
Abstract
Inflammatory damage plays an important role in cerebral ischemic pathogenesis and may represent a target for treatment. Evodiamine (Evo) has been proved to elicit a variety of biological effects through its anti-inflammatory property in the treatment of infectious disease, Alzheimer’s disease and hypoxia-induced inflammatory response. Whether this protective effect applies to cerebral ischemic injury, we therefore investigated the potential neuroprotective role of Evo and the underlying mechanisms. Male Institute of Cancer Research (ICR) mice were subjected to permanent middle cerebral artery occlusion (pMCAO) and randomly divided into five groups: Sham (sham-operated + 1 % DMSO + 0.5 % tween80), pMCAO (pMCAO + 0.9 % saline), Vehicle (pMCAO + 1 % DMSO + 0.5 % tween80), Evo-L (Vehicle + Evo 50 mg/kg) and Evo-H (Vehicle + Evo 100 mg/kg) groups. Evo was administered intragastrically twice daily for 3 days, and once again 30 min before mouse brain ischemia was induced by pMCAO. Neurological deficit, brain water content and infarct size were measured at 24 h after stroke. The expression of pAkt, pGSK3β, NF-κB and claudin-5 in ischemic cerebral cortex was analyzed by western blot and qRT-PCR. Compared with Vehicle group, Evo significantly ameliorated neurological deficit, brain water content and infarct size, upregulated the expression of pAkt, pGSK3β and claudin-5, and downregulated the nuclear accumulation of NF-κB (P < 0.05). Evo protected the brain from ischemic damage caused by pMCAO; this effect may be through upregulation of pAkt, pGSK3β and claudin-5, and downregulation of NF-κB expression.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Ischemic stroke remains a major medical problem due to the lack of effective treatment. Previous studies have indicated that inflammatory response is an important pathological mechanism in the pathogenesis of brain injury secondary to ischemia [1–3]. The close relationship between inflammatory response and cerebral ischemia has generated our considerable interest in seeking anti-inflammatory therapies to combat ischemia-induced damage [2, 4–6].
Ample evidences have suggested that Protein kinase B (PKB, also known as Akt)/glycogen synthase kinase (GSK) signaling pathway plays a central role in physical and pathological conditions to differently regulate inflammatory factors as well as boost survival [7]. Akt, is a serine/threonine kinase and plays a critical role in the modulation of cell development, growth, and survival. Akt phosphorylation is neuroprotective against ischemic injury. Akt is phosphorylated by its upstream kinases such as phosphoinositide dependent kinase 1 (PDK-1), which phosphorylates Akt at Thr308 [8] and mammalian target of rapamycin complex 2 that phosphorylates Akt at Ser473 [9]. Thus Akt is fully activated and then phosphorylates a diverse number of protein substrates including the constitutively active serine-threonine kinase glycogen synthase kinase 3-β (GSK3β) (Ser9), I κB kinase (IKK), Nuclear factor-kappa B (NF-κB) and so on. GSK3β, which has been reported to be involved in ischemic brain injury [10, 11], is particularly abundant in the central nervous system and is neuron-specific. And it’s activity has recently been identified in a number of studies as crucial in the regulation of the inflammatory response [12].
NF-κB is a family of transcription factors composed of five subunits, RelA/p65, c-Rel, RelB, p50 and p52 [13], and previous observation provides evidence that GSK3β regulated the inflammatory response by differentially affecting the nuclear amounts of transcription factors NF-κB subunit p65 [14]. Although NF-κB is essential for neuron survival and its activation may protect neurons against oxidative-stresses or ischemia-induced neurodegeneration, NF-κB activation can contribute to inflammatory reactions after brain injury and stroke [13, 15], which regulates the genes of a vast number of inflammatory mediators, such as IL-1, tumor necrosis factor-α (TNF-α), IL-6, inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2), all of which play a pivotal role in ischemic brain damage [16–18]. Previous studies have proved that NF-κB is activated in cerebral ischemia and inhibition of NF-κB reduces infarction volume and develops less ischemic damage in permanent ischemia especially [19–21].
Evodiamine (Evo) is an alkaloidal component extracted from the unripe fruit of Evodiae-fructus which is commonly used as an anti-inflammatory drug in traditional Chinese medicine. The structure of Evo is clear as shown in Fig. 1. With respect to the pharmacological actions of Evo, more attention has been paid to its beneficial effects involving anti-inflammatory [22, 23], immune modulation [24], anti-tumor [25–27] actions and retarding development of atherosclerosis [28, 29]. Furthermore, Evo exerts a protective effect on Alzheimer’s disease [23] and ischemia/reperfusion damage in heart [30]. However, there is still a paucity of data about the exact role of Evo on the brain parenchymatous tissue in the acute phase of cerebral ischemia.

The chemical structure of evodiamine
The aim of this study was to investigate whether Evo has neuroprotective effect on acute ischemic stroke, and the potential mechanisms for its neuroprotection in permanent middle cerebral artery occlusion (pMCAO) brain.
Materials and Methods
Experimental Animals
Male ICR mice (25–30 g, about 3 weeks, n = 120) were supplied by Vital River Laboratory Animal Technology Co. Ltd. The experimental protocol was approved by the institutional animal care and use committee and the local experimental ethics committee and conformed to internationally accept ethical standards. All mice were allowed to acclimatize to the new surrounding for at least 2 days ahead of any experimentation in a 12:12-h light/dark cycle.
Mouse Model of Permanent Focal Cerebral Ischemia
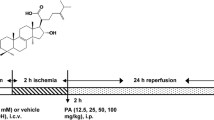
The procedure was performed as described previously [19, 31]. Briefly, animals were anesthetized with 10 % chloral hydrate (350 mg/kg) intraperitoneally. In anesthetized mice, body temperature was monitored and maintained at 36.5–37.5 °C. The right common carotid artery was exposed and isolated. The right middle cerebral artery (MCA) was occluded by inserting a monofilament nylon suture with a heat-rounded tip into the internal carotid artery, which was advanced further until it closed the origin of the MCA. Sham-operated control mice received the same surgical procedure without inserting a filament. During the experiments, MCA blood flow was monitored using a blood flow monitor (moor VMS-LDF, Moor Instruments Ltd., UK) with a fiber optic probe, adhered onto the skull surface of core area supplied by the MCA before and after clamping the MCA.
Groups and Drug Administration
Evo (Zelang Medical Technology Co. Ltd, Nanjing, China) with purity of more than 98 % was dissolved in dimethyl sulfoxide (DMSO) and tween-80 to prepare concentration of 5 mg/ml and 10 mg/ml. Evo was administered by gavage twice a day (once every 12 h) for 3 days before surgery and another dosage 30 min before operation. Sham operated group: mice received equal volume 0.9 % saline including 1 % DMSO and 0.5 % tween-80 (Sham); pMCAO group: mice received pMCAO and treated with equal volume 0.9 % saline (pMCAO); Vehicle group: mice received pMCAO and treated with equal volume 0.9 % saline including 1 % DMSO and 0.5 % tween-80 (Vehicle); low dose group: mice received pMCAO and treated with Evo at dose of 50 mg/kg (Evo-L); high dose group: mice received pMCAO and treated with Evo at dose of 100 mg/kg (Evo-H).
Neurological Function Assessment
A neurological test was carried out by an examiner blinded to the experimental groups before the mice (n = 16 per group) were killed at 24 h after pMCAO, following a modified scoring system based on that developed from Longa et al. [32] and our previous study [33, 34], as follows: (0) no deficits; (1) difficulty in fully extending the contralateral forelimb; (2) unable to extend the contralateral forelimb; (3) mild circling to the contralateral side; (4) severe circling; and (5) falling to the contralateral side. The higher the neurological deficit score, the more severe impairment of motor motion injury.
Measurement of Brain Water Content
Brain water content was observed at 24 h after pMCAO using the standard wet-dry method. 6 mice in each group were anesthetized with 10 % chloral hydrate and killed by decapitation. The brains were quickly removed and placed on a frozen surface. After dissecting free 2 mm of frontal pole, a coronal brain slice (about 3 mm thick) was cut and the slice was divided into the ipsilateral and contralateral hemispheres. The two hemisphere slices packaged with preweighed tin foil were immediately weighed on an electronic balance to obtain the wet weight, dried for 8 h in an oven at 120 °C, and then reweighed to obtain the dry weight. Brain water content was calculated with the equation as follows: Brain water content (%) = (wet weight − dry weight)/wet weight × 100 %.
Measurement of Infarct Volume
Infarct volume after pMCAO was determined by 2,3,5-triphenyltetrazolium chloride (TTC) at 24 h after pMCAO (n = 6 per group). Animals were reanesthetized and the brains were quickly collected. Then the brain tissue was sliced into five coronal section (1.5 mm thick each) and stained with 2 % solution of TTC at 37 °C for 15 min, followed by fixation with 4 % paraformalde-hyde. The normal tissue was stained red, while the infarct area was stained a pale gray color. TTC-stained sections were photographed and the digital images were analyzed using image analysis software (Image-Pro Plus 5.1). The lesion volume was calculated by multiplying the area by the thickness of slices. To compensate for the effect of brain edema, the percentage hemisphere lesion volume (%HLV) was calculated by the following formula [35]: %HLV = {[total infarct volume − (the volume of intact ipsilateral hemisphere − the volume of intact contralateral hemisphere)]/contralateral hemisphere volume} × 100 %.
Western Blotting
The cytosolic and nuclear protein and total protein were extracted respectively from mouse ischemic and control cortex following the manufacturer’s protocols (Applygen Technologies Inc., Beijing, China) at 24 h after pMCAO. About 30 mice were used for Western blot analysis (n = 6 per group). Protein concentration of the supernatant was determined using a BCA Protein Assay Reagent Kit (Novagen, Madison, WI, USA) with bovine serum albumin as the standard. An equivalent amount of 50 mg total protein samples, as well as 30 mg cytosolic or nuclear protein samples, was separated respectively, by sodium dodecyl sulfate-poly-acrylamide gels prior and transferred 2 h on to PVDF membranes (Millipore Corporation, USA). After blocking 1 h with 5 % non-fat dry milk in phosphate buffered saline (PBS), membranes were incubated overnight at 4 °C with rabbit polyclonal antibody anti-Akt (1:500, Cell Signaling Technology), anti-pAkt (Ser473) (1:500, Cell Signaling Technology), anti-GSK3β (1:500, Bioworld Technology, lnc.), anti-pGSK3β (Ser9) (1:500, Bioworld Technology, lnc.), claudin-5 (1:200, Santa Cruz Biotechnology), anti-β-actin (1:500, Santa Cruz Biotechnology); rabbit monoclonal antibody NF-κB (p65) (1:500, Santa Cruz Biotechnology) and mouse monoclonal antibody anti-β-actin (1:200, Santa Cruz Biotechnology). The second day, membranes were washed with PBS containing 0.1 % Tween-20 (TPBS) (10 min × 3) each time, and then incubated with fluorescent labeling second antibodies (IRDye® 800-conjugated goat anti-rabbit or anti-mouse IgG, 1:5,000 dilution, Rockland, Gilbertsville, PA) for 1 h at room temperature. Membranes were then again washed with TPBS (10 min × 3) and the relative density of bands was analyzed on an Odyssey infrared scanner (LI-COR Bioscience, USA). The densitometric values were normalized with respect to the values of β-actin immunoreactivity to correct for any loading and transfer differences between samples. This procedure was repeated 3 times each sample.
Real-Time PCR
30 mice were used in this part (n = 6 per group). Total mRNA was extracted by using the Trizol reagent (Invitrogen, Carlsbad, CA, USA) and was reverse-transcribed into cDNA using Revert Aid first Strand cDNA Synthesis Kit (Fermentas International Inc, Burlington, Canada) for Quantitative PCR (ABI7500, USA) in the presence of a fluorescent dye (SYBR Green I; Cwbio). Relative abundance of mRNA was calculated after normalization to β-actin ribosomal RNA. Results for each sample were collected at least three times. Real-time PCR was used to analyze the levels of Akt, GSK-3β, claudin-5 and NF-κB p65 mRNA at 24 h after pMCAO. The primers are as follows: Akt: F: 5′-GGCAGGATGTGTATGAGAAGAAG-3′, R: 5′-GAGTAGGAGAACGGGAAGT-3′; GSK3β: F: 5′-TCCTTATCCCTCCACATGCTCG-3′, R: 5′-CGTTATTGGTCTGT-CCACGGTCT-3′; NF-κB: F: 5′-GACCTGGAGCAAGCCATTAGC-3′, R: 5′-CGGT-TATCAAAAATCGGATGTGAG-3′; claudin-5: F: 5′-GGCGATTACGACAAGAAG-AACT-3′, R: 5′-TAGTGATGGTCAACGGACTCTG-3′; β-actin: F: 5′-GAGACCTT-CAACACCCGAGC-3′, R: 5′-ATGTCACGCACGATTTCCC-3′.
Data Analysis
Group data in this study was analyzed using SPSS 13.0 software. Quantitative data was represented as mean ± SD. Statistical analysis was performed by One-way ANOVA followed by LSD test for inter-group comparisons. For neurological deficits, Mann–Whitney U-test was used for comparison between two groups. Differences with P < 0.05 were considered statistically significant.
Results
Evo Reduced Neurological Deficits
Neurological deficit was examined and scored on a 6-point scale at 24 h after pMCAO. Compared with Vehicle group, the neurological deficit scores were significantly reduced in Evo-H and Evo-L group (Vehicle group vs. Evo-H group: 3.75 ± 0.93 vs. 2.13 ± 1.02, P < 0.05; Vehicle group vs. Evo-L group: 3.75 ± 0.93 vs. 2.94 ± 1.06, P < 0.05) (Fig. 2a).

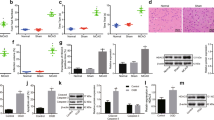
Effects of Evo administration on ischemic mouse brains. a Evo attenuated neurological deficit. Brain samples were obtained from neocortical infarction tissues of Sham group, Vehicle group, pMCAO group, Evo-L group and Evo-H group. The behavioral scores were significantly reduced in Evo-L group and Evo-H group (*P < 0.05) versus Vehicle group. Mann–Whitney test were used. b Evo decreased brain edema in ipsilateral hemisphere. Mice were respectively from five groups described above. The water content of ipsilateral hemisphere was reduced in Evo-L group and Evo-H group (*P < 0.05) versus Vehicle group; whereas no difference was found in contralateral hemispheres. Data are expressed as mean ± SD. One-way ANOVA followed by the LSD test. c Evo reduced the infarct volume in the brain. Samples were obtained from brain tissues of Sham group, pMCAO group, Vehicle group, Evo-L group and Evo-H group. TTC stain: comparison of infarct size between the Vehicle group and Evo treatment group at 24 h. The pale region was the infarct brain tissue and the red region was normal. d Evo reduced %HLV. The %HLV was significantly lower in Evo-L group and Evo-H group (*P < 0.05) than in Vehicle group. Results are mean ± SD of each group. One-way ANOVA followed by the LSD test
Evo Reduced the Brain Water Content
Ipsilateral brain water content of sham group was 78.68 ± 0.59 %. Compared with Vehicle control, Evo-L and Evo-H groups showed an intense decline in the percentage of brain water content (Vehicle group vs. Evo-L group: 84.25 ± 0.57 % vs. 83.16 ± 0.49 %, P < 0.05; Vehicle group vs. Evo-H group: 84.25 ± 0.57 % vs. 82.24 ± 0.31 %, P < 0.05) (Fig. 2b).
Evo Reduced the Infarct Volume
No infarction was observed in Sham group, while extensive lesion was developed in both striatum and lateral cortex in vehicle group. Compared with Vehicle group, the infarct size was significantly reduced both in Evo-H and Evo-L group (Vehicle group vs. Evo-H group: 43.90 ± 2.61 % vs. 32.78 ± 3.78 %, P < 0.05; Vehicle group vs. Evo-L group: 43.90 ± 2.61 % vs. 36.57 ± 3.36 %, P < 0.05) (Fig. 2c, d).
Evo Upregulated the Expression of pAkt, pGSK3β and Downregulated the Expression of NF-κB p65
The results showed that systemic administration of Evo to cerebral ischemic mouse significantly increased the expression of pAkt and pGSK3β at protein level (P < 0.05). While the expression of Akt and GSK3β at protein level was not significantly changed (P > 0.05) (Fig. 3a–d). In agreement with results of western blotting, Akt and GSK3β have the same expression at the mRNA level (P > 0.05) (Fig. 4a).

Immunoblots (a, c and e) and quantitative analysis (b, d and f) of pGSK3β (Ser9), pAkt (Ser473) and nuclear and cytoplasm NF-κB (P65). The expression of pAkt (Ser473) and pGSK3β (Ser9) were increased in Evo-L group and Evo-H group (*P < 0.05) versus Vehicle group, but the expression of Akt and GSK3β were constant in control and experimental groups. The expression of nuclear NF-κB (p65) was decreased in Evo-L group and Evo-H group (*P < 0.05) versus Vehicle group. On the contrary, the expression of cytosolic NF-κB (p65) was increased in Evo-L group and Evo-H group (*P < 0.05) versus Vehicle group. β-actin was used as an endogenous protein loading control. Data represent mean ± SD. One-way ANOVA followed by LSD test

qRT-PCR of Akt, GSK3β and NF-κB genes in the mice brain. The bar graph a showed the qRT-PCR of Evo induction (50 or 100 mg/kg, ig) Akt and GSK3β mRNA. In agreement with the result of Western Blotting, the expression of Akt and GSK3β were constant in control and experimental groups (P > 0.05). The bar graph b showed the qRT-PCR of Evo induction (50 or 100 mg/kg, ig) NF-κB (P65) mRNA. The expression of NF-κB mRNA was reduced in Evo-L group and Evo-H group (*P < 0.05) vs. Vehicle group. β-actin was used as qRT-PCR control. Data are mean ± SD. One-way ANOVA followed by LSD test
Western Blotting (Fig. 3e, f) suggested that the expression of NF-κB p65 nuclear was upregulated after ischemia, and was decreased in Evo-H and Evo-L groups at 24 h compared with Vehicle group (P < 0.05), while the NF-κB p65 cytosolic presented an opposite expression (P < 0.05). Meanwhile, qRT-PCR suggested that the total level of NF-κB was upregulated after ischemia, and was decreased in Evo-H and Evo-L groups at 24 h compared with Vehicle group (Fig. 4b) (P < 0.05).
Evo Protected the Structure and Function of Blood–Brain Barrier (BBB)
To further explore the protective mechanism of Evo in cerebral ischemia, the expression of tight junction protein claudin-5 was examined by western blot and qRT-PCR at 24 h after pMCAO. Compared with Sham group, claudin-5 was significantly downregulated in pMCAO and Vehicle group. Low and high doses of Evo significantly upregulated the reduced expression of claudin-5 at both protein and mRNA levels (both P < 0.05) (Fig. 5a–c).

Claudin-5 expression in mRNA and protein level. Western blotting photographs (a) and the respective quantity analysis bar chart (b) showed the Evo’s effect on claudin-5 protein level. The bar graph (c) showed Evo induction (50 or 100 mg/kg, ig) of claudin-5 mRNA. The expression of claudin-5 was significantly promoted both in mRNA and protein level in Evo-L group and Evo-H group (*P < 0.05) versus Vehicle group. β-actin was used as an endogenous protein loading control. The results were expressed normalized to the β-actin endogenous control in qRT-PCR. Data are mean ± SD. One-way ANOVA followed by LSD test
Discussion
The past decades have seen unprecedented advances in understanding of physiological processes in cerebral ischemia. To a large extent, it is due to the focus on pMCAO as a well-characterized and classical experimental model [36, 37]. Pro-inflammatory responses occur within minutes after the onset of cerebral ischemia and exacerbate the progression of brain damage [2, 38]. So the inhibition of inflammatory responses at the early stage of ischemia provides an attractive therapeutic strategy. In our study, pMCAO model is used to evaluate the expression of pAkt, pGSK-3β, NF-κB and claudin-5 in the acute focal cerebral ischemia and to explore the interrelation between Evo’s neuroprotection effect and the role of the Akt/GSK pathway in mediating the anti-inflammatory effects in vivo.
Evo has been reported to elicit a variety of biological effects in treatment of inflammatory diseases, atherosclerosis, tumors and others. In mouse model of Alzheimer’s disease, Evo improves cognitive abilities through its anti-inflammatory effects. Previous study has provided the evidence that Evo had a direct effect in the hypothalamus [39] and it can repress hypoxia-induced inflammatory response. Additionally, Evo exerts a protective effect on ischemia/reperfusion damage in heart [30]. These above observations raise the possibility that Evo may have protective effects on ischemic brain injury. Therefore, we tried to explore the neuroprotective properties of Evo at 24 h after ischemic stroke, and further investigated the potential mechanisms. Consistent with our hypothesis, Evo administration, both dosage of 50 and 100 mg/kg, could relieve neurological deficits, reduce brain edema, and decrease infarct size in a dose dependant manner, suggesting an effective protection of Evo in acute cerebral ischemia. In this study, we demonstrated that Evo induced rescue of Akt activity and consequently blocked GSK3β dephosphorylation after cerebral ischemia. Furthermore, Evo administration hampered NF-κB p65 shift from cytoplasm to nucleus. Thus, we speculated that activating Akt/GSK signal pathway may be an attractive candidate to explain protective effects of Evo in the acute stage of ischemic stroke.
The Akt/PKB signaling pathway is known as one of the most relevant pathways in regulating neuronal survival [40]. As a primary mediator of survival signals, Akt can phosphorylate and inactivate GSK3β which is a transducer of pro-inflammatory signals [41] at its N-terminus (at Ser9). In addition, Akt also has a role in modulating intracellular glucose metabolism, and consequently enhancing energy production after ischemia. Thus, Akt is an excellent therapeutic target for preserving neuron viability in the acute ischemic period. As recently shown, the inhibitory effect of Akt on inflammation is dependent on its downstream substrate GSK-3β. GSK3 has emerged as a key regulatory switch in the modulation of inflammation [42]. Unlike most kinases, GSK3 is constitutively active in cells and can be inactivated by phosphorylation [43]. Under stimulation, GSK3 is phosphorylated at serine 21 for GSK3α or serine 9 for GSK3β, resulting in the inhibition of GSK3 kinase activity. Under conditions where Akt activity is decreased such as after ischemia, GSK-3β can thus be activated. Functionally, active GSK-3β can enhance the operation of various pro-inflammatory signaling molecules, including NF-κB [42], IL-6, and MCP-1 [44]. In our study, Evo lowered the levels of active GSK-3β after stroke, thereby decreasing the ability of GSK-3β to augment the inflammatory response.
It is well known that NF-κB is critical effecter and regulator of inflammation response [15]. As a regulator of survival and death proteins, NF-κB plays a pivotal role of neuron survival in the central nervous system [45]. For pMCAO it was demonstrated that the role of NF-κB was detrimental [46], so inhibiting the activation of NF-κB is protective and may develops smaller infarct in the acute stage of ischemia [49]. In the current study, accumulation of nuclear NF-κB induced by ischemia was ameliorated after Evo administration, which indicated down-regulating NF-κB may be an attractive candidate to explain protective role of Evo in the acute stage of ischemic stroke.
Ischemia sets into motion a train of events to disrupt the BBB [47, 50]. After cerebral ischemia, BBB’s permeability was altered, peroxidase extravasation frequently involved arterioles, veins and venules surrounded by perivascular spaces [47] and considerable toxic materials enter into the brain, which results in tissue damage and vasogenic edema [48]. As a result, the regulation of BBB is one of the crucial points for the prevention of brain damage [51]. In the BBB, endothelial cells play the main barrier roles in excluding the toxic materials in order to maintain brain homeostasis [52]. Tight junctions (TJs) in endothelial cells are considered to determine vascular permeability. Claudin-5, expressed in large amounts especially in cerebral endothelial cells, is a major component of TJ strands [53], and the altered expression of claudin-5 can increase cerebral endothelial barrier permeability [54]. In our study, the expression of claudin-5 was decreased in the ischemic brain tissues whereas treatment of Evo increased the expression of claudin-5. It was suggested that Evo may play a role in ameliorating the permeability of BBB in cerebral ischemia; this effect may be through upregulating the expression of claudin-5.
In general, this study is the first time to give a scientific warrant to the therapeutic effect of Evo in the treatment of ischemic stroke. Although the approaches where Evo works may not be limited to one pathway, the results provided clear evidence that systemic administration of Evo could decrease neurological impairment and tissue injury under cerebral ischemic conditions and this effect may be through upregulating the expression of pAkt, pGSK3β, downregulating the expression of NF-κB, and ameliorating BBB permeability. This results provided some reasonable and basic research evidences for supporting a hypothesis that Evo may be a legitimate candidate for the treatment of cerebral ischemia and it’s therapeutical effect might be through upregulating Akt/GSK signaling pathway.
References
Fan H, Li L, Zhang X, Liu Y, Yang C, Yang Y, Yin J (2009) Oxymatrine downregulates TLR4, TLR2, MyD88, and NF-kappaB and protects rat brains against focal ischemia. Mediat Inflamm 2009:704–706
Pluta R, Ułamek M, Jabłoński M (2009) Alzheimer’s mechanisms in ischemic brain degeneration. Anat Rec 292:1863–1881
Xing Y, Zhang X, Zhao K, Cui L, Wang L, Dong L, Li Y, Liu Z, Wang C, Zhang X, Zhu C, Qiao H, Ji Y, Cao X (2012) Beneficial effects of sulindac in focal cerebral ischemia: a positive role in Wnt/β-catenin pathway. Brain Res 1482:71–80
Cui L, Zhang X, Yang R, Wang L, Liu L, Li M, Du W (2011) Neuroprotection and underlying mechanisms of oxymatrine in cerebral ischemia of rats. Neurol Res 33:319–324
Li L, Zhang X, Cui L, Wang L, Liu H, Ji H, Du Y (2013) Ursolic acid promotes the neuroprotection by activating Nrf2 pathway after cerebral ischemia in mice. Brain Res 1497:32–39
Zhu C, Zhang X, Qiao H, Wang L, Zhang X, Xing Y, Wang C, Dong L, Ji Y, Cao X (2012) The intrinsic PEDF is regulated by PPARγ in permanent focal cerebral ischemia of rat. Neurochem Res 37:2099–2107
Collino M, Aragno M, Castiglia S, Tomasinelli C, Thiemermann C, Boccuzzi G, Fantozzi R (2009) Insulin reduces cerebral ischemia/reperfusion injury in the hippocampus of diabetic rats: a role for glycogen synthase kinase-3beta. Diabetes 58:235–242
Sen P, Mukherjee S, Ray D, Raha S (2003) Involvement of the Akt/PKB signaling pathway with disease processes. Mol Cell Biochem 253:241–246
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307:1098–1101
Kelly S, Zhao H, Hua Sun G, Cheng D, Qiao Y, Luo J, Martin K, Steinberg GK, Harrison SD, Yenari MA (2004) Glycogen synthase kinase 3beta inhibitor Chir025 reduces neuronal death resulting from oxygen-glucose deprivation, glutamate excitotoxicity, and cerebral ischemia. Exp Neurol 188:378–386
Zhang X, Zhang X, Wang C, Li Y, Dong L, Cui L, Wang L, Liu Z, Qiao H, Zhu C, Xing Y, Cao X, Ji Y, Zhao K (2012) Neuroprotection of early and short-time applying berberine in the acute phase of cerebral ischemia: up-regulated pAkt, pGSK and pCREB, down-regulated NF-kappaB expression, ameliorated BBB permeability. Brain Res 1459:61–70
Kim HJ, Joe Y, Kong JS, Jeong SO, Cho GJ, Ryter SW, Chung HT (2013) Carbon monoxide protects against hepatic ischemia/reperfusion injury via ROS-dependent Akt signaling and inhibition of glycogen synthase kinase 3β. Oxid Med Cell Longev 2013:306421
Malek R, Borowicz KK, Jargiello M, Czuczwar SJ (2007) Role of nuclear factor kappaB in the central nervous system. Pharmacol Rep 59:25–33
Martin M, Rehani K, Jope RS, Michalek SM (2005) Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol 6:777–784
Ji H, Zhang X, Du Y, Liu H, Li S, Li L (2012) Polydatin modulates inflammation by decreasing NF-kappaB activation and oxidative stress by increasing Gli1, Ptch1, SOD1 expression and ameliorates blood-brain barrier permeability for its neuroprotective effect in pMCAO rat brain. Brain Res Bull 87:50–59
Pluta R, Jabłoński M, Ułamek-Kozioł M, Kocki J, Brzozowska J, Januszewski S, Furmaga-Jabłońska W, Bogucka-Kocka A, Maciejewski R, Czuczwar SJ (2013) Sporadic Alzheimer’s disease begins as episodes of brain ischemia and ischemically dysregulated Alzheimer’s disease genes. Mol Neurobiol 48:500–515
Yi JH, Park SW, Kapadia R, Vemuganti R (2007) Role of transcription factors in mediating post-ischemic cerebral inflammation and brain damage. Neurochem Int 50:1014–1027
Zheng Z, Yenari MA (2004) Post-ischemic inflammation: mole-cular mechanisms and therapeutic implications. Neurol Res 26:884–992
Chen L, Wang L, Zhang X, Cui L, Xing Y, Dong L, Liu Z, Li Y, Zhang X, Wang C, Bai X, Zhang J, Zhang L, Zhao X (2012) The protection by Octreotide against experimental ischemic stroke: up-regulated transcription factor Nrf2, HO-1 and down-regulated NF-kappaB expression. Brain Res 1475:80–87
Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M (1999) NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat Med 5:554–559
Wang L, Zhang X, Liu L, Cui L, Yang R, Li M, Du W (2010) Tanshinone II A down-regulates HMGB1, RAGE, TLR4, NF-kappaB expression, ameliorates BBB permeability and endothelial cell function, and protects rat brains against focal ischemia. Brain Res 1321:143–151
Liu Y, Zhang XJ, Yang CH, Fan HG (2009) Oxymatrine protects rat brains against permanent focal ischemia and downregulates NF-kappaB expression. Brain Res 1268:174–180
Yuan SM, Gao K, Wang DM, Quan XZ, Liu JN, Ma CM, Qin C, Zhang LF (2011) Evodiamine improves congnitive abilities in SAMP8 and APP(swe)/PS1(DeltaE9) transgenic mouse models of Alzheimer’s disease. Acta Pharmacol Sin 32:295–302
Chang CP, Chang JY, Wang FY, Tseng J, Chang JG (1995) The effect of Evodia rutaecarpa extract on cytokine secretion by human mononuclear cells in vitro. Am J Chin Med 23:173–180
Lee TJ, Kim EJ, Kim S, Jung EM, Park JW, Jeong SH, Park SE, Yoo YH, Kwon TK (2006) Caspase-dependent and caspase-independent apoptosis induced by evodiamine in human leukemic U937 cells. Mol Cancer Ther 5:2398–2407
Rasul A, Yu B, Zhong L, Khan M, Yang H, Ma T (2012) Cytotoxic effect of evodiamine in SGC-7901 human gastric adenocarcinoma cells via simultaneous induction of apoptosis and autophagy. Oncol Rep 27:1481–1487
Wei WT, Chen H, Wang ZH, Ni ZL, Liu HB, Tong HF, Guo HC, Liu DL, Lin SZ (2012) Enhanced antitumor efficacy of gemcitabine by evodiamine on pancreatic cancer via regulating PI3 K/Akt pathway. Int J Biol Sci 8:1–14
Heo SK, Yun HJ, Yi HS, Noh EK, Park SD (2009) Evodiamine and rutaecarpine inhibit migration by LIGHT via suppression of NADPH oxidase activation. J Cell Biochem 107:123–133
Wei J, Ching LC, Zhao JF, Shyue SK, Lee HF, Kou YR, Lee TS (2013) Essential role of transient receptor potential vanilloid type 1 in evodiamine-mediated protection against atherosclerosis. Acta Physiol (Oxf) 207:299–307
Rang WQ, Du YH, Hu CP, Ye F, Xu KP, Peng J, Deng HW, Li YJ (2004) Protective effects of evodiamine on myocardial ischemia-reperfusion injury in rats. Planta Med 70:1140–1143
Liu YN, Pan SL, Liao CH, Huang DY, Guh JH, Peng CY, Chang YL, Teng CM (2009) Evodiamine represses hypoxia-induced inflammatory proteins expression and hypoxia-inducible factor 1alpha accumulation in RAW264.7. Shock 32:263–269
Longa EZ, Weinstein PR, Carlson S, Cummins R (1989) Reversible middle cerebral arteryocclusion without craniectomy in rats. Stroke 20:84–91
Liu Z, He D, Zhang X, Li Y, Zhu C, Dong L, Zhang X, Xing Y, Wang C, Qiao H, Chen L (2012) Neuroprotective effect of early and short-time applying sophoridine in pMCAO rat brain: down-regulated TRAF6 and up-regulated p-ERK1/2 expression, ameliorated brain infaction and edema. Brain Res Bull 88:379–384
Wang C, Wang Z, Zhang X, Zhang X, Dong L, Xing Y, Li Y, Liu Z, Chen L, Qiao H, Wang L, Zhu C (2012) Protection by silibinin against experimental ischemic stroke: up-regulated pAkt, pmTOR, HIF-1α and Bcl-2, down-regulated Bax, NF-κB expression. Neurosci Lett 529:45–50
Tatlisumak T, Carano RA, Takano K, Opgenorth TJ, Sotak CH, Fisher M (1998) A novel endothelin antagonist, A-127722, attenuates ischemic lesion size in rats with temporary middle cerebral artery occlusion: a diffusion and perfusion MRI study. Stroke 29:850–857
Sun Y, Jin K, Xie L, Childs J, Mao XO, Logvinova A, Greenberg DA (2003) VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J Clin Invest 111:1843–1851
Yang C, Zhang X, Fan H, Liu Y (2009) Curcumin upregulates transcription factor Nrf2, HO-1 expression and protects rat brains against focal ischemia. Brain Res 1282:133–141
Ikeda K, Negishi H, Yamori Y (2003) Antioxidant nutrients and hypoxia/ischemia brain injury in rodents. Toxicology 189:55–61
Shi J, Yan J, Lei Q, Zhao J, Chen K, Yang D, Zhao X, Zhang Y (2009) Intragastric administration of evodiamine suppresses NPY and AgRP gene expression in the hypothalamus and decreases food intake in rats. Brain Res 1247:71–78
Song G, Ouyang G, Bao S (2005) The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med 9:59–71
Grimes CA, Jope RS (2001) The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol 65:391–426
Martinez A, Castro A, Dorronsoro I, Alonso M (2002) Glycogen synthase kinase 3 (GSK-3) inhibitors as new promising drugs for diabetes, neurodegeneration, cancer, and inflammation. Med Res Rev 22:373–384
Cohen P, Frame S (2001) The renaissance of GSK3. Nat Rev Mol Cell Biol 2:769–776
Steinbrecher KA, Wilson W 3rd, Cogswell PC, Baldwin AS (2005) Glycogen synthase kinase 3beta functions to specify gene-specific, NF-kappaB-dependent transcription. Mol Cell Biol 25:8444–8455
Duckworth EA, Butler T, Collier L, Collier S, Pennypacker KR (2006) NF-kappaB protects neurons from ischemic injury after middle cerebral artery occlusion in mice. Brain Res 1088:167–175
Nurmi A, Lindsberg PJ, Koistinaho M, Zhang W, Juettler E, Karjalainen-Lindsberg ML, Weih F, Frank N, Schwaninger M, Koistinaho J (2004) Nuclear factor-kappaB contributes to infarction after permanent focal ischemia. Stroke 35:987–991
Pluta R, Lossinsky AS, Wiśniewski HM, Mossakowski MJ (1994) Early blood-brain barrier changes in the rat following transient complete cerebral ischemia induced by cardiac arrest. Brain Res 633:41–52
Pluta R, Tomida S, Ikeda J, Nowak TS Jr, Klatzo I (1989) Cerebral vascular volume after repeated ischemic insults in the gerbil: comparison with changes in CBF and brain edema. J Cereb Blood Flow Metab 9:163–170
Bi X, Yan B, Fang S, Yang Y, He J, Li XM, Kong J (2009) Quetiapine regulates neurogenesis in ischemic mice by inhibiting NF-kappaB p65/p50 expression. Neurol Res 31:159–166
Yang Y, Rosenberg GA (2011) MMP-mediated disruption of claudin-5 in the blood-brain barrier of rat brain after cerebral ischemia. Methods Mol Biol 762:333–345
Torii H, Kubota H, Ishihara H, Suzuki M (2007) Cilostazol inhibits the redistribution of the actin cytoskeleton and junctional proteins on the blood-brain barrier under hypoxia/reoxygenation. Pharmacol Res 55:104–110
Gloor SM, Wachtel M, Bolliger MF, Ishihara H, Landmann R, Frei K (2001) Molecular and cellular permeability control at the blood-brain barrier. Brain Res Brain Res Rev 36:258–264
Morita K, Sasaki H, Furuse M, Tsukita S (1999) Endothelial claudin: claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J Cell Biol 147:185–194
Stamatovic SM, Keep RF, Andjelkovic AV (2011) Tracing the endocytosis of claudin-5 in brain endothelial cells. Methods Mol Biol 762:303–320
Acknowledgments
This work was funded by the National Natural Science Foundation of China (Grant No. 81371287) and Hebei Province (Grant No. C2010000564). We thank technicians Ruichun Liu and Hongran Wu for their technical assistance and Prof. Yansu Guo M.D. PhD. and Weisong Duan M.D. PhD. for providing valuable suggestions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhao, T., Zhang, X., Zhao, Y. et al. Pretreatment by Evodiamine is Neuroprotective in Cerebral Ischemia: Up-Regulated pAkt, pGSK3β, Down-Regulated NF-κB Expression, and Ameliorated BBB Permeability. Neurochem Res 39, 1612–1620 (2014). https://doi.org/10.1007/s11064-014-1356-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-014-1356-5