
Abstract
The reperfusion after an acute ischemic stroke can lead to a secondary injury, which is ischemia–reperfusion (I/R) injury. During ischemia, the reactive oxygen species (ROS) is over-produced, mostly from nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX). Besides, miRNAs are also associated with neuronal death in ischemic stroke. MiR-124-5p is selectively expressed within central nervous system (CNS) and is predicted to bind to NOX2 directly. Herein, we successfully set up cerebral I/R injury model in rats through middle cerebral artery occlusion (MCAO) surgery. After 12 h or 24 h of refusion, the superoxide dismutase (SOD) activity was significantly inhibited, accompanied by NOX2 protein increase within MCAO rat infarct area. In vitro, oxygen-glucose deprivation/refusion (OGD/R) stimulation on PC-12 cells significantly increased NOX2 protein levels, ROS production, and the cell apoptosis, while a significant suppression on SOD activity; OGD/R stimulation-induced changes in PC-12 cells described above could be significantly attenuated by NOX2 silence. In vivo, miR-124 overexpression improved, whereas miR-124 inhibition aggravated I/R injury in MCAO rats. miR-124-5p directly bound to the CYBB 3′-untranslated region (UTR) to negatively regulate CYBB expression and NOX2 protein level. In vitro, miR-124 overexpression improved, while NOX2 overexpression aggravated OGD/R-induced cellular injuries; NOX2 overexpression significantly attenuated the effects of miR-124 overexpression. Besides, miR-124 overexpression significantly repressed NF-κB signaling activation and TNFα and IL-6 production through regulating NOX2. In conclusion, miR-124-5p/NOX2 axis modulates NOX-mediated ROS production, the inflammatory microenvironment, subsequently the apoptosis of neurons, finally affecting the cerebral I/R injury.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute ischemic stroke is the second major cause of death worldwide [1,2,3,4]. Cerebral blood flow occlusion induces the depletion of energy and nutrients needed for cell metabolism, which results in osmotic gradient imbalance, depolarization of cell membranes and augmented intracellular calcium, thus leading to mitochondrial dysfunction. The reperfusion can be essential for the ischemic brain tissue to regain normal function, but it can also lead to secondary injury, known as ischemia–reperfusion (I/R) injury.
During ischemia, a large number of reactive oxygen species (ROS) can be produced, mostly from the nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX), which is a family of transmembrane proteins [5,6,7,8,9]. The excessive production of ROS leads to the oxidative stress, thus resulting in the activation of the NF-κB signaling and the synthesis of the pro-inflammatory cytokines, including nitric oxide (NO), tumor necrosis factor-alpha (TNF-α), and interleukin 6 (IL-6). Subsequently, enhanced neuroinflammation and matrix metalloproteinases-9 (MMP-9) participates in the destruction of extracellular matrix (ECM), as well as the degradation of tight junction proteins, causing blood-brain barrier injury [10, 11]. In theory, the balance between the production and elimination of ROS is maintained through multiple antioxidant defense mechanisms. The most critical of these mechanisms is the superoxide dismutase (SOD) family of enzymes whose activity levels indicate oxidative stress status [12]. Malondialdehyde (MDA) is considered as not only a product of lipid peroxidation but also a marker of oxidative stress to predict lipid peroxidation degree [13]. Activating this system can be essential to protect the brain against I/R injuries.
Of the seven members in the NOX family, NOX2 and NOX4 are considered the main sources of ROS 8–16 h after reperfusion in the stroke model [14]. NOX1 and 2 have been reported to express in neurons [15,16,17], smooth muscle cells [18, 19], and microglia [15, 16, 19,20,21,22]. Savchenko et al. [15] demonstrated that NOX1 and NOX2 expression might alter in microglia and neurons in response to stimulation in vitro; in addition, NOX2 expression is upregulated in microglia in response to activation in cases of multiple sclerosis [20], traumatic brain injury [23], and ischemia [16]. Moreover, NOX2 is increased within the spinal cord after damage [24,25,26]. Based on these previous findings, developing an in-depth understanding of the role of NOX2 and NOX-mediated ROS production during I/R injury, and searching for promising agents inhibiting NOX2 and ROS levels might provide novel strategies to prevent the patients from cerebral I/R injury.
miRNAs are considered short, non-coding RNAs exerting a regulatory effect on the vast majority of physiological and pathological processes contributing to multiple human diseases, such as stroke [27,28,29]. Via the specific base pairing to multicellular eukaryotic mRNAs in their 3′-untranslated region (UTR), miRNAs induce target mRNA degradation and/or inhibit their translation so as to suppress the expression of target genes [30], therefore regulating the proliferation, apoptosis, differentiation, invasion, migration, and development of cells [31]. Recently, several miRNAs have been demonstrated to be associated with neuronal death within the ischemic stroke. MiR-124 is almost selectively expressed within central nervous system (CNS), which makes it unique in miRNAs; miR-124 abundance in the CNS can exceed 100 times compared to that within other organs [32]. More importantly, the overexpression of miR-124 remarkably downregulated whereas the silence of miR-124 remarkably upregulated the infarct area of middle cerebral artery occlusion (MCAO) mice in vivo. In vitro, gain-of-function or loss-of-function of miR-124 could respectively lead to the reduction or increase of neuron apoptosis and death caused by oxygen-glucose deprivation/refusion (OGD/R), and the increase or reduction of anti-apoptosis protein (Bcl-2 and Bcl-xl) [33]. Interestingly, according to Targetscan prediction, miR-124 could directly bind to NOX2 (encoded by CYBB gene in rats). Thus, we speculate that miR-124 might target NOX2 to regulate NOX2 and NOX-mediated ROS production, finally improving MCAO/R- or OGD/R-induced injury in vivo and in vitro.
Herein, we established a cerebral ischemia–reperfusion model by MCAO operation in rats, evaluated the model, and examined SOD activity, MDA content, and NOX2 protein levels in rats’ brain. OGD/R model was then generated in PC-12 cells, and the cellular effects of NOX2 upon SOD activity, MDA content, ROS production, and cell apoptosis were examined. Next, gain or loss miR-124 function was detected in MCAO/R rats, and the putative binding of miR-124-5p to rat CYBB was examined. Afterward, the dynamic effects of miR-124 and NOX2 on SOD activity, MDA content, ROS production, cell apoptosis, as well as NF-κB signaling activation and TNFα and IL-6 production were examined within OGD/R-stimulated PC-12 cells. In summary, we attempted to propose a novel mechanism of miR-124/NOX2 axis modulating the ROS production and the inflammatory microenvironment during cerebral I/R injury, eventually protecting against cerebral I/R injury.
Materials and Methods
Establishment of MCAO/Reperfusion Model in Rats
Total 68 SD adult male rats (289 ± 10 g) were obtained from Chengdu Dossy Experimental Animals Co., Ltd. (Chengdu, China) and maintained in a constant 12-h dark/light cycle with standard laboratory chew and food-water available ad libitum. All experimental procedures were performed following the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996) and approved by the Animal Care and Use Ethics Committee of Shunyi District Hospital.
Animals were randomly divided into three groups in model establishment step: sham (n = 8), middle cerebral artery (MCAO) + 12 h refusion (n = 10), and MCAO + 24 h refusion (n = 50). Total 40 MCAO/R (24 h refusion) rats were randomly divided into four groups (n = 10 in each group): Agomir-NC (negative control), Agomir-124, Antagomir-NC, and Antagomir-124. Either the Agomir-NC, Agomir-124, Antagomir-NC, or Antagomir-124 (GenePharma, Shanghai, China) was intracerebroventricularly administrated 12 h before the MCAO operation.
Rats in the MCAO/R group were subjected to intraluminal occlusion of the right middle cerebral artery following the methods described previously [34,35,36]. After 90 min of occlusion, the suture was removed to restore blood flow and allowed reperfusion until 12 or 24 h. Rats in the sham group were subjected to suture insertion into the origin of the internal carotid artery, but not the middle cerebral artery, with the remaining procedures identical to those in the MCAO/R (24 h) injury group. Rat models were selected according to a previously described method [36]. All rats were sacrificed and the entire brain rapidly removed and incubated with triphenyl tetrazolium chloride (TTC) (Beijing Cinontech Co., Ltd.) with the ipsilateral cerebral cortex harvested for ELISA, Immunoblotting, and real-time PCR analyses.
miR-124 Administration
4.5 µl of 100 µM Agomir-NC, Agomir-124, Antagomir-NC, or Antagomir-124 were incubated with 1.5 µl siRNA-Mate transfection reagent (GenePharma) for 30 min, then stereotaxically delivered into the right lateral ventricle for more than10 min. The bone wound was closed with bone wax.
Cell Line and Cell Culture
PC-12 (ATCC® CRL-1721™, ATCC, Manassas, VA, USA) cell line was obtained and cultured in RPMI-1640 medium (Catalog No. 30-2001, ATCC) supplemented with 10% heat-inactivated horse serum and 5% fetal bovine serum (Invitrogen, Waltham, MA, USA). Cells were incubated in a humidified incubator with 5% CO2 at 37 °C, with the medium changed every 3 days. After 7 days in culture, the cells were used for subsequent OGD/R model analysis.
Cell Transfection
PC12 cells (2 × 105) were seeded into a six-wells plate. For NOX2 silence, 50 nmol/l the specific small interfering RNA of NOX2 (si-NOX2) or negative control (si-NC) using 8 µl siRNA-Mate transfection reagent (GenePharma, Co., Ltd., Shanghai, China). For NOX2 overexpression, 1 µg/ml NOX2 overexpression vector (NOX2 OE) or empty vector (NC) were transfected into PC12 cells with 8 µl Lipo2000 (Invitrogen, USA). For miR-124-5p overexpression or suppression, 50 nmol/L of Agomir-124 or Antagomir-124 were transfected into PC12 cells with 8 µl siRNA-Mate transfection reagent (GenePharma), The cells were harvested after 48 h. Western blot analyses or other experiments were performed.
OGD/R Model in PC-12 Cells
To mimic ischemic-like conditions in vitro, cell cultures were exposed to oxygen-glucose deprivation (OGD) for 4 h followed by culture in 5% CO2 and glucose-containing medium following the methods described previously [37]. PC-12 cells were transfected or non-transfected and transferred into a temperature controlled anaerobic chamber (Forma Scientific, Waltham, MA, USA) with 5% CO2 and 95% N2 (hypoxic condition) at 37 ℃ and cultured in deoxygenated glucose-free RPMI-1640 medium 4 h. After OGD, PC-12 cells were maintained in glucose-containing RPMI-1640 medium supplemented with 10% FBS under normoxia culture conditions for 12 h for reoxygenation. Cells cultured under normal conditions were used as a control. After treatments, the cells were observed under an optical microscope (Olympus, Tokyo, Japan) or collected for further experiments.
Neurological Function Evaluation
MCAO rats received a neurological function evaluation at 12 h or 24 h after the MCAO operation following a modified 6-point scoring system [38, 39]. A single observer blinded to group assignment performed neurological testing.
Triphenyl Tetrazolium Chloride (TTC) Staining and Quantitation of Infarct Volume
The whole brain tissue was harvested after 90 min of ischemia followed by 12 or 24 h of reperfusion, ensuring that upon removal, the integrity of the brain was maintained. The brains were rapidly frozen at − 20 °C for 20 min, sliced, and then incubated in 2% TTC solution at 37 °C for 30 min, avoiding light with a foil cover as described before [40], and later photographed using a digital camera. The infarct region appears white, while non-ischemic regions are red. The infarct volume was then calculated from digitized images using the ImageJ software package (NIH, Bethesda, MD, USA) following the methods described previously [41].
ELISA
The examination of SOD, MDA, TNF-α, and IL-6 was conducted by ELISA assay using rat SOD, MDA, TNF-α, and IL-6 ELISA kits according to the manufacturer’s instructions (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The specific binding optical density was assayed immediately at 450 nm with a spectrophotometer (Bio-Rad Laboratories, USA).
Protein Levels Determination by Immunoblotting
The protein levels of NOX2, p65, p-p65, and IκB were determined using Immunoblotting. Proteins were extracted and the concentrations of proteins were analyzed using the bicinchoninic acid (BCA) protein assay kit (Beyotime Institute of Biotechnology, China). After that, the protein samples were loaded onto an SDS-PAGE minigel for protein separation and transferred onto a PVDF membrane. Afterward, the membrane was probed with the antibodies (dilution 1:1000) listed below: anti-NOX2 (ab80508, Abcam, Cambridge, MA, USA), anti-p-p65 (ab86299, Abcam), anti-p65 (ab16502, Abcam), and anti-IκB (ab32518, Abcam) at 4 °C overnight. Then, the blots were incubated with anti-rabbit or anti-mouse HRP-conjugated secondary antibody (dilution 1:5000, ab205718 and ab205719). Signals visualization was conducted by ECL Substrates (Millipore, MA, USA) using β-actin (ab8226, Abcam) as an endogenous protein for normalization. The gray intensity analysis was performed using ImageJ software (NIH, MD, USA).
Real-Time PCR
Total RNA was extracted from targeted cells using Trizol reagent (Invitrogen, CA, USA) and the expression of miR-124-5p was determined using PCR-based analyses following the methods described previously [42]. The specific primers were: reverse transcription primer: 5′-GTC GTA TCC AGT GCG TGT CGT GGA GTC GGC AAT TGC ACT GGA TAC GAC ATC AAG-3′; forward: 5′-CGT GTT CAC AGC GGA C-3′; reverse: 5′-CAG TGC GTG TCG TGG A-3′. Hairpin-it TM miRNAs qPCR kit (Genepharma, Shanghai, China) were used. RNU6B was employed as endogenous controls for miRNA expression determination (forward:5’-CCT GCT TCG GCA GCA CA-3’; Reverse: 5′-AAC GCT TCA CGA ATT TGC GT-3′). The data were processed using a 2−ΔΔCT method.
Luciferase Reporter Assay
For miR-124-5p binding to rat CYBB (gene encoding NOX2 protein in rats) 3′-UTR, the fragment of CYBB 3′-UTR was cloned to the downstream of the Renilla psiCHECK2 vector (Promega, Madison, WI, USA) and named wt-CYBB 3′-UTR. The mutant type (mut-CYBB 3′-UTR) was mutated in the putative miR-124 binding sites. 293T cells were co-transfected with these reporter vectors and agomir-124 or antagomir-124, respectively; the luciferase activity was detected using the Dual-Luciferase Reporter Assay System (Promega). Renilla luciferase activity was normalized to Firefly luciferase activity for each transfected well.
Flow Cytometry
For intracellular ROS level measurement, cells were incubated with 10 µmol/l DCFH-DA (Sigma, USA) in the dark for 20 min at 37 °C prior to harvest, and then washed with PBS. The fluorescence intensity of the cells was determined by flow cytometry (BD, CA,USA). For apoptosis assay, cells were harvested and incubated with 5 µl annexinV-FITC solution and 5 µl propidium iodide solution (Keygene, Nanjing, China) in the dark for 10 min at room temperature and determined by flow cytometry (BD, CA,USA).
Transferase-Mediated Deoxyuridine Triphosphate-Biotin Nick End-Labeling (TUNEL) Assay
TUNEL assay was performed to detect OGD/R-mediated apoptosis in PC-12 cells using TUNEL analysis kit (Roche, Basel, Switzerland). The cell nucleus was presented as blue color, and the positive nucleus was indicated as red color. The number of positive nuclei was determined by manually counting (magnification × 400) the positively labeled nuclei in five randomly selected fields under a microscope. Apoptosis rate was considered as the percentage of TUNEL-positive cells from the total cell number. The positive control and negative control for TUNEL assay were shown in Fig.S1.
Statistical Analysis
Results from at least three independent experiments are expressed as mean ± standard deviation (S.D.). Statistical significance was evaluated by one-way analysis of variance (ANOVA) followed by Tukey's multiple comparison test or by Student's t test. A P value of < 0.05 indicates statistical significance.
Results
NOX2 is Up-Regulated in the Cerebral Ischemia–Reperfusion Model (I/R) in Rats
Firstly, I/R model was established within adult SD rats and examined. After minutes of ischemia and then 12 or 24 h of reperfusion, the infarct (white region) and non-ischemic (red region) regions in sham, MCAO + 12 h refusion, and MCAO + 24 h refusion groups were analyzed by TTC staining followed by the quantitative analyses. Figure 1a, b showed that MCAO operation remarkably increased the infarct volume after 12 and 24 h of refusion. Consistently, the neurological function scores of MCAO rats were significantly increased after 12 and 24 h of refusion (Fig. 1c), indicating the existence of ischemic injury.

NOX2 is up-regulated in the ischemia reperfusion model (I/R) in rats. a The infarct (white region) and non-ischemic (red region) regions in sham, MCAO + 12 h refusion, and MCAO + 24 h refusion groups revealed by TTC staining (n = 5). b The quantitative analysis on infarct volumes in three groups by using the ImageJ software package. n = 5. c MCAO rats underwent neurological function evaluation at 12 h or 24 h after MCAO surgical procedure following a modified 6-point scoring system n = 10. d, e SOD activity and MDA content in three groups determined by ELISA. n = 5. f NOX2 protein levels in three groups determined by Immunoblotting. n = 5. All results were represented as means ± SD from three independent experiments. **P < 0.01.
NOX [43] and NOX-mediated reactive oxygen species (ROS) could react with lipids, proteins, and DNA during ischemia, thus activating apoptosis pathways, and eventually exacerbating cerebral ischemic damage [44, 45]. Thus, we also monitored the SOD activity, MDA content, and NOX2 protein levels in three groups. Consistent with previous studies, SOD activity was significantly impaired in MCAO rats after 12 and 24 h of refusion (Fig. 1d); in the meantime, MDA content and NOX2 protein levels were significantly increased in MCAO rats after 12 and 24 h of refusion (Fig. 1e, f), indicating the increase of NOX and NOX-mediated ROS.
Cellular Effects of NOX2 on OGD/R Model Within PC-12 Cells
After confirming increases in NOX2 and NOX-mediated ROS during ischemic injury, next, we further investigated the cellular effects of NOX2 on OGD/R model established within PC-12 cells. We transfection si-NC or si-NOX2 to conduct NOX2 silence within OGD/R-stimulated PC-12 cells, and performed Immunoblotting to verify the transfection efficiency (Fig. 2a). PC-12 cells cultured in normal condition were taken as a control. NOX2 protein levels could be significantly induced by OGD/R stimulation while decreased by si-NOX2 transfection (Fig. 2a). Consistently, SOD activity was significantly inhibited by OGD/R stimulation while rescued by NOX2 silence (Fig. 2b). In the meantime, the content of MDA and the production of ROS could be significantly enhanced within OGD/R-stimulated PC-12 cells while suppressed by NOX2 silence (Fig. 2c, d). As a result of ROS increase, the apoptosis of PC-12 cells was significantly enhanced upon OGD/R stimulation while suppressed by NOX2 silence (Fig. 2e, f). Consistently, the morphology change of PC12 cells shown that OGD/R stimulation reduced the cell numbers while NOX2 silence partly reversed the reduction effect of OGD/R stimulation to some extent (Fig. 2g). These data indicate that NOX2 and NOX-mediated ROS could be significantly induced upon OGD/R stimulation, which further results in enhanced PC-12 cell apoptosis and cellular damage; NOX2 silence remarkably reverses OGD/R stimulation-induced damages within PC-12 cells.

Cellular effects of NOX2 on OGD/R model in PC-12 cells. PC-12 cells were not transfected or transfected with si-NC (negative control) or si-NOX2, subjected to OGD/R stimulation, and then examined for a NOX2 protein levels by Immunoblotting; n = 4. b, c SOD activity and MDA content in three groups by ELISA; n = 4. d ROS production by Flow cytometry; n = 4. e, f cell apoptosis by TUNEL staining and flow cytometry n = 4. (scale bar: 50 µm). g The morphology change of PC12 cells was observed by optical microscope (scale bar: 200 µm) n = 3. All results were represented as means ± SD from three independent experiments. *P < 0.05, **P < 0.01, compared to control group; #P < 0.05, ##P < 0.01, compared to OGD/R + si-NC group.
Protective Effects of miR-124 on MCAO Rats
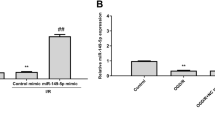
Since miR-124 can not only improve I/R-induced cerebral damage and dysfunction via modulating Ku70 [46], but also be predicted to target NOX2-encoding gene, CYBB; here, we hypothesize that miR-124 might also exert its protective effect on MCAO rats via regulating NOX2. To validate this hypothesis, we first examined miR-124 expression in rats received sham or MCAO operation and then 24 h-refusion. Figure 3a showed that miR-124 expression could be remarkably suppressed within MCAO rats, indicating the effect of miR-124 during ischemic injury. Next, we conducted miR-124 overexpression/inhibition within MCAO rats via administrating the Agomir-NC (negative control), Agomir-124, Antagomir-NC (negative control), or Antagomir-124 with intracerebroventricularly injection, and performed real-time PCR to verify the efficiency (Fig. 3b). Figure 3c, d showed that MCAO/R-induced increases in the infarct volumes were dramatically decreased via the overexpression of miR-124 while further increased via the inhibition of miR-124. Consistently, neurological function scores of MCAO/R rats were dramatically decreased via the overexpression of miR-124 while increased via the inhibition of miR-124 (Fig. 3e). Meanwhile, miR-124 overexpression significantly promoted SOD activity and inhibited MDA content; miR-124 inhibition inhibited SOD activity and promoted MDA content (Fig. 3f, g). In summary, miR-124 overexpression can protect MCAO/R rats in a ROS-related manner.

Protective effects of miR-124 on MCAO rats. a Rats were subjected to sham or MCAO operation followed by refusion for 24 h and examined for miR-124 expression by real-time PCR. n = 4. b miR-124 overexpression or miR-124 inhibition was conducted in MCAO rats via administrating the Agomir-NC (negative control), Agomir-124, Antagomir-NC (negative control), or Antagomir-124 (50 nmol/kg) by intracerebroventricularly injection at 12 h before MCAO operation, as confirmed by real-time PCR. (n = 5). c The infarct (white region) and non-ischemic (red region) regions in miR-124-overexpressed or miR-124-inhibited MCAO rats’ brains revealed by TTC staining. n = 5. d The quantitative analysis on infarct volumes in three groups by using the ImageJ software package. n = 5. e MCAO rats in four groups underwent neurological function evaluation at 24 h after MCAO surgical procedure following a modified 6-point scoring system. n = 10. f, g SOD activity and MDA content in four groups determined by ELISA. n = 5. All results were represented as means ± SD from three independent experiments. *P < 0.05, **P < 0.01.
miR-124 Targets NOX2 to Inhibit Its Expression in PC-12 Cells
Next, we validated the putative binding of miR-124-5p to CYBB 3′-UTR. We examined miR-124 expression after OGD/R stimulation on PC-12 cells; As shown in Fig. 4a, the expression of miR-124 is remarkably decreased within OGD/R PC-12 cells. Next, we transfected Agomir-NC (negative control), Agomir-124, Antagomir-NC (negative control), or Antagomir-124 to conduct miR-124 overexpression/inhibition in OGD/R model within PC-12 cells, and performed real-time PCR to verify the transfection efficiency (Fig. 4b). Within OGD/R PC-12 cells, the protein levels of NOX2 could be significantly downregulated via the overexpression of miR-124 while upregulated via the inhibition of miR-124 (Fig. 4c).

miR-124 targets NOX2 to inhibit its expression in PC-12 cells. a PC-12 cells were subjected to OGD/R stimulation and examined for miR-124 expression. n = 6. b miR-124 overexpression or miR-124 inhibition was conducted in OGD/R model in PC-12 cells by transfection of Agomir-NC (negative control), Agomir-124, Antagomir-NC (negative control), or Antagomir-124, as confirmed by real-time PCR. n = 4. c NOX2 protein levels were determined in miR-124-overexpressed or miR-124-inhibited OGD/R model in PC-12 cells by Immunoblotting. n = 4. d A schematic diagram showing the predicted miR-124 binding site in rat CYBB 3′-UTR. Two types of CYBB 3′-UTR luciferase reporter vectors, wild- and mutant-type, were constructed as described in the Materials and methods section and co-transfected in PC-12 cells with Agomir-NC, Agomir-124, Antagomir-NC, or Antagomir-124; the luciferase activity was determined. n = 4. All results were represented as means ± SD from three independent experiments. **P < 0.01.
To validate the predicted binding, we constructed two types of CYBB 3′-UTR luciferase reporter vectors, wild-type and mutant-type (wt-CYBB 3′-UTR and mut-CYBB 3′-UTR), which contain wild or mutated miR-124 binding site (Fig. 4d). We co-transfected these luciferase reporter vectors in PC-12 cells with Agomir-NC, Agomir-124, Antagomir-NC, or Antagomir-124 and validated the luciferase activity. The overexpression of miR-124 dramatically suppressed, while the inhibition of miR-124 enhanced the luciferase activity of wt-CYBB 3′-UTR; mutating the putative miR-124 binding site could abolish the alterations in luciferase activity (Fig. 4d). In summary, miR-124 could target CYBB 3′-UTR to reduce NOX2 protein level in PC-12 cells.
miR-124 Inhibits NOX2 to Suppress ROS Production in OGD/R-Stimulated PC-12 Cells
To confirm whether miR-124 exerts its protective effects on ischemic injury via NOX2 and NOX-mediated ROS, we determined the dynamic effects of miR-124 and NOX2 upon the ROS production within OGD/R PC-12 cells. These cells were co-transfected with Agomir-124 and NOX2-overexpressing vector (NOX2 OE) and examined for NOX2 protein levels; within OGD/R-stimulated PC-12 cells, the protein levels of NOX2 could be significantly suppressed via the overexpression of miR-124 while enhanced via NOX2 OE transfection. NOX2 overexpression significantly attenuated the effects of miR-124 overexpression (Fig. 5a). SOD activity could be enhanced via the overexpression of miR-124 whereas inhibited via the overexpression of NOX2; MDA content and ROS production were reduced via the overexpression of miR-124 whereas increased via the overexpression of NOX2; NOX2 overexpression could dramatically attenuate the effects of miR-124 overexpression (Fig. 5b–d), indicating that miR-124 suppresses ROS production via downregulating NOX2. Consistently, OGD/R PC-12 cell apoptosis was significantly suppressed via the overexpression of miR-124 whereas enhanced via the overexpression of NOX2; NOX2 overexpression could remarkably attenuate the effects of miR-124 overexpression (Fig. 5e, g). The optical microscope results further confirmed that overexpression of miR-124 reduced cell numbers while NOX2 overexpression partly blocked the reduction effect under OGD/R stimulation. These results suggested that miR-124 exerts its protective effects on PC-12 cell upon OGD/R stimulation via inhibiting NOX2 and NOX-mediated ROS.

miR-124 inhibits NOX2 to suppress ROS production in OGD/R PC-12 cells PC-12 cells were co-transfected with Agomir-124 and NOX2-overexpressing vector and subjected to OGD/R operation and examined for a NOX2 protein levels by Immunoblotting; b, c SOD activity and MDA content in three groups by ELISA; d ROS production by Flow cytometry; e, f) cell apoptosis by TUNEL staining and flow cytometry n = 3. (scale bar:50 µm). g The morphology change of PC12 cells was observed by optical microscope (scale bar: 200 µm) n = 3. All results were represented as means ± SD from three independent experiments. *P < 0.05, **P < 0.01, compared to control group; #P < 0.05, ##P < 0.01, compared to NOX2 + Agomir-NC group.
miR-124/NOX2 Axis Modulates the Inflammatory Microenvironment in OGD/R PC-12 Cells
The excessive production of ROS leads to oxidative stress, thus activating the nuclear factor kappa-beta (NF-κB) signaling pathway to synthesize pro-inflammatory cytokines, including the tumor necrosis factor-alpha (TNF-α) and interleukin 6 (IL-6) that enhance neuroinflammation [10, 11]. To further investigate the molecular mechanism, we co-transfected OGD/R-stimulated or non-stimulated PC-12 cells with Agomir-124 and NOX2-overexpressing vector and examined p-p65, p65, and IκB protein levels. Consistent with previous studies, OGD/R stimulation dramatically enhanced p-p65/p65 ratio while suppressed IκB expression (Fig. 6a). OGD/R stimulation-induced increases in p-p65/p65 ratio and suppression on IκB protein could be significantly attenuated via the overexpression of miR-124 whereas enhanced via the overexpression of NOX2; NOX2 overexpression could remarkably attenuate the effects of miR-124 overexpression (Fig. 6a). Consistently, the production of TNFα and IL-6 was induced by OGD/R stimulation (Fig. 6b, c). OGD/R stimulation-induced TNFα and IL-6 production could be suppressed via the overexpression of miR-124 whereas enhanced via the overexpression of NOX2; NOX2 overexpression could dramatically attenuate the effects of miR-124 overexpression (Fig. 6b–c). In summary, miR-124 might improve inflammatory microenvironment within OGD/R-stimulated PC-12 cells via inhibiting NOX2 and NOX-mediated ROS.

miR-124/NOX2 axis modulates the inflammatory microenvironment in OGD/R PC-12 cells. PC-12 cells were co-transfected with Agomir-124 and NOX2-overexpressing vector and subjected to OGD/R operation or non-operation and examined for a the protein levels of p-p65, p65 and IκB by Immunoblotting; n = 4. b, c the production of TNFα and IL-6 by ELISA. n = 4. All results were represented as means ± SD from three independent experiments. *P < 0.05, **P < 0.01, compared to control group; ##P < 0.01, compared to Agomir-NC + NC group; &P < 0.05, &&P < 0.01, compared to Agomir-NC + NOX2 group.
Discussion
Herein, we successfully set up a cerebral I/R injury model in rats through MCAO surgery and evaluated the model. After 12 h or 24 h of refusion, the SOD activity was significantly inhibited, while the MDA content and NOX2 protein levels could be dramatically upregulated within MCAO rat infarct area. OGD/R stimulation on PC-12 cells significantly increased NOX2 protein levels, MDA content, ROS production, and the cell apoptosis, while a significant suppression on SOD activity; OGD/R stimulation-induced changes in PC-12 cells described above could be significantly attenuated by NOX2 silence. In vivo, the overexpression of miR-124 improved, whereas the inhibition of miR-124 aggravated I/R injury in MCAO rats. In PC-12 cells, miR-124 directly bound to the CYBB 3′-UTR to negatively regulate CYBB expression and NOX2 protein level. In vitro, miR-124 overexpression also improved, while NOX2 overexpression aggravated OGD/R-induced cellular injuries; NOX2 overexpression significantly attenuated the effects of miR-124 overexpression. In addition, miR-124 overexpression significantly repressed NF-κB signaling activation and TNFα and IL-6 production, which was promoted by NOX2 overexpression; similarly, the effects of miR-124 overexpression were significantly reversed by NOX2 overexpression.
Ischemia induces complex metabolic issues which might result in non-reversible damage and death of neuronal cells within the ischemia nuclear. The cerebral blood flow recovery (reperfusion) leads to an increase in blood oxygen concentration and ROS production. In turn, the occurrence of oxidative stress following ischemic reperfusion is the main reason for aggravating the process of I/R injury [47]. The overproduction of ROS may subsequently lead to the peroxidation of lipid, protein, and nucleic acids [48], further decrease Bcl-2/Bax ratio and finally result in cell apoptosis [49]. SOD is an endogenous oxygen free radical scavenger and its level could reflect the ability of eliminating free radicals [50]. On the contrary, MDA is a stable end product of lipid peroxidations and could be determined to evaluate the degree of cell damage [51]. In the present study, we generated a cerebral I/R model in adult rats by MCAO operation in which we observed not only the upregulation of infarct areas but also significantly decreased SOD activity and increased MDA content, indicating the existence of ROS-mediated oxidative stress during the cerebral I/R injury.
NOX represents an important ROS source which can mediate oxidative stress in the process of heart, brain, lung and renal I/R injury [52, 53]. Previous studies have shown that the increased expression and activation of Nox induced the massive production of ROS in cerebral I/R injury [16]. Nox activation and ROS production may lead to activation of microglia and production of inflammatory mediators, such as IL-1β and TNF-α in a mice model with focal cerebral I/R injury [15]. Among the members of Nox family, NOX2 may play greater roles than other members in cerebral I/R injury [22]. NOX2 increases from 24 to 72 h after reperfusion in endothelial cells and microglia of the penumbra in a mice model of MCAO [54]. Consistent with these previous studies, NOX2 protein levels were significantly increased after 12 h and 24 h of refusion in MCAO rats, suggesting the specific role of NOX2 in cerebral I/R injury.
It has been previously reported that NOX2 inhibition might not only impair ROS production and lipid peroxidation, but also reduce infarction area [14]. Pretreatment with NOX2-specific inhibitor gp91ds-tat or potent ROS scavenger melatonin could reduce the infarct sizes in brain tissues of rats and effectively suppress I/R-induced increase in ROS levels, neuronal apoptosis and degeneration [55, 56]. However, these previous studies mainly addressed the in vivo effects of NOX2 on cerebral I/R injury. To confirm the cellular effects and mechanism of NOX2, we set up an OGD/R model within PC-12 cells and conducted NOX2 silence in OGD/R-stimulated PC-12 cells. It has been predicted that NOX2 silence significantly promoted the activity of SOD while suppressed MDA content and the production of ROS within PC-12 cells suffered to OGD/R injury. As previously mentioned, SOD family exerts ROS scavenging capacity [12] and MDA is regarded as a marker of oxidative stress to predict lipid peroxidation degree [13], here, NOX2 silence indeed attenuates ROS production. More importantly, the apoptosis of nerve cells represents an essential part of the pathophysiology of brain stroke [57]; here, NOX2 silence significantly inhibited OGD/R-induced cell apoptosis and cell number reduction within PC-12 cells, indicating that inhibiting NOX2 might be a potent strategy for improving cerebral I/R injury.
There is currently evidence that miRNAs exert a critical effect on modulating neuronal death upon ROS-mediated oxidative stress during cerebral ischemic stroke [58]. NOX family members have also been reported to be targets of miRNAs [59,60,61]. miR-124 is regarded as a critical miRNA in CNS which can protect MCAO mice in vivo and OGD/R neurons in vitro [33]. Herein, we observed significantly downregulated miR-124 expression in both MCAO rats and OGD/R-stimulated PC-12 cells. Regarding the functions, early miR-124 injection in MCAO mice has been reported to result in a significantly increased neuronal survival [62]. In vivo, miR-124 overexpression significantly decreased the infarct area of MCAO mice. In vitro, the gain of miR-124 function resulted in reduced neuron apoptosis and death induced by OGD, and increased anti-apoptosis protein, Bcl-2 and Bcl-xl, respectively [33]. Consistent with the previous studies, miR-124 overexpression significantly decreased MCAO-induced upregulation in infarct area and ROS production in vivo and miR-124 overexpression attenuated OGD/R-induced ROS production and cell apoptosis in vitro, indicating the protective role of miR-124 against I/R injury. As for the molecular mechanism, via direct binding to rat CYBB in its 3′-UTR, miR-124 can suppress its expression and decrease NOX2 protein level. More importantly, NOX2 overexpression exerted opposing effects on OGD/R-treated PC-12 cells by enhancing OGD/R-induced ROS production and cell apoptosis in vitro, and significantly reversed the effects of miR-124 overexpression. In summary, miR-124 exerts its protective effects against MCAO- and OGD/R-induced injuries via suppressing NOX2 and NOX-mediated ROS production.
To provide further evidence, we also examined the dynamic effects of miR-124 and NOX2 upon NF-κB signaling pathway activation and TNFα and IL-6 production. Accordingly, oxidative stress caused by ROS overproduction induced NF-κB signaling pathway activation, resulting in pro-inflammatory cytokine release, including IL-1β, IL-6, and TNFα [63, 64], thus leading to blood-brain barrier disruption and angioedema. Herein, miR-124 overexpression inhibited the phosphorylation of p65 while increased the protein levels of IκB, as well as reduced the production of both TNFα and IL-6, indicating that miR-124 overexpression improved the inflammatory microenvironment caused by OGD/R stimulation in PC-12 cells. Conversely, NOX2 overexpression enhanced OGD/R-induced activation of NF-κB signaling and cytokines release, while significantly reversed the effects of miR-124 overexpression.
In conclusion, miR-124/NOX2 axis modulates NOX-mediated ROS production, the inflammatory microenvironment, subsequently the apoptosis of neurons, finally affecting the cerebral I/R injury. Our findings provide a rationale for further studies on the potential therapeutic benefits of miRNA/mRNA axis in ischemic stroke treatment.
References
Strong K, Mathers C, Bonita R (2007) Preventing stroke: saving lives around the world. Lancet Neurol 6:182–187
Johnston SC, Mendis S, Mathers CD (2009) Global variation in stroke burden and mortality: estimates from monitoring, surveillance, and modelling. Lancet Neurol 8:345–354
Murray CJ, Ezzati M, Flaxman AD, Lim S, Lozano R, Michaud C, Naghavi M, Salomon JA, Shibuya K, Vos T, Wikler D, Lopez AD (2012) GBD 2010: design, definitions, and metrics. Lancet 380:2063–2066
Pundik S, Xu K, Sundararajan S (2012) Reperfusion brain injury: focus on cellular bioenergetics. Neurology 79:S44–S51
Lott C, Hennes HJ, Dick W (1999) Stroke–a medical emergency. J Accid Emerg Med 16:2–7
Chrissobolis S, Miller AA, Drummond GR, Kemp-Harper BK, Sobey CG (2011) Oxidative stress and endothelial dysfunction in cerebrovascular disease. Front Biosci (Landmark Ed) 16:1733–1745
Vidale S, Consoli A, Arnaboldi M, Consoli D (2017) Postischemic inflammation in acute stroke. J Clin Neurol 13:1–9
Zhang Y, Wang T, Yang K, Xu J, Wu JM, Liu WL (2016) NADPH oxidase 2 does not contribute to early reperfusion-associated reactive oxygen species generation following transient focal cerebral ischemia. Neural Regen Res 11:1773–1778
Haslund-Vinding J, McBean G, Jaquet V, Vilhardt F (2017) NADPH oxidases in oxidant production by microglia: activating receptors, pharmacology and association with disease. Br J Pharmacol 174:1733–1749
Lakhan SE, Kirchgessner A, Hofer M (2009) Inflammatory mechanisms in ischemic stroke: therapeutic approaches. J Transl Med 7:97
Yang Y, Rosenberg GA (2011) Blood-brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke 42:3323–3328
Adibhatla RM, Hatcher JF (2010) Lipid oxidation and peroxidation in CNS health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal 12:125–169
Kelly PJ, Morrow JD, Ning M, Koroshetz W, Lo EH, Terry E, Milne GL, Hubbard J, Lee H, Stevenson E, Lederer M, Furie KL (2008) Oxidative stress and matrix metalloproteinase-9 in acute ischemic stroke: the Biomarker Evaluation for Antioxidant Therapies in Stroke (BEAT-Stroke) study. Stroke 39:100–104
Qin YY, Li M, Feng X, Wang J, Cao L, Shen XK, Chen J, Sun M, Sheng R, Han F, Qin ZH (2017) Combined NADPH and the NOX inhibitor apocynin provides greater anti-inflammatory and neuroprotective effects in a mouse model of stroke. Free Radic Biol Med 104:333–345
Savchenko VL (2013) Regulation of NADPH oxidase gene expression with PKA and cytokine IL-4 in neurons and microglia. Neurotox Res 23:201–213
McCann SK, Dusting GJ, Roulston CL (2008) Early increase of Nox4 NADPH oxidase and superoxide generation following endothelin-1-induced stroke in conscious rats. J Neurosci Res 86:2524–2534
Dugan LL, Ali SS, Shekhtman G, Roberts AJ, Lucero J, Quick KL, Behrens MM (2009) IL-6 mediated degeneration of forebrain GABAergic interneurons and cognitive impairment in aged mice through activation of neuronal NADPH oxidase. PLoS ONE 4:e5518
Ambasta RK, Kumar P, Griendling KK, Schmidt HH, Busse R, Brandes RP (2004) Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J Biol Chem 279:45935–45941
Pendyala S, Natarajan V (2010) Redox regulation of Nox proteins. Respir Physiol Neurobiol 174:265–271
Fischer MT, Sharma R, Lim JL, Haider L, Frischer JM, Drexhage J, Mahad D, Bradl M, van Horssen J, Lassmann H (2012) NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain 135:886–899
Jaquet V, Scapozza L, Clark RA, Krause KH, Lambeth JD (2009) Small-molecule NOX inhibitors: ROS-generating NADPH oxidases as therapeutic targets. Antioxid Redox Signal 11:2535–2552
Kim D, You B, Jo EK, Han SK, Simon MI, Lee SJ (2010) NADPH oxidase 2-derived reactive oxygen species in spinal cord microglia contribute to peripheral nerve injury-induced neuropathic pain. Proc Natl Acad Sci USA 107:14851–14856
Dohi K, Ohtaki H, Nakamachi T, Yofu S, Satoh K, Miyamoto K, Song D, Tsunawaki S, Shioda S, Aruga T (2010) Gp91phox (NOX2) in classically activated microglia exacerbates traumatic brain injury. J Neuroinflammation 7:41
Byrnes KR, Garay J, Di Giovanni S, De Biase A, Knoblach SM, Hoffman EP, Movsesyan V, Faden AI (2006) Expression of two temporally distinct microglia-related gene clusters after spinal cord injury. Glia 53:420–433
Byrnes KR, Washington PM, Knoblach SM, Hoffman E, Faden AI (2011) Delayed inflammatory mRNA and protein expression after spinal cord injury. J Neuroinflammation 8:130
Pajoohesh-Ganji A, Knoblach SM, Faden AI, Byrnes KR (2012) Characterization of inflammatory gene expression and galectin-3 function after spinal cord injury in mice. Brain Res 1475:96–105
Henninger N, Mayasi Y (2019) Nucleic acid therapies for ischemic stroke. Neurotherapeutics. 16(2):299–313
Martinez B, Peplow PV (2016) Blood microRNAs as potential diagnostic and prognostic markers in cerebral ischemic injury. Neural Regen Res 11:1375–1378
Yang Y, Sandhu HK, Zhi F, Hua F, Wu M, Xia Y (2015) Effects of hypoxia and ischemia on microRNAs in the brain. Curr Med Chem 22:1292–1301
Ameres SL, Zamore PD (2013) Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol 14:475–488
Pal AS, Kasinski AL (2017) Animal models to study microRNA function. Adv Cancer Res 135:53–118
Mishima T, Mizuguchi Y, Kawahigashi Y, Takizawa T, Takizawa T (2007) RT-PCR-based analysis of microRNA (miR-1 and – 124) expression in mouse CNS. Brain Res 1131:37–43
Sun Y, Gui H, Li Q, Luo ZM, Zheng MJ, Duan JL, Liu X (2013) MicroRNA-124 protects neurons against apoptosis in cerebral ischemic stroke. CNS Neurosci Ther 19:813–819
Sun J, Tong L, Luan Q, Deng J, Li Y, Li Z, Dong H, Xiong L (2012) Protective effect of delayed remote limb ischemic postconditioning: role of mitochondrial K(ATP) channels in a rat model of focal cerebral ischemic reperfusion injury. J Cereb Blood Flow Metab 32:851–859
Min XL, Wang TY, Cao Y, Liu J, Li JT, Wang TH (2015) MicroRNAs: a novel promising therapeutic target for cerebral ischemia/reperfusion injury? Neural Regen Res 10:1799–1808
Longa EZ, Weinstein PR, Carlson S, Cummins R (1989) Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20:84–91
Tang J, Hu Z, Tan J, Yang S, Zeng L (2016) Parkin protects against oxygen-glucose deprivation/reperfusion insult by promoting Drp1 degradation. Oxid Med Cell Longev 2016:8474303
Brait VH, Jackman KA, Walduck AK, Selemidis S, Diep H, Mast AE, Guida E, Broughton BR, Drummond GR, Sobey CG (2010) Mechanisms contributing to cerebral infarct size after stroke: gender, reperfusion, T lymphocytes, and Nox2-derived superoxide. J Cereb Blood Flow Metab 30:1306–1317
Li P, Shen M, Gao F, Wu J, Zhang J, Teng F, Zhang C (2017) An antagomir to MicroRNA-106b-5p ameliorates cerebral ischemia and reperfusion injury in rats via inhibiting apoptosis and oxidative stress. Mol Neurobiol 54:2901–2921
Kam KY, Yu SJ, Jeong N, Hong JH, Jalin AM, Lee S, Choi YW, Lee CK, Kang SG (2011) p-Hydroxybenzyl alcohol prevents brain injury and behavioral impairment by activating Nrf2, PDI, and neurotrophic factor genes in a rat model of brain ischemia. Mol Cells 31:209–215
Selvamani A, Sathyan P, Miranda RC, Sohrabji F (2012) An antagomir to microRNA Let7f promotes neuroprotection in an ischemic stroke model. PLoS ONE 7:e32662
Liu Y, Wang Y, He X, Zhang S, Wang K, Wu H, Chen L (2018) LncRNA TINCR/miR-31-5p/C/EBP-alpha feedback loop modulates the adipogenic differentiation process in human adipose tissue-derived mesenchymal stem cells. Stem Cell Res 32:35–42
Bedard K, Krause KH (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87:245–313
Nathan C, Ding A (2010) SnapShot: reactive oxygen intermediates (ROI). Cell 140:951–951 e952
Wu LJ, Wu G, Akhavan Sharif MR, Baker A, Jia Y, Fahey FH, Luo HR, Feener EP, Clapham DE (2012) The voltage-gated proton channel Hv1 enhances brain damage from ischemic stroke. Nat Neurosci 15:565–573
Zhu F, Liu JL, Li JP, Xiao F, Zhang ZX, Zhang L (2014) MicroRNA-124 (miR-124) regulates Ku70 expression and is correlated with neuronal death induced by ischemia/reperfusion. J Mol Neurosci 52:148–155
Sinning C, Westermann D, Clemmensen P (2017) Oxidative stress in ischemia and reperfusion: current concepts, novel ideas and future perspectives. Biomark Med 11:11031–11040
Andreadou I, Iliodromitis EK, Farmakis D, Kremastinos DT (2009) To prevent, protect and save the ischemic heart: antioxidants revisited. Expert Opin Ther Targets 13:945–956
Huttemann M, Lee I, Grossman LI, Doan JW, Sanderson TH (2012) Phosphorylation of mammalian cytochrome c and cytochrome c oxidase in the regulation of cell destiny: respiration, apoptosis, and human disease. Adv Exp Med Biol 748:237–264
Ciancarelli I, Di Massimo C, De Amicis D, Pistarini C, Tozzi Ciancarelli MG (2015) Uric acid and Cu/Zn superoxide dismutase: potential strategies and biomarkers in functional recovery of post-acute ischemic stroke patients after intensive neurorehabilitation. Curr Neurovasc Res 12:120–127
Mishra S, Mishra BB (2017) Study of lipid peroxidation, nitric oxide end product, and trace element status in Type 2 diabetes mellitus with and without complications. Int J Appl Basic Med Res 7:88–93
Kleikers PW, Wingler K, Hermans JJ, Diebold I, Altenhofer S, Radermacher KA, Janssen B, Gorlach A, Schmidt HH (2012) NADPH oxidases as a source of oxidative stress and molecular target in ischemia/reperfusion injury. J Mol Med (Berl) 90:1391–1406
Kahles T, Brandes RP (2013) Which NADPH oxidase isoform is relevant for ischemic stroke? The case for nox 2. Antioxid Redox Signal 18:1400–1417
Green SP, Cairns B, Rae J, Errett-Baroncini C, Hongo JA, Erickson RW, Curnutte JT (2001) Induction of gp91-phox, a component of the phagocyte NADPH oxidase, in microglial cells during central nervous system inflammation. J Cereb Blood Flow Metab 21:374–384
Wang J, Liu Y, Shen H, Li H, Wang Z, Chen G (2020) Nox2 and Nox4 participate in ROS-induced neuronal apoptosis and brain injury during ischemia-reperfusion in rats. Acta Neurochir Suppl 127:47–54
Li H, Wang Y, Feng D, Liu Y, Xu M, Gao A, Tian F, Zhang L, Cui Y, Wang Z, Chen G (2014) Alterations in the time course of expression of the Nox family in the brain in a rat experimental cerebral ischemia and reperfusion model: effects of melatonin. J Pineal Res 57:110–119
Quillinan N, Herson PS, Traystman RJ (2016) Neuropathophysiology of brain injury. Anesthesiol Clin 34:453–464
Barca-Mayo O, De Pietri Tonelli D (2014) Convergent microRNA actions coordinate neocortical development. Cell Mol Life Sci 71:2975–2995
Varga ZV, Kupai K, Szucs G, Gaspar R, Paloczi J, Farago N, Zvara A, Puskas LG, Razga Z, Tiszlavicz L, Bencsik P, Gorbe A, Csonka C, Ferdinandy P, Csont T (2013) MicroRNA-25-dependent up-regulation of NADPH oxidase 4 (NOX4) mediates hypercholesterolemia-induced oxidative/nitrative stress and subsequent dysfunction in the heart. J Mol Cell Cardiol 62:111–121
Yang J, Brown ME, Zhang H, Martinez M, Zhao Z, Bhutani S, Yin S, Trac D, Xi JJ, Davis ME (2017) High-throughput screening identifies microRNAs that target Nox2 and improve function after acute myocardial infarction. Am J Physiol Heart Circ Physiol 312:H1002–H1012
Gordillo GM, Biswas A, Khanna S, Pan X, Sinha M, Roy S, Sen CK (2014) Dicer knockdown inhibits endothelial cell tumor growth via microRNA 21a-3p targeting of Nox-4. J Biol Chem 289:9027–9038
Hamzei Taj S, Kho W, Riou A, Wiedermann D, Hoehn M (2016) MiRNA-124 induces neuroprotection and functional improvement after focal cerebral ischemia. Biomaterials 91:151–165
Surh YJ (2002) More than spice: capsaicin in hot chili peppers makes tumor cells commit suicide. J Natl Cancer Inst 94:1263–1265
Tang J, Luo K, Li Y, Chen Q, Tang D, Wang D, Xiao J (2015) Capsaicin attenuates LPS-induced inflammatory cytokine production by upregulation of LXRalpha. Int Immunopharmacol 28:264–269
Funding
This work was supported by the China Scholarship Council Grant (Number 201906375044).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical Approval
All experimental procedures were performed following the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85 − 23, revised 1996) and approved by the Animal Care and Use Ethics Committee of Shunyi District Hospital.
Informed Consent
Consent for publication was obtained from the participants.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wu, Y., Yao, J. & Feng, K. miR-124-5p/NOX2 Axis Modulates the ROS Production and the Inflammatory Microenvironment to Protect Against the Cerebral I/R Injury. Neurochem Res 45, 404–417 (2020). https://doi.org/10.1007/s11064-019-02931-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-019-02931-0