
Abstract
The present study was designed to evaluate the potential role of miR-93 in cerebral ischemic/reperfusion (I/R) injury in mice. The stroke model was produced in C57BL/6 J mice via middle cerebral artery occlusion (MCAO) for 1 h followed by reperfusion. And miR-93 antagomir was transfected to down-regulate the miR-93 level. Our results showed that miR-93 levels in the cerebral cortex of mice increased at 24 and 48 h after reperfusion. Importantly, in vivo study demonstrated that treatment with miR-93 antagomir reduced cerebral infarction volume, neural apoptosis and restored the neurological scores. In vitro study demonstrated that miR-93 antagomir attenuated hydrogen peroxide (H2O2)-induced injury. Moreover, miR-93 antagomir suppressed oxidative stress in I/R brain and H2O2 treated cortical neurons. Furthermore, we founded that down-regulation of miR-93 increased the expression of nuclear factor erythroid 2-related factor (Nrf2) and heme oxygenase-1 (HO-1) and the luciferase reporter assay confirmed that miR-93 directly binds to the predicted 3′-UTR target sites of the nrf2 gene. Finally, we found that knockdown of Nrf2 or HO-1 abolished miR-93 antagomir-induced neuroprotection against oxidative stress in H2O2 treated neuronal cultures. These results suggested that miR-93 antagomir alleviates ischemic injury through the Nrf2/HO-1 antioxidant pathway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
It is known that stroke is a serious damage to human health worldwide, has high incidence, high morbidity and high mortality characteristics. The pathophysiological mechanisms of stroke-induced neuronal death are complex cellular biochemical events, involving oxidative stress-related pathways [1]. It is already well accepted that overproduction of reactive oxygen species (ROS) after stroke, such as hydrogen peroxide (H2O2), leads to the oxidation of proteins, lipids, and DNA and drive neuronal cell death [2]. Nuclear factor erythroid 2-related factor 2 (Nrf2) is important in the protection of cells against oxidative stress, that activates the transcription of antioxidant stress genes, including heme oxygenase-1 (HO-1) [3, 4]. The Nrf2/HO-1 pathway has been shown to play important neuroprotective role in brain injury after ischemic stroke [5–7].
MicroRNAs(miRs) are small noncoding single-stranded RNA molecules of 21–23 nucleotides that modulate gene expression by binding to the complementary seed sequences in the 3′-UTRs of mRNAs [8, 9]. MiRNAs are evolutionarily conserved, and play a critical role in a variety of normal biological processes including cell differentiation and development, metabolism, proliferation, and apoptotic cell death [10, 11]. Accumulating evidences have linked dysregulated miRs expression in brain to the pathological process of ischemic stroke [12–14]. The miR-106b-25 cluster, a paralog of the miR-17-92 cluster, which all resides in the thirteenth intron of the MCM7 gene, contains three miRs, miR-25, miR-93 and miR-106b. Until recently, only a few studies have assessed the effects of the miR-106b-25 cluster on ischemic stroke [15]. Notably, miR-93 was related to inflammation, oxidative stress, and cell apoptosis [16–18]. One previous expression analysis has revealed that miR-93 is upregulated in the postischemic brain, suggesting that miR-93 may act as a potential therapeutic target for acute ischemic stroke [12]. However, the functional significance and clear mechanism of miR-93 in cerebral ischemic injury have yet to be clarified.
In this study, we examined the role of miR-93 in cerebral ischemic/reperfusion (I/R) injury in mice and in H2O2-treated primary cortical neurons. We demonstrated that miR-93 might be a critical player in the regulation of neural ischemic injury and able to influence oxidative stress though the Nrf2/HO-1 defense pathway.
Methods
Animal Model
All procedures in this study were conducted according to the guidelines set by the Animal Care and Use Committee of Liaoning Medical University and are consistent with the NIH Guide for the care and use of laboratory animals. Adult male C57BL/6 J mice (weighing 22–25 g) purchased from Experimental Animal Center of Chinese Academy of Medical Sciences, PR China were maintained in temperature controlled rooms (20 ± 2 °C) with access to food and water ad libitum. The mouse model of ischemic stroke was prepared as described before [19, 20]. In brief, after anesthetized with pentobarbital sodium (60 mg/kg i.p.), the right common and the right external carotid artery were exposed and focal cerebral ischemia was produced by 1 h of middle cerebral artery occlusion (MCAO) with a 6-0 surgical nylon monofilament (0.23 mm in diameter) followed by 24 h of reperfusion. Sham-operated mice underwent an identical procedure, without inserting the suture.
Measurement of Neurological Deficit and Infarct Volume
Mice were tested for neurological deficits at 24 h after 1 h MCAO according to neurological disability status scale (NDSS) reported by Rodriguez et al. [21]. NDSS has ten progressive steps beyond zero (normal), extending to status ten (death). Briefly, the six major steps indicate: 0 represents no neurological dysfunction; 2 represents slight decrease in mobility and the presence of passivity; 4 represents moderate neurological dysfunction and including additional alterations, such as flattened posture, hunched back, lateralized posture, ataxic gait, moderate hypomobility, decreased muscular strength and body tone and slight motor incoordination; 6 corresponding to more handicapped animals but still able to walk, with more tremor, jerks and/or convulsions, marked hypomobility, forelimb flexion, circling and moderate motor incoordination; 8 corresponding to respiratory distress, and total incapacity to move/coordinate. If the criteria for the accurate grade were not met, the nearest appropriate number was utilized: 1, 3, 5, 7 and 9.
Infarct volumes were determined by 2,3,5-triphenyltetrazolium chloride (TTC) staining. Mice were killed after 24 h reperfusion. The brain was quickly removed and cut into 1.0 mm thickness coronal sections. The sections were incubated for 15 min in a solution of 0.5 % TTC (Sigma-Aldrich, St. Louis, MO, USA) at 37 °C, and then the sections were scanned into a computer. The images of stained sections were analyzed using Image Pro Plus1 6.0 (Media Cybernetics, Silver Spring, MD).
Intracerebroventricular Injection of miR-93 Antagomir
MiR-93 antagomir and nonsensical negative control were purchased from GenePharma (Shanghai, China). miR-93 antagomir (100 μm) or the control (100 μm) were mixed with Lipofectamine RNAiMAX Transfection Reagent (Invitrogen, Carlsbad, CA) and incubated at room temperature for 30 min; 7 μL of the mixture was injected into the right lateral ventricle and the needle was kept in the place for 10 min before the MCAO [22]. The injection position was at the point of 0.5 mm posterior to bregma, 1.0 mm lateral to bregma, and 3.5 mm below the skull surface.
Primary Neuronal Cell Culture and Treatment
Primary cortical neurons obtained from postnatal day 1 C57BL/6 J mice were cultured in neurobasal medium (Gibco Inc., Grand Island, USA) supplemented with 2 % B27 (Gibco Inc., Grand Island, USA) and maintained at 37 °C in a 5 % CO2/air environment. The miR-93 antagomir (GenePharma, Shanghai, PR China) and small interference RNA (siRNA) targeting to Nrf2 or HO-1 (GenePharma, Shanghai, PR China) were transfected into primary cortical neurons for 48 h using Lipofectamine RNAiMAX Transfection Reagent (Invitrogen Tec., Carlsbad, CA, USA) after 6 days as manufacturer’s instruction. To initiate oxidative stress, primary cortical neurons were stimulated by 200 μm H2O2 for 6 h. Then neurons and supernatant were collected for measurements.
Real-Time PCR
Total RNA including miRNAs was isolated from cerebral tissue with the NucleoSpin® miRNA kit (Macherey–Nagel, Germany) according to the manufacturer’s protocol. Reverse transcription were performed by using the miRCURY LNATM Universal RT microRNA PCR (Exiqon A/S, Vedbaek, Denmark). The real-time PCR amplification was performed with an Mx3000PTM system (Agilent Technologies, CA 95051, USA) using Brilliant II SYBR® Green QPCR master mix (Agilent Technologies, CA 95051, USA) according to the manufacturer’s protocol, with U6 used as an internal control.
Cell Viability Assessment
The viability of cortical neurons was evaluated by using thiazolyl blue tetrazolium bromide (MTT, 0.5 mg/mL; Applichem Inc., Omaha, NE, USA) and the CytoTox 96® Non-Radioactive Cytotoxicity Assay (lactate dehydrogenase; LDH; Promega Cor, Madison, WI, USA) following the manufacturer’s instructions.
TUNEL Staining
Cell apoptosis was assessed by using the terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL) staining kit (TMR Red in situ cell death detection kit; Roche Diagnostics, Indianapolis, IN), according to the manufacturer’s instructions. Briefly, fix cell samples with a freshly prepared fixation solution for 1 h and then rinse slides with phosphate-buffered saline. Cells were incubated in permeabilization solution for 2 min on ice, and then incubated with TUNEL reaction mixture in a humidified atmosphere for 1 h at 37 °C in the dark. MAP2 (1:500; RRID: AB_776174; Abcam, Cambridge, United Kingdom) and 4′,6-diamidino-2-phenylindole (DAPI) were used to visualize the neurons and neural nuclei, respectively. Images were acquired by a fluorescence microscope system (DM4000B; Leica, Wetzlar, Germany).
Western Blot Analysis
The ipsilateral cortices were collected at 24 h after reperfusion and processed for Western blot as described previously [23, 24]. Antibodies in this study were rabbit anti-HO-1 (1:1000; Abcam, Cambridge, UK), anti-Nrf2 (1:1000; Abcam, Cambridge, UK), β-actin antibody (1:3000; Sigma-Aldrich Corp. St. Louis, MO 63103, USA), and corresponding secondary antibodies (Stressgen Biotechnologies Corporation, Victoria, BC, Canada). The Enhanced Chemiluminescence (ECL) kit (GE Healthcare, UK) was employed to detect the signals. Protein levels were quantified by densitometry and normalized to β-actin as a loading control.
Luciferase Assays
The 3′-UTR of the Nrf2 was amplified by PCR using primers (forward: 5′-atttaggaggatttgacc-3′; reverse: 5′-tttttgccagagctaaacaattt-3′) and cloned into the pmiR-RB-REPORTTM luciferase reporter vector (RiboBio, Guangzhou, People’s Republic of China). The mutant 3′-UTR of the Nrf2 gene was constructed with QuikChange II Site-Directed Mutagenesis (Stratagene, USA). Mouse N2A cells were plated at 0.5 × 105 cells per well in 24-well plates. The following day, cells were co-transfected pmiR-RB-ReportTM vector (RIBOBIO, Guangzhou, China), including the 3′-UTR of Nrf2 (with either wild-type or mutant miR-93 binding sites), and miR-93 agomir or miR control plasmid using Lipofectamine RNAiMAX Transfection Reagent (Invitrogen Tec., Carlsbad, CA, USA). Luciferase assays were performed 48 h after transfection by using Dual-Luciferase® Reporter Assay System (Promega Cor, Madison, WI, USA) according to the manufacturer’s protocols.
Determination of Oxidative Stress
Brain homogenates and primary cortical neurons were prepared with cold phosphate-buffered saline. The superoxide dismutase (SOD) activity, malondialdehyde (MDA) and ROS levels were assessed using SOD, MDA and ROS detection kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China), respectively, following the manufacturer’s instructions.
Statistical Analysis
We performed quantitative analysis of Western blot by using GelDoc-2000 Imagine System. For protein expression level, the protein ratio (band density of protein/band density of β-actin) in the control group was expressed as one, and then the other groups were expressed as percentage of that from control group. Statistical analysis was performed with the t test and by one-way analysis of variance (ANOVA) followed by all pairwise multiple comparison procedures using Bonferroniʼs test. Data are expressed as mean ± SEM, and differences were considered significant at P < 0.05.
Results
The Expressions of miR-93 were Upregulated in MCAO-Induced Ischemic Stroked Mice
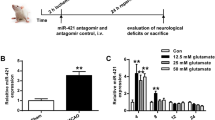
To determine the expression of miR-93 in MCAO-induced ischemic stroke mice, we examined miR-93 levels using quantitative real-time reverse transcription-polymerase chain reaction (RT-PCR) in the mouse cerebral cortex. Our results showed that miR-93 levels on the ipsilateral side of the brain significantly increased at the 24 and 48 h reperfusion time point after 1 h MCAO compared with the sham group (Fig. 1). MiR-93 expression was not significantly changed at 6 and 12 h.

The expression of miR-93 in MCAO-induced ischemic stroked mice. MiR-93 was detected by RT-PCR in mice that treated with MCAO for 1 h and reperfusion for 6–48 h. Compared with the sham group, miR-93 levels were increased at 24 and 48 h in the cerebral cortex of mice after 1 h MCAO-induced ischemic stroke values present as mean ± SEM (n = 8 per group). *P < 0.05 vs. sham group
MiR-93 Antagomir Ameliorated Ischemic Brain Damage
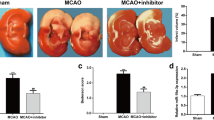
To evaluate the role of miR-93 in ischemic brain injury in vivo, the miR-93 antagomir was administrated via intracerebroventricular injection. The down-regulated miRNA-93 levels in ischemic cortex of MCAO mice were confirmed by using quantitative RT-PCR (Fig. 2a). TTC staining showed that miR-93 antagomir effectively attenuated cerebral infarction, as compared with those in the I/R group (Fig. 2b). In addition, the western blotting results showed the miR-93 antagomir significantly reduced the activated caspase-3 levels in the cortex of mice relative to the I/R group (Fig. 2c). Results of the neurological score indicated that miR-93 antagomir could improve the neurological deficit of mice with ischemic stroke (Fig. 2d). These results suggested that down-regulation of miR-93 could attenuate cerebral I/R injury in mice.

MiR-93 antagomir protects against cerebral I/R injury in mice. a The RT-PCR results showed that intracerebroventricular injection of miR-93 antagomir could effectively down-regulated miRNA-93 expression level in ischemic cortex of 1 h MCAO/24 h reperfusion treated mice. b Cerebral infarct volume evaluated by TTC-stained cerebral coronal sections. c The apoptosis in the ipsilateral cortex as detected by the activated caspase-3 levels using western blotting. d Results of the neurological score. Values present as mean ± SEM (n = 10 per group). *P < 0.05 vs. sham group; # P < 0.05 vs. I/R + miR control group
MiR-93 Antagomir Attenuated Oxidative Stress in Transient MCAO Mouse Model
To investigate the influence of miR-93 on cerebral I/R-induced oxidative stress in vivo, SOD activity and the levels of MDA and ROS in the cortex were evaluated. It was shown that SOD activity which was decreased in the ischemic brain of I/R mice compared with the sham group, was significantly upregulated by miR-93 antagomir treatment compared with the I/R group (Fig. 3a). MDA levels increased in the I/R group compared with the sham group, but were down-regulated by miR-93 antagomir treatment (Fig. 3b). I/R similarly increased ROS production in the cortex relative to the sham group, but the level was suppressed by miR-93 antagomir treatment (Fig. 3c).

Effect of miR-93 antagomir on oxidative stress induced by I/R in mice. a SOD activity in the cortex was detected by biochemical kit. b MDA content in the cortex was detected by biochemical kit. c ROS level in the cortex was detected by biochemical kit. Values present as mean ± SEM (n = 10 per group). *P < 0.05 vs. sham group; # P < 0.05 vs. I/R + miR control group
MiR-93 Antagomir Induced HO-1 Expression Through Nrf2
Using bioinformatics analysis, we found a binding site for miR-93 at the 3′-UTR of Nrf2 mRNA with a high possibility ranking, implying that Nrf2 may be one of the possible target genes of miR-93. Nrf2, a member of the transcription factor family, functions in HO-1 induction. As shown in Fig. 4a, miR-93 antagomir treatment effectively increased the expression of Nrf2 relative to the I/R group. In addition, miR-93 antagomir treatment also induced an upregulation in HO-1 expression in the cerebral cortex of I/R mice (Fig. 4b).

Effect of miR-93 antagomir on Nrf2/HO-1 levels. a Effect of miR-93 antagomir of Nrf2 protein level in cerebral cortex, as determined by western blotting (n = 10 per group). b HO-1 level in cerebral cortex as determined by western blotting (n = 10 per group). c Showed the design of a miR-93 reporter vector containing a T7-driven-luciferase cDNA fused to Nrf2 3′-UTR or mutated Nrf2 3′-UTR. The putative binding position between miR-93 and the 3′-UTR of Nrf2 mRNA was also shown. d Luciferase reporter assay was performed by cotransfection of luciferase reporter containing Nrf2 3′-UTR or mutated Nrf2 3′-UTR with miR-93 agomir or its control into N2A cells. (n = 6 per group). Values present as mean ± SEM. *P < 0.05 vs. sham group or miR control; # P < 0.05 vs. I/R + miR control group
To test whether miRNA-93 directly targeted Nrf2 by binding to the 3′-UTR sequence, we cloned a luciferase reporter vector in which T7 driven-luciferase cDNA was followed by a fragment of the Nrf2 3′-UTR or the miR-93 binding site mutated Nrf2 3′-UTR (Fig. 4c). The luciferase activity assay indicated that miR-93 agomir significantly decreased luciferase activity of the reporter vector containing miR-93 binding sequences of Nrf2 3′-UTR but not the mutant 3′-UTR vector (Fig. 4d). These results suggested that miRNA-93 could directly target the 3′-UTR of Nrf2 to negatively regulate Nrf2-protein levels in cortical neurons.
MiR-93 Antagomir Alleviated H2O2-Induced Oxidative Stress Damage in Primary Cortical Neurons Through the Nrf2/HO-1 Pathway
To confirm that miR-93 regulates H2O2-induced cell death in vitro, we transfected primary cortical neurons with miR-93 antagomir. H2O2-induced cell death was simultaneously assayed by MTT, LDH and TUNEL methods. As demonstrated in Fig. 5a–c, treatment with miR-93 antagomir effectively reduces H2O2-induced cell death compared to control groups. To demonstrate that antioxidants contribute to the effects of miR-93 in H2O2-induced cell death, we co-transfected Nrf2 or HO-1 siRNA with miR-93 antagomir in primary cortical neurons, and then detected the cell death rate. The results indicated that suppressing Nrf2 or HO-1 had the ability to block the protective effect of miR-93 antagomir in response to H2O2 stress.

MiR-93 antagomir inhibits primary cortical neurons death after H2O2 treatment. a Neurons viability was detected by MTT. b LDH assay in cultured cortical neurons was conducted. c Representative microscopic image of the neuronal apoptosis detected by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and DAPI double staining (scale bar 100 μm). Values present as mean ± SEM (n = 8 per group).*P < 0.05 vs. control; # P < 0.05 vs. control + H2O2, & P < 0.05 vs. miR-93 antagomir + H2O2
To further confirm whether silencing of miR-93 increased anti-oxidative ability of neurons, SOD activity and MDA content in primary cortical neurons were detected. It was shown that SOD activity which was reduced in primary cortical neurons upon H2O2 exposure, was elevated by miR-93 antagomir treatment (Fig. 6a). The effect of miR-93 antagomir on SOD activity was blocked by Nrf2 or HO-1 knockdown treatment (Fig. 6a). Additionally, MDA level which was increased in primary cortical neurons was blocked by miR-93 antagomir treatment; this effect was inhibited by both Nrf2 depletion and HO-1 depletion (Fig. 6b). These results demonstrate that miR-93 plays an important role in the modulation of oxidative stress tolerance in primary cortical neurons upon H2O2 stimulation.

MiR-93 antagomir attenuates oxidative stress damage in primary cortical neurons. Effect of miR-93 antagomir on SOD activity (a) and MDA content (b) in primary cortical neurons were evaluated by biochemical kit. Values present as mean ± SEM (n = 8 per group).*P < 0.05 vs. control; # P < 0.05 vs. control + H2O2 and & P < 0.05 vs. miR-93 antagomir + H2O2
Discussion
In this study, we investigated the role of miR-93 in mice with transient focal I/R injury and in primary cortical neurons subjected to oxidative stress induced by H2O2. MiR-93 levels increased in a time-dependent manner in the cerebral cortex following ischemia and reperfusion in mice. Importantly, intracerebroventricular injection of miR-93 antagomir reduced cerebral infarct volume, neural cell apoptosis, and oxidative stress after transient MCAO. Meanwhile, miR-93 antagomir increased HO-1 and Nrf2 expression levels. Further studies revealed that the mechanisms of miR-93 in ischemic cerebral injury may involve the Nrf2/HO-1 defense pathway by directly targeting Nrf2. Taken together, our data demonstrated that miR-93 down-regulation protects against cerebral I/R injury by stimulating antioxidant responses in neurons, providing a new therapeutic target in acute ischemic stroke.
Until now, studies of miR-93 have been focused mainly on its involvement in many types of human cancers, such as glioblastoma, gastric cancer, breast carcinoma, and others. miR-93 is overexpressed in a number of types of human cancers and functions as an oncogene by targeting important cancer-related genes [25–29], whereas, in some types of cancers miR-93 was down-regulated and repressed proliferation paradoxically [30, 31]. However, the present study is the first to investigate its role in ischemia-induced cerebral injury. Our results showed that miR-93 was up-regulated at 24 h and persistently increased until 48 h in the cerebral cortex of mice after 1 h MCAO-induced ischemic stroke in vivo, which is comparable to the results reported by Dharap et al. [12]. These data suggest that miR-93 might be a critical role in the pathogenesis of ischemic stroke, proposing the potential of miR-93 down-regulation as a neuroprotective strategy for cerebral ischemia injury. Follow-up in vivo studies indicated that treatment of C57BL/6 J mice with intracerebroventricular injection of miR-93 antagomir reduced infarct volume, neural apoptosis, and oxidative stress in the brain, and improved behavioral outcome of mice with ischemic stroke. Taken together, these results demonstrate that downregulation of miR-93 protects the brain from focal cerebral ischemia injury.
To further elucidate the mechanism of miR-93 on alleviating MCAO-induced cerebral ischemic injury, the present study investigated the effects of miR-93 on the levels of oxidative stress markers and antioxidants. Oxidative stress is a core pathological component of brain ischemia-reperfusion injury causing neuronal malfunction and cell death [32–34]. Several miRNAs have been reported to be involved in the cellular response to oxidative stress via the antioxidant defense system [22, 24]. As expected, we found that treatment with miR-93 antagomir reduced ROS and MDA levels in the ischemic brain of I/R mice, and increased the SOD activity as well as the expression of HO-1 and Nrf2.
The Nrf2/HO-1 pathway is an important cellular defense mechanism against I/R-induced oxidative stress [35, 36]. To further directly determine the role of the Nrf2 antioxidant pathway in miR-93 antagomir-mediated neuroprotection, the present study utilized H2O2 treatment of primary neurons to stimulate oxidative stress in vitro. The results showed that miR-93 significantly regulated Nrf2/HO-1 levels in ischemic model. Furthermore, luciferase reporter assay also confirmed that Nrf2 is a direct target of miR-93. In primary cortical cultures, miR-93 antagomir treatment alleviated H2O2-induced injury, as evidenced by increased cell viability and SOD activity, and decreased lactate dehydrogenase leakage and MDA production, which is one of the classic oxidative stress markers to directly reflect the rate and extent of lipid peroxidation. The current study also shows that knockdown of Nrf2 or HO-1 can block miR-93 antagomir-mediated neuroprotection against H2O2-induced cortical neuron injury and oxidative stress. These results suggest that miR-93 antagomir could upregulate Nrf2/HO-1 expression levels, thereby enhancing the protective defense mechanisms through anti-oxidative pathway.
In summary, the present study demonstrated that the down-regulation of miR-93 could alleviate neural ischemic injury through activating Nrf2/HO-1 antioxidant pathway, raising an interesting prospect for using miR-93 as a therapeutic target for ischemic stroke. However, it is far away from the clinical application. There are some severe problems waiting to solve, including tissue and cell-specific delivery in vivo, degradation avoidance, and target specificity. How to bring the miRNA into the brain is an important problem. Obviously, intracerebroventricular injection would be limited in the patients for the injury caused by the surgery. Delivery systems for miRNA to efficiently cross the blood brain barrier and target brain tissue will be the subject of future investigations.
References
Moskowitz MA, Lo EH, Iadecola C (2010) The science of stroke: mechanisms in search of treatments. Neuron 67:181–198
Jung JE, Kim GS, Chen H, Maier CM, Narasimhan P, Song YS, Niizuma K, Katsu M, Okami N, Yoshioka H, Sakata H, Goeders CE, Chan PH (2010) Reperfusion and neurovascular dysfunction in stroke: from basic mechanisms to potential strategies for neuroprotection. Mol Neurobiol 41:172–179
Nguyen T, Nioi P, Pickett CB (2009) The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem 284:13291–13295
Keum YS, Choi BY (2014) Molecular and chemical regulation of the Keap1-Nrf2 signaling pathway. Molecules 19:10074–10089
Shah ZA, Li RC, Ahmad AS, Kensler TW, Yamamoto M, Biswal S, Dore S (2010) The flavanol (−)-epicatechin prevents stroke damage through the Nrf2/HO1 pathway. J Cereb Blood Flow Metab 30:1951–1961
Ding Y, Chen M, Wang M, Wang M, Zhang T, Park J, Zhu Y, Guo C, Jia Y, Li Y, Wen A (2014) Neuroprotection by acetyl-11-keto-beta-Boswellic acid, in ischemic brain injury involves the Nrf2/HO-1 defense pathway. Sci Rep 4:7002
Alfieri A, Srivastava S, Siow RC, Cash D, Modo M, Duchen MR, Fraser PA, Williams SC, Mann GE (2013) Sulforaphane preconditioning of the Nrf2/HO-1 defense pathway protects the cerebral vasculature against blood–brain barrier disruption and neurological deficits in stroke. Free Radic Biol Med 65:1012–1022
Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136:215–233
Fasanaro P, Greco S, Ivan M, Capogrossi MC, Martelli F (2010) microRNA: emerging therapeutic targets in acute ischemic diseases. Pharmacol Ther 125:92–104
Huang Y, Shen XJ, Zou Q, Zhao QL (2010) Biological functions of microRNAs. Bioorg Khim 36:747–752
Zhao Y, Srivastava D (2007) A developmental view of microRNA function. Trends Biochem Sci 32:189–197
Dharap A, Bowen K, Place R, Li LC, Vemuganti R (2009) Transient focal ischemia induces extensive temporal changes in rat cerebral microRNAome. J Cereb Blood Flow Metab 29:675–687
Tan JR, Koo YX, Kaur P, Liu F, Armugam A, Wong PT, Jeyaseelan K (2011) microRNAs in stroke pathogenesis. Curr Mol Med 11:76–92
Rink C, Khanna S (2011) MicroRNA in ischemic stroke etiology and pathology. Physiol Genomics 43:521–528
Zhang JF, Shi LL, Zhang L, Zhao ZH, Liang F, Xu X, Zhao LY, Yang PB, Zhang JS, Tian YF (2016) MicroRNA-25 negatively regulates cerebral ischemia/reperfusion injury-induced cell apoptosis through Fas/FasL pathway. J Mol Neurosci. doi:10.1007/s12031-016-0712-0
Fabbri E, Borgatti M, Montagner G, Bianchi N, Finotti A, Lampronti I, Bezzerri V, Dechecchi MC, Cabrini G, Gambari R (2014) Expression of microRNA-93 and Interleukin-8 during Pseudomonas aeruginosa-mediated induction of proinflammatory responses. Am J Respir Cell Mol Biol 50:1144–1155
Singh B, Ronghe AM, Chatterjee A, Bhat NK, Bhat HK (2013) MicroRNA-93 regulates NRF2 expression and is associated with breast carcinogenesis. Carcinogenesis 34:1165–1172
Rouse M, Rao R, Nagarkatti M, Nagarkatti PS (2014) 3,3′-Diindolylmethane ameliorates experimental autoimmune encephalomyelitis by promoting cell cycle arrest and apoptosis in activated T cells through microRNA signaling pathways. J Pharmacol Exp Ther 350:341–352
Han RQ, Ouyang YB, Xu L, Agrawal R, Patterson AJ, Giffard RG (2009) Postischemic brain injury is attenuated in mice lacking the beta2-adrenergic receptor. Anesth Analg 108:280–287
Xiong X, Barreto GE, Xu L, Ouyang YB, Xie X, Giffard RG (2011) Increased brain injury and worsened neurological outcome in interleukin-4 knockout mice after transient focal cerebral ischemia. Stroke 42:2026–2032
Rodriguez R, Santiago-Mejia J, Gomez C, San-Juan ER (2005) A simplified procedure for the quantitative measurement of neurological deficits after forebrain ischemia in mice. J Neurosci Methods 147:22–28
Liu P, Zhao H, Wang R, Wang P, Tao Z, Gao L, Yan F, Liu X, Yu S, Ji X, Luo Y (2015) MicroRNA-424 protects against focal cerebral ischemia and reperfusion injury in mice by suppressing oxidative stress. Stroke 46:513–519
Wang P, Liang J, Li Y, Li J, Yang X, Zhang X, Han S, Li S, Li J (2014) Down-regulation of miRNA-30a alleviates cerebral is-chemic injury through enhancing beclin 1-mediated autophagy. Neurochem Res 39:1279–1291
Zhao H, Tao Z, Wang R, Liu P, Yan F, Li J, Zhang C, Ji X, Luo Y (2014) MicroRNA-23a-3p attenuates oxidative stress injury in a mouse model of focal cerebral ischemia-reperfusion. Brain Res 1592:65–72
Wang L, Wang Q, Li HL, Han LY (2013) Expression of MiR200a, miR93, metastasis-related gene RECK and MMP2/MMP9 in human cervical carcinoma–relationship with prognosis. Asian Pac J Cancer Prev 14:2113–2118
Kim BH, Hong SW, Kim A, Choi SH, Yoon SO (2013) Prognostic implications for high expression of oncogenic microRNAs in advanced gastric carcinoma. J Surg Oncol 107:505–510
Ohta K, Hoshino H, Wang J, Ono S, Iida Y, Hata K, Huang SK, Colquhoun S, Hoon DS (2015) MicroRNA-93 activates c-Met/PI3K/Akt pathway activity in hepatocellular carcinoma by directly inhibiting PTEN and CDKN1A. Oncotarget 6:3211–3224
Li G, Ren S, Su Z, Liu C, Deng T, Huang D, Tian Y, Qiu Y, Liu Y (2015) Increased expression of miR-93 is associated with poor prognosis in head and neck squamous cell carcinoma. Tumour Biol 36:3949–3956
Montanini L, Lasagna L, Barili V, Jonstrup SP, Murgia A, Pazzaglia L, Conti A, Novello C, Kjems J, Perris R, Benassi MS (2012) MicroRNA cloning and sequencing in osteosarcoma cell lines: differential role of miR-93. Cell Oncol (Dordr) 35: 29–41
Yang IP, Tsai HL, Hou MF, Chen KC, Tsai PC, Huang SW, Chou WW, Wang JY, Juo SH (2012) MicroRNA-93 inhibits tumor growth and early relapse of human colorectal cancer by affecting genes involved in the cell cycle. Carcinogenesis 33:1522–1530
Tang Q, Zou Z, Zou C, Zhang Q, Huang R, Guan X, Li Q, Han Z, Wang D, Wei H, Gao X, Wang X (2015) MicroRNA-93 suppress colorectal cancer development via Wnt/beta-catenin pathway downregulating. Tumour Biol 36:1701–1710
Tasoulis MK, Douzinas EE (2016) Hypoxemic reperfusion of ischemic states: an alternative approach for the attenuation of oxidative stress mediated reperfusion injury. J Biomed Sci 23:7
Halladin NL (2015) Oxidative and inflammatory biomarkers of ischemia and reperfusion injuries. Dan Med J 62:B5054
Manzanero S, Santro T, Arumugam TV (2013) Neuronal oxidative stress in acute ischemic stroke: sources and contribution to cell injury. Neurochem Int 62:712–718
Jiang S, Deng C, Lv J, Fan C, Hu W, Di S, Yan X, Ma Z, Liang Z, Yang Y (2016) Nrf2 Weaves an Elaborate Network of Neuroprotection Against Stroke. Mol Neurobiol. doi:10.1007/s12035-016-9707-7
Alfieri A, Srivastava S, Siow RC, Modo M, Fraser PA, Mann GE (2011) Targeting the Nrf2-Keap1 antioxidant defence pathway for neurovascular protection in stroke. J Physiol 589:4125–4136
Acknowledgments
This work was supported by Grants from the National Natural Science Foundation of China (NSFC) (Grant No. 81501017), Innovation Foundation for the Unversity Students (Grant No. 201510160000013) and the President Foundation of Jinzhou Medical University (Grant No. XZJJ20140105, XZJJ2015012). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors confirm that there are no conflicts.
Rights and permissions
About this article

Cite this article
Wang, P., Liang, X., Lu, Y. et al. MicroRNA-93 Downregulation Ameliorates Cerebral Ischemic Injury Through the Nrf2/HO-1 Defense Pathway. Neurochem Res 41, 2627–2635 (2016). https://doi.org/10.1007/s11064-016-1975-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-016-1975-0