C, Hayashizaki Y et al (2003) The PDZ protein tax-interacting protein-1 inhibits beta-catenin transcriptional activity and growth of colorectal cancer cells. J Biol Chem 278:38758–38764
Kaneko T (2000) Enzymes responsible for glutamate synthesis and degradation. In: Ottersen OP, Storm-Mathisen J (eds) Handbook of chemical neuroanatomy. Elsevier, Amsterdam, pp 203–230
Kaneko T, Mizuno N (1988) Immunohistochemical study of glutaminase-containing neurons in the cerebral cortex and thalamus of the rat. J Comp Neurol 267:590–602
Kaneko T, Mizuno N (1992) Mosaic distribution of phosphate-activated glutaminase-like immunoreactivity in the rat striatum. Neuroscience 49:329–345
Kaneko T, Mizuno N (1994) Glutamate-synthesizing enzymes in GABAergic neurons of the neocortex: a double immunofluorescence study in the rat. Neuroscience 61:839–849
Kaneko T, Mizuno N (1996) Spiny stellate neurons in layer VI of the rat cerebral cortex. Neuroreport 7:2331–2335
Kaneko T, Urade Y, Watanabe Y, Mizuno N (1987) Production, characterization, and immunohistochemical application of monoclonal antibodies to glutaminase purified from rat brain. J Neurosci 7:302–309
Kaneko T, Itoh K, Shigemoto R, Mizuno N (1989) Glutaminase-like immunoreactivity in the lower brainstem and cerebellum of the adult rat. Neuroscience 32:79–98
Kaneko T, Akiyama H, Nagatsu I, Mizuno N (1990) Immunohistochemical demonstration of glutaminase in catecholaminergic and serotoninergic neurons of rat brain. Brain Res 507:151–154
Kaneko T, Nakaya Y, Mizuno N (1992) Paucity of glutaminase-immunoreactive nonpyramidal neurons in the rat cerebral cortex. J Comp Neurol 322:181–190
Kaneko T, Kang Y, Mizuno N (1995) Glutaminase-positive and glutaminase-negative pyramidal cells in layer VI of the primary motor and somatosensory cortices: a combined analysis by intracellular staining and immunocytochemistry in the rat. J Neurosci 15:8362–8377
Kausalya PJ, Phua DC, Hunziker W (2004) Association of ARVCF with zonula occludens (ZO)-1 and ZO-2: binding to PDZ-domain proteins and cell-cell adhesion regulate plasma membrane and nuclear localization of ARVCF. Mol Biol Cell 15:5503–5515
Koehler RC, Gebremedhin D, Harder DR (2006) Role of astrocytes in cerebrovascular regulation. J Appl Physiol 100:307–317
Kornau HC, Schenker LT, Kennedy MB, Seeburg PH (1995) Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science 269:1737–1740
Kovacevic Z, McGivan JD (1983) Mitochondrial metabolism of glutamine and glutamate and its physiological significance. Physiol Rev 63:547–605
Krebs HA (1935) The synthesis of glutamine from glutamate and ammonia, and the enzymatic hydrolysis of glutamine in animal tissues. Biochem J 29:1951–1969
Kvamme E (1984) Enzymes of cerebral glutamine metabolism. In: Häussinger D, Sies H (eds) Glutamine metabolism in mammalian tissues. Springer, Berlin, pp 32–48
Kvamme E (1998) Synthesis of glutamate and its regulation. Prog Brain Res 116:73–85
Kvamme E, Olsen BE (1979) Evidence for two species of mammalian phosphate-activated glutaminase having different regulatory properties. FEBS Lett 107:33–36
Kvamme E, Olsen BE (1981) Evidence for compartmentation of synaptosomal phosphate-activated glutaminase. J Neurochem 36:1916–1923
Kvamme E, Tveit B, Svenneby G (1970) Glutaminase from pig renal cortex. I. Purification and general properties. J Biol Chem 245:1871–1877
Kvamme E, Svenneby G, Hertz L, Schousboe A (1982) Properties of phosphate activated glutaminase in astrocytes cultured from mouse brain. Neurochem Res 7:761–770
Kvamme E, Svenneby G, Torgner IA (1983) Calcium stimulation of glutamine hydrolysis in synaptosomes from rat brain. Neurochem Res 8:25–38
Kvamme E, Torgner IA, Roberg B (1991) Evidence indicating that pig renal phosphate-activated glutaminase has a functionally predominant external localization in the inner mitochondrial membrane. J Biol Chem 266:13185–13192
Kvamme E, Roberg B, Torgner IA (2000) Phosphate-activated glutaminase and mitochondrial glutamine transport in the brain. Neurochem Res 25:1407–1419
Laake JH, Slyngstad TA, Haug FM, Ottersen OP (1995) Glutamine from glial cells is essential for the maintenance of the nerve terminal pool of glutamate: immunogold evidence from hippocampal slice cultures. J Neurochem 65:871–881
Laake JH, Takumi Y, Eidet J, Torgner IA, Roberg B, Kvamme E, Ottersen OP (1999) Postembedding immunogold labelling reveals subcellular localization and pathway-specific enrichment of phosphate activated glutaminase in rat cerebellum. Neuroscience 88:1137–1151
Lebon V, Petersen KF, Cline GW, Shen J, Mason GF, Dufour S et al (2002) Astroglial contribution to brain energy metabolism in humans revealed by 13C nuclear magnetic resonance spectroscopy: elucidation of the dominant pathway for neurotransmitter glutamate repletion and measurement of astrocytic oxidative metabolism. J Neurosci 22:1523–1531
Lee Y-Z, Yang C-W, Chang HY, Hsu H-Y, Chen I-S, Chang H-S et al (2014) Discovery of selective inhibitors of Glutaminase-2, which inhibit mTORC1, activate autophagy and inhibit proliferation in cancer cells. Oncotarget 5:6087–6101
Lohmann R, Souba WW, Bode BP (1999) Rat liver endothelial cell glutamine transporter and glutaminase expression contrast with parenchymal cells. Am J Physiol 276:G743–G750
Magistretti PJ, Pellerin L (1999) Astrocytes couple synaptic activity to glucose utilization in the brain. News Physiol Sci 14:177–182
Magistretti PJ, Pellerin L, Rothman DL, Shulman RG (1999) Energy on demand. Science 283:496–497
Magnusson KR, Larson AA, Madl JE, Altschuler RA, Beitz AJ (1986) Co-localization of fixative-modified glutamate and glutaminase in neurons of the spinal trigeminal nucleus of the rat: an immunohistochemical and immunoradiochemical analysis. J Comp Neurol 247:477–490
Manns ID, Mainville L, Jones BE (2001) Evidence for glutamate, in addition to acetylcholine and GABA, neurotransmitter synthesis in basal forebrain neurons projecting to the entorhinal cortex. Neuroscience 107:249–263
Márquez J, López de la Oliva AR, Matés JM, Segura JA, Alonso FJ (2006) Glutaminase: a multifaceted protein not only involved in generating glutamate. Neurochem Int 48:465–471
Márquez J, Tosina M, de la Rosa V, Segura JA, Alonso FJ, Matés JM et al (2009) New insights into brain glutaminases: beyond their role on glutamatergic transmission. Neurochem Int 55:64–70
Márquez J, Matés JM, Alonso FJ, Martín-Rufián M, Lobo C, Campos-Sandoval JA (2015) Canceromics studies unravel tumor’s glutamine addiction after metabolic reprogramming. In: Mazurek S, Shoshan M (eds) Tumor cell metabolism. Pathways, regulation and biology. Springer, Wien, pp 257–287
Martín-Rufián M, Tosina M, Campos-Sandoval JA, Manzanares E, Lobo C, Segura JA et al (2012) Mammalian glutaminase Gls2 gene encodes two functional alternative transcripts by a surrogate promoter usage mechanism. PLoS One 7, e38380
Masson J, Darmon M, Conjard A, Chuhma N, Ropert N, Thoby-Brisson M et al (2006) Mice lacking brain/kidney phosphate-activated glutaminase have impaired glutamatergic synaptic transmission, altered breathing, disorganized goal-directed behavior and die shortly after birth. J Neurosci 26:4660–4671
Matés JM, Segura JA, Campos-Sandoval JA, Lobo C, Alonso L, Alonso FJ, Márquez J (2009) Glutamine homeostasis and mitochondrial dynamics. Int J Biochem Cell Biol 41:2051–2061
Matés JM, Segura JA, Martín-Rufián M, Campos-Sandoval JA, Alonso FJ, Márquez J (2013) Glutaminase isoenzymes as key regulators in metabolic and oxidative stress against cancer. Curr Mol Med 13:514–534
McGivan JD, Bradford NM (1983) Characteristics of the activation of glutaminase by ammonia in sonicated rat liver mitochondria. Biochim Biophys Acta 759:296–302
McKenna MC (2007) The glutamate-glutamine cycle is not stoichiometric: fates of glutamate in brain. J Neurosci Res 85:3347–3358
McKenna MC, Sonnewald U, Huang X, Hopkins IB (1996) Exogenous glutamate concentration regulates the metabolic fate of glutamate in astrocytes. J Neurochem 66:386–393
Medina MA, Quesada AR, Márquez FJ, Sánchez-Jiménez F, Núñez de Castro I (1988) Inorganic phosphate and energy charge compartmentation in Ehrlich ascites tumor cells in the presence of glucose and/or glutamine. Biochem Int 16:713–718
Meertens LM, Miyata KS, Cechetto JD, Rachubinski RA, Capone JP (1998) A mitochondrial ketogenic enzyme regulates its gene expression by association with the nuclear hormone receptor PPARα. EMBO J 17:6972–6978
Mock B, Kozak C, Seldin MF, Ruff N, D’Hoostelaere L, Szpirer C, Levan G, Seuanez H, O’Brien S, Banner C (1989) A glutaminase (gls) gene maps to mouse chromosome 1, rat chromosome 9, and human chromosome 2. Genomics 5:291–297
Morehouse RF, Curthoys NP (1981) Properties of rat renal phosphate-dependent glutaminase coupled to Sepharose. Evidence that dimerization is essential for activation. Biochem J 193:709–716
Nagase T, Ishikawa K, Suyama M, Kikuno R, Hirosawa M, Miyajima N et al (1998) Prediction of the coding sequences of unidentified human genes. XII. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res 5:355–364
Nedergaard M, Takano T, Hansen AJ (2002) Beyond the role of glutamate as a neurotransmitter. Nat Rev Neurosci 3:748–755
Neely JD, Amiry-Moghaddam M, Ottersen OP, Froehner SC, Agre P, Adams ME (2001) Syntrophin-dependent expression and localization of aquaporin-4 water channel protein. Proc Natl Acad Sci U S A 98:14108–14113
Nicklas WJ, Zeevalk G, Hyndman A (1987) Interactions between neurons and glia in glutamate/glutamine compartmentation. Biochem Soc Trans 15:208–210
Nimmo GA, Tipton KF (1979) The distribution of soluble and membrane-bound forms of glutaminase in pig brain. J Neurochem 33:1083–1084
Nimmo GA, Tipton KF (1980) Purification of soluble glutaminase from pig brain. Biochem Pharmacol 29:359–367
Nimmo GA, Tipton KF (1981) Kinetic comparisons between soluble and membrane-bound glutaminase preparations from pig brain. Eur J Biochem 117:57–64
Norenberg MD, Martínez-Hernández A (1979) Fine structural localization of glutamine synthetase in astrocytes of rat brain. Brain Res 161:303–310
Olalla L, Aledo JC, Bannenberg G, Márquez J (2001) The C-terminus of human glutaminase L mediates association with PDZ domain-containing proteins. FEBS Lett 488:116–122
Olalla L, Gutiérrez A, Campos JA, Khan ZU, Alonso F, Segura JA, Márquez J, Aledo JC (2002) Nuclear localization of L-glutaminase in mammalian brain. J Biol Chem 277:38939–38944
Olalla L, Gutiérrez A, Jiménez AJ, López-Téllez JF, Khan ZU, Pérez J, Alonso FJ, de la Rosa V, Campos-Sandoval JA, Segura JA, Aledo JC, Márquez J (2008) Expression of scaffolding PDZ protein GIP (glutaminase-interacting-protein) in mammalian brain. J Neurosci Res 86:281–292
Patel M, McGivan JD (1984) Partial purification and properties of rat liver glutaminase. Biochem J 220:583–590
Perera SY, Voith DM, Curthoys NP (1991) Biosynthesis and processing of mitochondrial glutaminase in HTC hepatoma cells. Biochem J 273:265–270
Pérez-Gómez C, Matés JM, Gómez-Fabre PM, del Castillo-Olivares A, Alonso FJ, Márquez J (2003) Genomic organization and transcriptional analysis of the human L-glutaminase gene. Biochem J 370:771–784
Pérez-Gómez C, Campos-Sandoval JA, Alonso FJ, Segura JA, Manzanares E, Ruiz-Sánchez P, González ME, Márquez J, Matés JM (2005) Co-expression of glutaminase K and L isoenzymes in human tumour cells. Biochem J 386:535–542
Pilowsky P, Sun QJ, Llewellyn-Smith I, Arnolda L, Chalmers J, Minson J (1997) Phosphate-activated glutaminase immunoreactivity in brainstem respiratory neurons. J Auton Nerv Syst 63:85–90
Porter LD, Ibrahim H, Taylor L, Curthoys NP (2002) Complexity and species variation of the kidney-type glutaminase gene. Physiol Genomics 9:57–66
Quesada AR, Sánchez-Jiménez F, Pérez-Rodríguez J, Márquez J, Medina MA, Núñez de Castro I (1988) Purification of phosphate-dependent glutaminase from isolated mitochondria of Ehrlich ascites-tumour cells. Biochem J 255:1031–1035
Ramos M, del Arco A, Pardo B, Martínez-Serrano A, Martínez-Morales JR, Kobayashi K et al (2003) Developmental changes in the Ca2+-regulated mitochondrial aspartate–glutamate carrier aralar1 in brain and prominent expression in the spinal cord. Dev Brain Res 143:33–46
Resendes MC, Dobransky T, Ferguson SS, Rylett RJ (1999) Nuclear localization of the 82-kDa form of human choline acetyltransferase. J Biol Chem 274:19417–19421
Roberg B, Torgner IA, Kvamme E (1995) The orientation of phosphate activated glutaminase in the inner mitochondrial membrane of synaptic and non-synaptic rat brain mitochondria. Neurochem Int 27:367–376
Roberg B, Torgner IA, Kvamme E (1999) Inhibition of glutamine transport in rat brain mitochondria by some amino acids and tricarboxylic acid cycle intermediates. Neurochem Res 24:811–816
Robinson MM, McBryant SJ, Tsukamoto T, Rojas C, Ferraris DV, Hamilton SK et al (2007) Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem J 406:407–414
Rothman DL, Sibson NR, Hyder F, Shen J, Behar KL, Shulman RG (1999) In vivo nuclear magnetic resonance spectroscopy studies of the relationship between the glutamate-glutamine neurotransmitter cycle and functional neuroenergetics. Phil Trans R Soc Lond B 354:1165–1177
Sachdev S, Hoffmann A, Hannink M (1998) Nuclear localization of IkappaB alpha is mediated by the second ankyrin repeat: the IkappaB alpha ankyrin repeats define a novel class of cis-acting nuclear import sequences. Mol Cell Biol 18:2524–2534
Satrústegui J, Contreras L, Ramos M, Marmol P, del Arco A, Saheki T et al (2007) Role of aralar, the mitochondrial transporter of aspartate–glutamate, in brain N-acetylaspartate formation and Ca2+ signaling in neuronal mitochondria. J Neurosci Res 85:3359–3366
Schalk AM, Nguyen HA, Rigouin C, Lavie A (2014) Identification and structural analysis of an L-asparaginase enzyme from guinea pig with putative tumor cell killing properties. J Biol Chem 289:33175–33186
Schousboe A, Hertz L, Svenneby G, Kvamme E (1979) Phosphate activated glutaminase activity and glutamine uptake in primary cultures of astrocytes. J Neurochem 32:943–950
Sedgwick SG, Smerdon SJ (1999) The ankyrin repeat: a diversity of interactions on a common structural framework. Trends Biochem Sci 24:311–316
Segura JA, Aledo JC, Gómez-Biedma S, Núñez de Castro I, Márquez J (1995) Tumor glutaminase purification. Protein Expr Purif 6:343–351
Senba E, Kaneko T, Mizuno N, Tohyama M (1991) Somato-, branchio- and viscero-motor neurons contain glutaminase-like immunoreactivity. Brain Res Bull 26:85–97
Shapiro RA, Morehouse RF, Curthoys NP (1982) Inhibition by glutamate of phosphate-dependent glutaminase of rat kidney. Biochem J 207:561–566
Shapiro RA, Haser WG, Curthoys NP (1985) The orientation of phosphate-dependent glutaminase on the inner membrane of rat renal mitochondria. Arch Biochem Biophys 243:1–7
Shapiro RA, Haser WG, Curthoys NP (1987) Immunoblot analysis of glutaminase peptides in intact and solubilized mitochondria isolated from various rat tissues. Biochem J 242:743–747
Shapiro RA, Farrell L, Srinivasan M, Curthoys NP (1991) Isolation, characterization, and in vitro expression of a cDNA that encodes the kidney isoenzyme of the mitochondrial glutaminase. J Biol Chem 266:18792–18796
Shen J, Petersen KF, Behar KL, Brown P, Nixon TW, Mason GF, Petroff OA, Shulman GI, Shulman RG, Rothman DL (1999) Determination of the rate of the glutamate/glutamine cycle in the human brain by in vivo 13C NMR. Proc Natl Acad Sci U S A 96:8235–8240
Siesjö BK (1978) Brain energy metabolism. Wiley, New York
Smith EM, Watford M (1988) Rat hepatic glutaminase: purification and immunochemical characterization. Arch Biochem Biophys 260:740–751
Smith EM, Watford M (1990) Molecular cloning of a cDNA for rat hepatic glutaminase. Sequence similarity to kidney-type glutaminase. J Biol Chem 265:10631–10636
Snodgrass PJ, Lund P (1984) Allosteric properties of phosphate-activated glutaminase of human liver mitochondria. Biochim Biophys Acta 798:21–27
Soghomonian JJ, Martin DL (1998) Two isoforms of glutamate decarboxylase: why? Trends Pharmacol Sci 19:500–505
Sonnewald U (2014) Glutamate synthesis has to be matched by its degradation—where do all the carbons go? J Neurochem 131:399–406
Sonnewald U, Westergaard N, Petersen SB, Unsgard G, Schousboe A (1993) Metabolism of [U-13C]glutamate in astrocytes studied by 13C NMR spectroscopy: incorporation of more label into lactate than into glutamine demonstrates the importance of the tricarboxylic acid cycle. J Neurochem 61:1179–1182
Stalnecker CA, Ulrich SM, Li Y, Ramachandran S, McBrayer MK, DeBerardinis RJ, Cerione RA et al (2015) Mechanism by which a recently discovered allosteric inhibitor blocks glutamine metabolism in transformed cells. Proc Natl Acad Sci U S A 112:394–399
Svenneby G (1971) Activation of pig brain glutaminase. J Neurochem 18:2201–2208
Svenneby G (1972) Time- and temperature-dependent activation of pig brain glutaminase. J Neurochem 19:165–174
Svenneby G, Torgner I, Kvamme E (1973) Purification of phosphate-dependent pig brain glutaminase. J Neurochem 20:1217–1224
Svenneby G, Roberg B, Hogstad S, Torgner IAA, Kvamme E (1986) Phosphate-activated glutaminase in the crude mitochondrial fraction (P2 fraction) from human brain cortex. J Neurochem 47:1351–1355
Szeliga M, Matyja E, Obara M, Grajkowska W, Czernicki T, Albrecht J (2008) Relative expression of mRNAs coding for glutaminase isoforms in CNS tissues and CNS tumors. Neurochem Res 33:808–813
Szeliga M, Obara-Michlewska M, Matyja E, Lazarczyk M, Lobo C, Hilgier W et al (2009) Transfection with liver-type glutaminase (LGA) cDNA alteres gene expression and reduces viability, migration and proliferation of T98G glioma cells. Glia 57:1014–1023
Takeda K, Ishida A, Takahashi K, Ueda T (2012) Synaptic vesicles are capable of synthesizing the VGLUT substrate glutamate from α-ketoglutarate for vesicular loading. J Neurochem 121:184–196
Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, Kuno R et al (2006) Tumor necrosis factor-a induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem 281:21362–21368
Thangavelu K, Pan CQ, Karlberg T, Balaji G, Uttamchandani M, Suresh V et al (2012) Structural basis for the allosteric inhibitory mechanism of human kidney-type glutaminase (KGA) and its regulation by Raf-Mek-Erk signaling in cancer cell metabolism. Proc Natl Acad Sci U S A 109:7705–7710
Thanki CM, Sugden D, Thomas AJ, Bradford HF (1983) In vivo release from cerebral cortex of [14C]glutamate synthesized from [U-14C]glutamine. J Neurochem 41:611–617
Thomas AG, Rojas C, Tanega C, Shen M, Simeonov A, Boxer MB et al (2013) Kinetic characterization of ebselen, chelerythrine and apomorphine as glutaminase inhibitors. Biochem Biophys Res Commun 438:243–248
Turman JE Jr, Chandler SH (1994) Immunohistochemical localization of glutamate and glutaminase in guinea pig trigeminal premotoneurons. Brain Res 634:49–61
Turner A, McGivan JD (2003) Glutaminase isoform expression in cell lines derived from human colorectal adenomas and carcinomas. Biochem J 370:403–408
Van der Gucht E, Jacobs S, Kaneko T, Vandesande F, Arckens L (2003) Distribution and morphological characterization of phosphate-activated glutaminase-immunoreactive neurons in cat visual cortex. Brain Res 988:29–42
Velletri T, Romeo F, Tucci P, Peschiaroli A, Annicchiarico-Petruzzelli M, Niklison-Chirou MV et al (2013) GLS2 is transcriptionally regulated by p73 and contributes to neuronal differentiation. Cell Cycle 12:1–10
Verhoeven AJ, van Iwaarden JF, Joseph SK, Meijer AJ (1983) Control of rat-liver glutaminase by ammonia and pH. Eur J Biochem 133:241–244
Waagepetersen HS, Qu H, Sonnewald U, Shimamoto K, Schousboe A (2005) Role of glutamine and neuronal glutamate uptake in glutamate homeostasis and synthesis during vesicular release in cultured glutamatergic neurons. Neurochem Int 47:92–102
Wallace DR, Dawson R Jr (1992) Ammonia regulation of phosphate-activated glutaminase displays regional variation and impairment in the brain of aged rats. Neurochem Res 17:1113–1122
Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R et al (2010) Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 18:207–219
Wang Y, Huang Y, Zhao L, Li Y, Zheng J (2014) Glutaminase 1 is essential for the differentiation, proliferation, and survival of human neural progenitor cells. Stem Cells Dev 23:2782–2790
Ward HK, Bradford HF (1979) Relative activities of glutamine synthetase and glutaminase in mammalian synaptosomes. J Neurochem 33:339–342
Wenthold RJ, Skaggs KK, Altschuler RA (1986) Immunocytochemical localization of aspartate aminotransferase and glutaminase immunoreactivities in the cerebellum. Brain Res 363:371–375
Whitelaw BS, Robinson MB (2013) Inhibitors of glutamate dehydrogenase block sodium-dependent glutamate uptake in rat brain membranes. Front Endocrinol 4:123
Willoughby D, Schwiening CJ (2002) Electrically evoked dendritic pH transients in rat cerebellar Purkinje cells. J Physiol 544:487–499
Würdig S, Kugler P (1991) Histochemistry of glutamate metabolizing enzymes in the rat cerebellar cortex. Neurosci Lett 130:165–168
Yamaguchi S, Jeenes DJ, Archer DB (2001) Protein-glutaminase from Chryseobacterium proteolyticum, an enzyme that deamidates glutaminyl residues in proteins. Purification, characterization and gene cloning. Eur J Biochem 268:1410–1421
Yezierski RP, Kaneko T, Miller KE (1993) Glutaminase-like immunoreactivity in rat spinomesencephalic tract cells. Brain Res 624:304–308
Yudkoff M, Zaleska MM, Nissim I, Nelson D, Erecinska M (1989) Neuronal glutamine utilization: pathways of nitrogen transfer studied with [15N]glutamine. J Neurochem 53:632–640
Zhang A, Yeung PL, Li CW, Tsai SC, Dinh GK, Wu X et al (2004) Identification of a novel family of ankyrin repeats containing cofactors for p160 nuclear receptor coactivators. J Biol Chem 279:33799–33805
Acknowledgements
We are grateful to everybody in the Canceromics group for creating most of the own work described in this chapter, particularly to our colleagues F.J. Alonso, J.A. Segura, M. Martín-Rufián, C. Cardona, C. Lobo, A. Peñalver, and former members L. Olalla, P. Gómez-Fabre, C. Pérez-Gómez, and J.C. Aledo. The excellent collaboration of Dr. Antonia Gutiérrez and her team is also gratefully acknowledged. We are in debt with Dr. Alfonso Gutiérrez-Adán, Dr. Fernando Rodríguez de Fonseca, and Dr. Emilio Ambrosio for helpful discussions and collaborations with animal models. This work was supported by Excellence Grant CVI-6656 (Regional Andalusian government) and Grant RD12/0028/0013 of the RTA RETICS network from the Spanish Health Institute Carlos III. Thanks are due to Mrs. Kristina Pačesaitė for the design of the graphic work and to Mrs. Laura Castilla for excellent technical work.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Márquez, J., Matés, J.M., Campos-Sandoval, J.A. (2016). Glutaminases. In: Schousboe, A., Sonnewald, U. (eds) The Glutamate/GABA-Glutamine Cycle. Advances in Neurobiology, vol 13. Springer, Cham. https://doi.org/10.1007/978-3-319-45096-4_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-45096-4_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-45094-0
Online ISBN: 978-3-319-45096-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)