
Abstract
Numerous evidences have highlighted the efficient role of dexmedetomidine (DEX) in multi-organ protection. In the present study, the neuroprotective role of DEX on cerebral ischemic injury and the underlining signaling mechanisms were explored. In order to simulate cerebral ischemic injury, we performed middle cerebral artery occlusion in mice and oxygen-glucose deprivation in neurons. Immunohistochemistry, Western blot analysis, and RT-qPCR were used to examine expression of HDAC5, NPAS4, MDM2, and PSD-95 in hippocampus tissues of MCAO mice and OGD-treated neurons. MCAO mice received treatment with DEX and sh-PSD-95, followed by neurological function evaluation, behavioral test, infarct volume detection by TTC staining, and apoptosis by TUNEL staining. Additionally, gain- and loss-of-function approaches were conducted in OGD-treated neuron after DEX treatment. Cell viability and apoptosis were assessed with the application of CCK-8 and flow cytometry. The interaction between MDM2 and PSD-95 was evaluated using Co-IP assay, followed by ubiquitination of PSD-95 detection. As per the results, HDAC5 and MDM2 were abundantly expressed, while NPAS4 and PSD-95 were poorly expressed in hippocampus tissues of MCAO mice and OGD-treated neurons. DEX elevated viability, and reduced LDH leakage rate and apoptosis rate of OGD-treated neurons, which was reversed following the overexpression of HDAC5. Moreover, HDAC5 augmented MDM2 expression via NPAS4 inhibition. MDM2 induced PSD-95 ubiquitination and degradation. In MCAO mice, DEX improved neurological function and behaviors and decreased infarct volume and apoptosis, which was negated as a result of PSD-95 silencing. DEX plays a neuroprotective role against cerebral ischemic injury by disrupting MDM2-induced PSD-95 ubiquitination and degradation via HDAC5 and NPAS4.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ischemia is a pathological condition characterized by the deficiency of blood supply to a limited area of tissue, leading to the subsequent mass production of reactive oxygen species and oxidative damage [1]. Cerebral ischemia may originate from cardiac arrest or stroke, both of which contribute to the insufficient blood supply to the brain parenchyma [2]. Cerebral ischemic injury has a high incidence rate, accounting for 1–4 new cases per 1000 live births, and is the leading cause of neurological disabilities [3]. Despite the advances in currently available therapies in improving patients’ survival rate over the past decades, permanent disability in the form of cognitive, learning, motor, and communication impairments is inevitable in most survivors [4].
Dexmedetomidine (DEX) has demonstrated multiple neuroprotective actions serving as a clinical anesthetic agent; for instance, DEX possesses neuroprotective potentials in ischemia stroke via GLT-1 upregulation and PI3K/AKT signaling pathway activation [5]. In addition, DEX alleviates hypoxia/reoxygenation (H/R)-induced brain injury and protects hippocampal neurons against H/R-induced apoptosis through the activation of the HIF-1α/p53 signaling pathway [6]. The protective role of DEX in the neurons against β-amyloid (Aβ) cytotoxicity by inhibiting the accumulation of histone deacetylase 5 (HDAC5) has been highlighted in a previous study [7]. HDAC5 is a member of the histone deacetylases (HDACs); some isoforms of HDACs can protect brain cells against ischemic injury, while others can promote their death [8]. Concurrently, HDAC5 can impair the inhibited neuronal apoptosis induced by MRTF-A during cerebral ischemia/reperfusion (I/R) injury [9]. HDAC5 has been shown to have a negative correlation with the expression of Neuronal Per-Arnt-Sim domain protein 4 (NPAS4) by binding to its enhancer [10]. NPAS4 is known to be an activity-dependent transcription factor expressed in various brain insults (including cerebral ischemia) and can play a key role in protecting neurons against cerebral ischemia [11]. Overexpression of NPAS4 in olfactory bulb granule cells results in the downregulation in the expression of murine double minute 2 (MDM2) which is considered a p53-specific E3 ubiquitin ligase [12]. Based on previous findings, the MDM2-p53 pathway plays a key role in preconditioning-induced neuroprotection against cerebral ischemic injury and flagged MDM2 to be an essential target in ischemic tolerance [13]. In Fmr1 knockout (KO) neurons, dephosphorylation of MDM2 has been shown to salvage the ubiquitination, degradation, and synapse elimination of postsynaptic density protein 95 (PSD-95) [14]. PSD-95 is regarded as a neuronal plasticity marker, and its protein levels have been found to be decreased in bilateral common carotid artery occlusion (CCAO) models of cerebral ischemia [15]. The main objective for this study is to explore the neuroprotective role of DEX in cerebral ischemic injury along the potential mechanism involving the HDAC5/NPAS4/MDM2/PSD-95 signaling in the established middle cerebral artery occlusion (MCAO) mouse model and oxygen-glucose deprivation (OGD) cell model.
Materials and Methods
Ethics Statement
This study was conducted with the approval of the ethics committee of Shanghai Medical College of Fudan University and in strict accordance with the Guide for the Care and Use of Laboratory animals published by the US National Institutes of Health, with extensive efforts made to minimize animal suffering.
Establishment of MCAO-Induced Mouse Cerebral Ischemic Injury Models
Healthy adult male C57BL/6 J mice weighing 20–28 g purchased from Shanghai Experimental Animal Center (Shanghai, China) were housed at a temperature of 22 °C with 70% humidity under a 12-h light/dark cycle and given ad libitum access to food and water. DEX (SML0956, Sigma-Aldrich Chemical Company, St Louis, MO, USA) was dissolved in normal saline. Then the mice were grouped into the control with 10 mice, sham operation with 10 mice, and MCAO treatment with 60 mice. The MCAO mice received further treatment with normal saline, DEX, normal saline + short hairpin RNA negative control (sh-NC), DEX + sh-NC, and DEX + sh-PSD-95. Two hours before MCAO treatment, the corresponding adenovirus was injected into the mice via the cerebral ventricles, and the mice received intraperitoneal injections with DEX at a dose of 25 μg/kg. Following the administration of anesthesia, the mice were placed in a prostrate position in a stereotactic head frame, and an incision was made along the midline of the scalp, with a hole (0.5 mm posterior and 1.0 mm lateral to the bregma) drilled on the right side of the skull, and 4 μL adenovirus (0.2 μL/min) was then injected into the ventricle (2.5 mm vertically) with a Hamilton syringe through a micro-injection pump (KDS310, KD Scientific Inc., Holliston, MA). The syringe was kept 5 min after injection to prevent leakage. Next, the syringe was removed, and the hole was sealed. Thereafter, the incision was sutured to allow the recovery of the mice. After 2 h, the mice were anesthetized by means of intraperitoneal injections of 1% sodium pentobarbital. The scalp of mice was cut off, after which a thread was connected to monitor cerebral blood flow on the exposed ear skull. Next, a 1-cm incision was made from the angle of the mandible down to the sternum whereupon the left carotid sheath, the common carotid artery, the external carotid artery, and the internal carotid artery were localized. The proximal end of the common carotid artery was ligated twice, and the external carotid artery was separated and ligated, after which the internal carotid artery was separated, and the internal carotid artery was clamped with use of a micro-clamp. A small opening was made on the wall of the external carotid artery, and the head of the thread (head diameter, 0.23 mm; main diameter, 0.18 mm) was inserted into the common carotid artery, after which the foramen of external carotid artery was ligated with use of 5/0 suture. Upon the removal of the clamp, the thread was inserted to the internal carotid artery and up to the middle cerebral artery (about 12.0 mm depth), after which blood flow signal (decreased to about 20% as the indicator) was observed. Absorbent cotton that was soaked in 0.9% sodium chloride injection solution was used to cover the wound. After 1 h of blocking, the thread was pulled out, and double ligation was performed between the thread entrance to the external carotid artery and the bifurcation of the internal carotid artery. Then the thread knot on the common carotid artery was untied to restore the blood flow from the common carotid artery to the internal carotid artery. When the blood flow returned to 100%, the skin was sutured. Thereafter, the artery of sham-operated mice was not plugged. During the operation, the body temperature of mice was monitored, and the mice were given ad libitum access to water and food after the operation. All adenoviruses were purchased from Sangon Biotech Co., Ltd. (Shanghai, China), and primer sequences and plasmids were also constructed by the company.
Isolation and Culture of Neurons
The cerebral cortex was extracted from 1-day-old C57BL/6 J mice and placed in Hank’s balanced salt solution (HBSS) without Ca2+ and Mg2+ but containing 1-mm sodium pyruvate and 10-mm 4-(2-hydroxyethyl)-1-piperazineëthanesulfonic acid (HEPES) buffer. Then the hippocampus tissue was isolated in HBSS solution containing 0.125% trypsin at 37 °C for 10 min. Subsequently, the tissue was triturated and dispersed to single cells, and the digestion was terminated with Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum (FBS, Thermo Fisher Scientific, Waltham, MA, USA). The scattered tissue was allowed to stand for 3 min. Thereafter, the supernatant was transferred to a new centrifuge tube and underwent centrifugation at 2000 rpm for 2 min, after which the precipitate was added to the Neurobasal medium containing B-27, 0.5 mm/L-glutamine, and 20 IU/mL penicillin/streptomycin. Thereafter, the cells were seeded in a 6-well plate coated with poly-D-lysine (PDL) at a density of 4 × 106 cells/well (100 μg/mL) and cultured in a 5% CO2 incubator at 37 °C. Every 2–3 days, half of the fresh medium without glutamic acid was renewed. The purity of neurons was about 95%. After 7 days of culture, the following experiments were carried out.
Neuron Treatment
The neurons were treated with 1-μM DEX for 30 min and infected with lentiviruses for 6 h. Then 18–24 h of culture was conducted in a renewed medium, after which the cells were treated with OGD to simulate hypoxia-ischemia of neurons in vitro. Briefly, the original medium was discarded and washed with phosphate-buffered saline (PBS), after which the cells were added with glucose-free DMEM and cultured in 95% N2 and 5% CO2 anaerobic chamber for 4 h. Conversely, the control cells were cultured in DMEM for 4 h. After OGD treatment, all cells were cultured in the Neurobasal medium in a 5% CO2 incubator at a temperature of 37 °C for 24 h. OGD-treated cells were subsequently treated with normal saline, DEX, DEX + overexpression (oe)-HDAC5, DEX + sh-PSD-95, sh-HDAC5-1, sh-HDAC5-2, sh-NPAS4-1, sh-NPAS4-2, sh-HDAC5 + sh-NPAS4, sh-MDM2-1, sh-MDM2-2, oe-MDM2 + MG132- (infected with oe-MDM2 lentivirus without MG132 treatment), sh-MDM2 + MG132-, oe-MDM2 + MG132+ (infected with oe-MDM2 and treated with MG132), sh-MDM2 + MG132 +, sh-PSD-95-1, and sh-PSD-95-2 as well as their corresponding controls. All lentiviruses were attained from Sangon Biotech Co., Ltd. (Shanghai, China), and primer sequences and plasmids were also constructed by the company.
Evaluation of Neurological Function
At 24 h post-MCAO modeling, the nervous system was examined by two blinded investigators, and the scoring system consists of 7 sub-tests: symmetry of limb movement, spontaneous activity, forepaw extension, climbing, body proprioception, beam walking, and vibration tactile response. Neurological scores ranged from 3 (the most severe defect) to 21 (normal).
Behavioral Test
Pole-climbing test was first conducted. In brief, 24 h after the operation, with head up, mice were put on a vertical wooden pole (length of 50 cm, diameter of 8 mm) with a rough surface. The test indexes include the time of mice spent in turning completely head downwards (Tturn) and the total time it spent in descending down and reach the floor with its front paws (Ttotal). The average value was obtained after 3 tests.
Subsequently, foot fault test was performed as follows: 24 h after the operation, the mice were placed on the horizontal grid floor above the ground surface, and subsequently they were allowed to walk for 2 min. When the mice’s foot stepped on the wrong grid and fell down from the opening between the grids, it would appear foot faults. The number of foot faults on the right injured limb was recorded and analyzed.
Hematoxylin-Eosin Staining
Hippocampus tissues were fixed using 10% neutral formalin for 24 h, followed by gradient alcohol dehydration and clearing using xylene. Tissues were embedded, followed by section. Each of the section was 20-μM-thick coronal brain sections and was cut serially at 400-μM distance, then cleared in xylene, and dehydrated by gradient alcohol. Then, the sections were stained by hematoxylin for 3 min, differentiated by 0.5% hydrochloric acid alcohol for 10 s and turned back to blue for 10 min. Eosin was utilized to stain the sections for 5 min, followed by routine dehydration, clearing and mounting using neutral gum. Finally, the sections were observed using an optical microscope (XP-330; Shanghai Bingyu Optical Instruments Co., Ltd., Shanghai, China) for histological changes of hippocampus tissues.
Terminal Deoxynucleotidyl Transferase-Mediated dUTP-Biotin Nick End Labeling Assay
Cell apoptosis was measured using Apoptosis Assay Kit (C1098, Beyotime Biotechnology Co., Shanghai, China). Cells were dewaxed twice with xylene (5 min/time); rehydrated with absolute alcohol for 5 min, 90% ethanol for 2 min, and 70% ethanol for 2 min; washed with distilled water for 2 min; and treated with 20-μg/mL protease K without DNase at a temperature of 37 °C for 15 min followed by 3 PBS washes. TUNEL assay solution was prepared, 50 μL of which was added to the sample, and incubation was carried out at 37 °C under dark conditions for 60 min. After 3 PBS washes, the cells were blocked with 4′,6-diamidino-2-phenylindole (DAPI) and observed under a microscope (BX63, Olympus, Japan), with apoptosis rate calculated.
2,3,5-Triphenyl Tetrazolium Chloride Staining
One day after model construction, the brain tissue was collected and cut into coronal sections every 1 mm. The sections were then stained by means of TTC at a temperature of 37 °C for 20 min and fixed with 2% paraformaldehyde. The infarcted area was white and the non-infarcted area was pink. The sections were photographed by means of a digital camera (IXUS175, Canon, Japan) and quantified using ImageJ software. Infarct volume and infarct volume percentage were calculated as follows: Infarct volume = total contralateral hemispheral volume − non ischemic ipsilateral hemispheral volume and infarct volume percentage = infarct volume/total contralateral hemispheral volume.
RNA Isolation and Quantitation
Total RNA was extracted from tissues and cells using the TRIzol reagent (16096020, Thermo Fisher Scientific Inc., Waltham, MA, USA), whereupon reverse transcription into complementary DNA (cDNA) was implemented with the use of the First Strand cDNA Synthesis Kit (D7168L, Shanghai Beyotime Biotechnology Co., Ltd., Shanghai, China). Subsequently, reverse transcription quantitative polymerase chain reaction (RT-qPCR) was conducted as per the instructions of RT-qPCR assay kit (Q511-02, Vazyme Biotech, Nanjing, China). β-Actin served as the internal reference for HDAC5, NPAS4, MDM2, and PSD-95 primers. Sangon Biotech Co., Ltd. (Shanghai, China) was authorized to design and synthesize all primer sequences (Table 1). The fold changes were calculated using the 2-ΔΔCt method.
Western Blot Analysis
Total protein was extracted from tissues and cells by lysing with radio-immunoprecipitation assay (RIPA) lysis buffer (P0013B, Shanghai Beyotime Biotechnology Co. Ltd., Shanghai, China) containing phenylmethylsulphonyl fluoride (PMSF) for 5 min. The protein concentration was then determined with a bicinchoninic acid (BCA) kit. After separation by means of sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), the protein was transferred onto a polyvinylidene fluoride membrane. Next, the membrane received treatment with 5% skim milk powder or 5% bovine serum albumin (BSA), followed by overnight incubation at 4 °C with primary rabbit anti-mouse antibodies: β-actin (ab8227, 1:5000), HDAC5 (ab1439, 1:1000), NPAS4 (ab76758, 1:1000), MDM2 (ab16895, 1:5000), PSD-95 (PPS059, 1:1000), cleaved caspase-3 (ab49822, 1:1000), and caspase-3 (ab13847, 1:1000). The following day, the membrane was re-probed with horseradish peroxidase-labeled secondary goat anti-rabbit immunoglobulin G (IgG) (ab6721, 1:5000) or rabbit anti-mouse IgG (ab6728, 1:5000) for 1 h at room temperature post 3 TBST washes. The aforementioned antibodies were from Abcam Inc. (Cambridge, UK) except PSD-95 (R&D Systems, Minneapolis, MN, USA). Afterwards, the membrane was visualized by means of enhanced chemiluminescence (ECL) solution, and the protein bands were quantified by Image Quant LAS 4000C. Protein expression was determined as follows: protein expression = the ratio of the gray value of the target band/the gray value of β-actin band.
Cell Counting Kit-8 Assay
The proliferation of OGD-treated neurons was detected as per the instructions provided on the CCK-8 kit (GK10001, GLPBIO, USA). Each well was added with 10 μL CCK-8 reagents, mixed and incubated in a cell incubator for 1 h. The absorbance value was measured at 450 nm and the cell proliferation curve was drawn.
Flow Cytometry
Cell apoptosis was detected by means of the fluorescein isothiocyanate (FITC) and propidium iodide (PI) kit (C1062L, Beyotime Institute of Biotechnology, Shanghai, China). The cells following varied treatments were cultured in the incubator for 48 h and were collected in 200 μL buffer. Next, the cells were added with 10 μL Annexin V-FITC and 5 μL PI and reacted at room temperature under dark conditions for 15 min, followed by the addition of 300 μL buffer. The apoptosis was finally detected by a flow cytometer, and the apoptosis rate was calculated.
Determination of Lactate Dehydrogenase Leakage Rate
LDH leakage rate was detected by the LDH cytotoxicity kit (CYTODET-RO, Sigma-Aldrich Chemical Company, St Louis, MO, USA). Briefly, the culture medium of neurons exposed to OGD was collected and subjected to centrifugation in order to obtain the supernatant. Next, the cells were lysed using 1% Triton X-100 (T9284, Sigma-Aldrich Chemical Company, St Louis, MO, USA), and cell debris was removed, and cell lysate was obtained with the application of centrifugation. Cell supernatant and cell lysate were respectively subjected to incubation with LDH reaction mixture at a temperature of 37 °C for 15 min. Thereafter, the absorbance value was measured using a microplate reader at 490 nm, which was expressed as the percentage of LDH release in total LDH.
Co-Immunoprecipitation Assay
The interaction between endogenous MDM2 and PSD-95 protein was detected by Co-IP. Cells were lysed with Pierce IP buffer (1% Triton X-100, 1-mm ethylenediamine tetraacetic acid [EDTA], 150-mm NaCl, 25-mm Tris HCl, pH = 7.5) and added with protease and phosphatase inhibitors. Overnight incubation on cell lysate was implemented at a temperature of 4 °C with mouse antibody to PSD-95 (ab2723, 1:50, Abcam Inc., Cambridge, UK) and mouse antibody to IgG (3420, 1:50, Cell Signaling Technology, Beverly, MA, USA). The sample was then added with protein G beads (Dynabeads, Thermo Fisher Scientific Inc., Waltham, MA, USA), slowly rotated at 4 °C for 8 h, and finally analyzed by Western blot analysis.
In Vivo Ubiquitination Experiments
Endogenous PSD-95 ubiquitination was determined. In short, the cells were incubated with 10-μM MG132 (HY-13259, MedChemExpress, USA) for 6 h and then infected with viruses. Cells were subjected to lysing in 1% SDS RIPA buffer and sonication. IP reaction was then carried out. Briefly, the diluted cell lysate (0.1% SDS) underwent incubation overnight at a temperature of 4 °C with mouse anti-mouse PSD-95 (ab2723, 1:50, Abcam Inc.), after which the protein G beads were added for an additional 8 h of incubation at 4 °C. After 3 washes in IP buffer, Western blot analysis was employed to detect the ubiquitination of PSD-95 with the use of rabbit anti-mouse ubiquitin (ab7780, 1:1000, Abcam Inc.).
Immunohistochemistry
Tissue sections were dewaxed and rehydrated. Slides underwent incubation in citrate solution for 10 min for antigen retrieval. The sections were blocked with 1% BSA at room temperature for 2 h and then probed with primary antibodies: rabbit anti-mouse HDAC5 (ab1439, 1:100, Abcam Inc.), rabbit anti-mouse NPAS4 (SAB4501155, 1:100, Sigma-Aldrich Chemical Company, St Louis, MO, USA), mouse anti-mouse MDM2 (ab16895, 1:100, Abcam Inc.), and mouse anti-mouse PSD-95 (ab2723, 1:100, Abcam Inc.) overnight at a temperature of 4 °C. The next day, the sections were re-probed with secondary goat anti-rabbit (ab6721, 1:500, Abcam Inc.) or rabbit anti-mouse (ab6728, 1:500, Abcam Inc.) at room temperature for 45 min. The 3,3′-diaminobenzidine (DAB) kit was applied for development. The sections were finally analyzed under a microscope (BX63, Olympus, Japan)
Statistical analysis
All data were processed with the use of the SPSS 21.0 software (IBM Corp. Armonk, NY, USA). Measurement data were expressed as mean ± standard deviation. Two-group comparisons were analyzed using unpaired t-test, while multiple-group comparisons were performed by means of one-way analysis of variance (ANOVA) with Tukey’s tests. A value of p < 0.05 denotes statistical significance.
Results
HDAC5 Was Highly Expressed in MCAO Mice and OGD-Treated Neurons
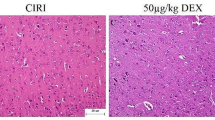
MCAO-induced mouse and OGD-provoked cell cerebral ischemic injury models were initially developed to investigate the mechanism of neuronal damage of hdac5 secondary to cerebral ischemic injury. As shown in Fig. 1a, neurological score presented without obvious changes in the sham-operated mice and normal mice (p > 0.05). MCAO mice had a lower neurological score in comparison with sham-operated mice (p < 0.05). In addition, MCAO mice had a higher number of foot faults than sham-operated mice (p < 0.05; Fig. 1b). As depicted in Fig. 1c, Tturn and Ttotal of MCAO mice were prolonged compared to sham-operated mice (p < 0.05). Histopathological analysis on cerebral tissues was conducted via HE staining, the results of which suggested sparse pyramidal cells with disordered arrangement, neuron loss, pyknosis and edema, deep staining, and unclear nucleolus in MCAO mice compared with sham-operated mice (Fig. 1d). TUNEL results showed augmented neuron apoptosis rate of MCAO mice relative to the sham-operated mice (p < 0.05; Fig. 1e). Furthermore, the results of TTC staining revealed that the volume of cerebral infarction of MCAO mice was increased versus sham-operated mice (p < 0.05; Fig. 1f). These results were indicative of the successful establishment of the MCAO mouse model. The histopathological changes of cerebral tissues following cerebral ischemia included atrophy of cortex and degeneration of neurons in the cortex and hippocampus. Thus, we employed immunohistochemistry to measure HDAC5 expression in hippocampus tissues of MCAO mice, the results of which showed that the positive expression rate of HDAC5 both at mRNA and protein levels presented with an upward trend in hippocampus tissues of MCAO mice (p < 0.05; Fig. 1g). RT-qPCR and Western blot analysis yielded similar results in the mRNA and protein expression of HDAC5 in in hippocampus tissues of MCAO mice (p < 0.05; Fig. 1g).

Abundant HDAC5 expression in MCAO mice and OGD-treated cells. a Scatter diagram of mouse neurological score. b Scatter diagram of mouse foot faults. c Scatter diagram of mouse pole-climbing test. d HE staining analysis (× 400) of mouse cerebral tissues. e Cell apoptosis of mouse cerebral tissues measured by TUNEL staining. f TTC staining analysis of mouse cerebral infarction. g Expression of HDAC5 in mouse hippocampus tissues measured by immunohistochemistry, RT-qPCR, and Western blot analysis. h Cell viability upon OGD induction detected by CCK-8 assay. i LDH leakage rate in cells upon OGD induction. j Cell apoptosis upon OGD induction measured by flow cytometry. k Western blot analysis of cleaved caspase-3 and caspase-3 proteins in OGD-treated cells. l mRNA expression of HDAC5 in OGD-treated cells detected by RT-qPCR. m Western blot analysis of HDAC5 protein in OGD-treated cells. *p < 0.05 vs. sham-operated mice or control cells. n = 10 for mice in the normal, sham, and MCAO group. Data were analyzed by unpaired t-test. Cell experiment was conducted in triplicate
Subsequent results illustrated a reduction in neuron viability (Fig. 1h), as well as an increase in LDH leakage rate (Fig. 1i) and apoptosis rate (Fig. 1j) following OGD treatment (all p < 0.05). Western blot analysis showed elevated expression of cleaved caspase-3 in OGD-treated cells versus control cells (p < 0.05), while caspase-3 expression showed no alterations (p > 0.05; Fig. 1k). In addition, OGD-treated cells presented with upregulated mRNA and protein expression of HDAC5 (p < 0.05; Fig. 1l,m). The aforementioned results suggested that HDAC5 was extensively expressed in mice following cerebral ischemic injury and OGD-treated cells, which might be correlated to neuron apoptosis.
DEX Inhibited HDAC5 Expression in MCAO Mice and OGD-Treated Neurons
Next, we aimed to determine the interaction between DEX and HDAC5 as DEX has been reported to be protective against cerebral ischemic injury [16]. The hippocampus tissues obtained from MCAO and OGD-treated were added with DEX to detect the expression of HDAC5 using immunohistochemistry. The results of immunohistochemistry showed a decreased positive expression rate of HDAC5 protein in MCAO mice treated with DEX (p < 0.05; Fig. 2a). Moreover, the results of RT-qPCR and Western blot analysis demonstrated that HDAC5 mRNA and protein expression were downregulated in hippocampus tissues of MCAO mice treated with DEX (p < 0.05; Fig. 2b,c). In addition, the results of RT-qPCR and Western blot analysis also revealed a decline in HDAC5 expression in OGD-treated neurons treated with DEX (p < 0.05; Fig. 2d,e). Collectively, DEX had the potential to downregulate the expression of HDAC5 in MCAO mice and OGD-treated neurons.

DEX downregulates HDAC5 expression in MCAO mice and OGD-treated neurons. MCAO mice and OGD-treated cells were treated with normal saline or DEX. a Positive expression rate of HDAC5 protein in mouse cerebral tissues measured by immunohistochemistry. b mRNA expression of HDAC5 in mouse hippocampus tissues detected by RT-qPCR. c Western blot analysis of HDAC5 protein in mouse hippocampus tissues. d mRNA expression of HDAC5 in OGD-treated cells detected by RT-qPCR. e Western blot analysis of HDAC5 protein in OGD-treated cells. *p < 0.05 vs. MCAO mice treated with normal saline or OGD cells treated with normal saline. n = 10 for mice following each treatment. Data were analyzed by one-way ANOVA with Tukey’s tests. Cell experiment was conducted in triplicate
Overexpression of HDAC5 Abolished the Protective Potential of DEX on OGD-Treated Neurons
Neurons treated with OGD and DEX were infected with lentiviruses carrying overexpression of HDAC5 to further elucidate whether DEX affects the cerebral infarction of MACO mice through HDAC5. RT-qPCR and Western blot analysis revealed an increase in HDAC5 expression upon treatment with oe-HDAC5 in the presence of DEX (p < 0.05; Fig. 3a,b). In addition, DEX treatment resulted in an increase in neuron viability (Fig. 3c) and a reduction in LDH leakage rate (Fig. 3d) and apoptosis rate (Fig. 3e), while dual treatment with DEX and oe-HDAC5 diminished cell viability and potentiated LDH leakage rate and apoptosis rate (all p < 0.05). Then, Western blot analysis results showed downregulated expression of cleaved caspase-3 in OGD cells treated with DEX, which was reversed following dual treatment with DEX and oe-HDAC5 (p < 0.05), while no alterations were observed in caspase-3 expression in cells upon any treatment (p > 0.05; Fig. 3f). Taken together, the protective effect of DEX on OGD-treated neurons could be reversed through the upregulation of HDAC5.

Upregulated HDAC5 undermines the protective effect of DEX on OGD-treated neurons. OGD-treated cells were treated with normal saline, DEX, DEX + oe-NC, or DEX + oe-HDAC5. a mRNA expression of HDAC5 in OGD-treated cells detected by RT-qPCR. b Western blot analysis of HDAC5 protein in OGD-treated cells. *p < 0.05 vs. OGD cells treated with DEX + oe-NC. c Cell viability detected by CCK-8 assay. d LDH leakage rate in OGD-treated cells. e Cell apoptosis measured by flow cytometry. f Western blot analysis of cleaved caspase-3 and caspase-3 proteins in OGD-treated cells. *p < 0.05 vs. OGD cells treated with normal saline. #p < 0.05 vs. OGD cells treated with DEX + oe-NC. n = 10 for mice following each treatment. Data were analyzed by one-way ANOVA with Tukey’s tests. Cell experiment was conducted in triplicate
HDAC5 Promoted MDM2 Expression by Inhibiting NPAS4 in OGD-Treated Neurons
Next, we sought to dissect out the mechanism of HDAC5 in neuron injury. Firstly, the expression of NPAS4 and MDM2 in hippocampus tissues of MCAO mice was detected with the application of immunohistochemistry, the results of which showed decreased positive expression of NPAS4 and upregulated MDM2-positive expression in hippocampus tissues of MCAO mice (p < 0.05; Fig. 4a). Meanwhile, the results of RT-qPCR and Western blot analysis displayed that NPAS4 expression was decreased, while MDM2 expression was elevated in hippocampus tissues of MCAO mice (p < 0.05; Fig. 4b,c).

HDAC5 upregulates MDM2 expression via NPAS4 downregulation in OGD-treated neurons. a Positive expression of NPAS4 and MDM2 proteins in MCAO mice detected by immunohistochemistry. *p < 0.05 vs. sham-operated mice. b mRNA expression of NPAS4 and MDM2 in mouse hippocampus tissues detected by RT-qPCR. c Western blot analysis of NPAS4 and MDM2 proteins in mouse hippocampus tissues. d mRNA expression of NPAS4 and MDM2 in OGD-treated neurons detected by RT-qPCR. e Western blot analysis of NPAS4 and MDM2 proteins in OGD-treated cells. f Silencing efficiency of HDAC5 and NPAS4 in OGD-treated neurons evaluated by RT-qPCR. g Silencing efficiency of HDAC5 and NPAS4 in OGD-treated neurons evaluated by Western blot analysis. h Western blot analysis of HDAC5, NPAS4, and MDM2 proteins in OGD cells treated with sh-HDAC5 or sh-HDAC5 + sh-NPAS4. *p < 0.05 vs. control cells or OGD cells treated with sh-NC. #p < 0.05 vs. OGD cells treated with sh-HDAC5 + sh-NC. Data in (a)–(c) were analyzed by analyzed unpaired t-test and data in (d)–(f) by one-way ANOVA with Tukey’s tests. Cell experiment was conducted in triplicate
Additionally, RT-qPCR and Western blot analysis revealed a decline in NPAS4 expression, while MDM2 expression was elevated in OGD-treated cells (p < 0.05; Fig. 4d,e). The above data suggested that MCAO mice and OGD-treated neurons have poor NPAS4 expression and high MDM2 expression.
To further detect whether HDAC5 can promote the expression of MDM2 by inhibiting NPAS4, we first performed RT-qPCR and Western blot analysis, which demonstrated that the expression of HDAC5 was reduced in response to sh-HDAC5-1 or sh-HDAC5-2, and the expression of NPAS4 was also decreased upon sh-NPAS4-1 and sh-NPAS4-2 treatments. sh-HDAC5-1 and sh-NPAS4-1 were observed to have improved silencing efficiency (p < 0.05; Fig. 4f,g) and therefore were used for subsequent experiments. Subsequently, the results of Western blot analysis presented with downregulated expression of HDAC5 and MDM2 and upregulated NPAS4 expression in the absence of si-HDAC5 (p < 0.05). Moreover, treatment with both sh-HDAC5 and sh-NPAS4 led to reduced NPAS4 expression (p < 0.05) and augmented MDM2 expression (p < 0.05) in addition to no difference in HDAC5 expression (p > 0.05; Fig. 4h). The aforementioned data supported that HDAC5 might affect cell biological functions via the inhibition of NPAS4 and enhancement of MDM2 expression in OGD-treated neurons.
MDM2 Ubiquitinates and Degrades PSD-95 in OGD-Treated Neurons
The regulatory role of MDM2 in the ubiquitination of PSD-95 was investigated in neurons. As shown in Fig. 5a, immunohistochemistry staining revealed that the expression of PSD-95 was reduced in hippocampus tissues of MCAO mice (p < 0.05). Besides, RT-qPCR and Western blot analysis revealed a decline in PSD-95 expression in hippocampus tissues of MCAO mice (p < 0.05) (Fig. 5b,c). Moreover, in OGD-treated neurons, PSD-95 expression was downregulated as illustrated by RT-qPCR and Western blot analysis (p < 0.05) (Fig. 5d,e). Next, RT-qPCR and Western blot analysis confirmed the efficiency of MDM2 overexpression or silencing in neurons, which was evidenced by increased expression of MDM2 following treatment with oe-MDM2 and decreased MDM2 expression upon sh-MDM2-1 and sh-MDM2-2 treatments (p < 0.05; Fig. 5f,g). Afterwards, sh-MDM2-1 was found to have the superior silencing effect and was selected for subsequent experiments.

MDM2 induces PSD-95 ubiquitination and degradation in OGD-treated neurons. a Positive expression of PSD-95 protein in MCAO mice detected by immunohistochemistry. *p < 0.05 vs. sham-operated mice. b mRNA expression of PSD-95 in mouse hippocampus tissues detected by RT-qPCR. c Western blot analysis of PSD-95 protein in mouse hippocampus tissues. d mRNA expression of PSD-95 in OGD-treated neurons detected by RT-qPCR. e Western blot analysis of PSD-95 protein in OGD-treated cells. *p < 0.05 vs. control cells. f Overexpression efficiency of MDM2 in neurons evaluated by RT-qPCR. g Silencing efficiency of MDM2 in neurons evaluated by Western blot analysis. h Western blot analysis of PSD-95 protein in OGD cells treated with oe-MDM2, sh-MDM2, or in combination with MG132-. *p < 0.05 vs. oe-NC-treated cells. #p < 0.05 vs. sh-NC-treated cells. i Interaction between PSD-95 and MDM2 detected by Co-IP assay. j Representative images of PSD-95 ubiquitination and quantitation. k Western blot analysis of PSD-95 protein expression in the presence of DEX or oe-HDAC5. *p < 0.05 vs. OGD cells treated with normal saline. #p < 0.05 vs. OGD cells treated with DEX + oe-NC. Data in (b) and (c) were analyzed by analyzed unpaired t-test and data in (a), (d)–(g), and (i) by one-way ANOVA with Tukey’s tests
Western blot analysis showed a decreased expression of PSD-95 protein in the presence of oe-MDM2 versus the treatment of oe-NC (p < 0.05), while increased PSD-95 protein expression was observed in response to treatment with sh-MDM2 in contrast to the treatment of sh-NC (p < 0.05). After MG132 was supplemented, no significant difference was detected in the PSD-95 protein expression following each treatment (p > 0.05; Fig. 5h). The results of Co-IP assay illustrated an interaction between MDM2 and PSD-95 (Fig. 5i). In addition, the ubiquitination of PSD-95 was enhanced secondary to treatment with MDM2 overexpression (p < 0.05), while it was attenuated upon MDM2 knockdown (p < 0.05; Fig. 5j). The subsequent objective was to further study the effect of HDAC5 on the expression of PSD-95 through the NPAS4/MDM2 axis. As shown in Fig. 5k, the protein expression of PSD-95 was upregulated in DEX-treated OGD cells (p < 0.05), which was negated by overexpression of HDAC5 (p < 0.05). In summary, MDM2 enhanced the ubiquitination and degradation of PSD-95 in OGD-treated neurons.
PSD-95 Knockdown Reversed the Neuroprotection of DEX on OGD-Treated Neurons
Next, a series of experiments were conducted to further verify whether DEX could promote the expression of PSD-95 through the HDAC5/NPAS4/MDM2 axis and protect the OGD-induced neurons. First, RT-qPCR and Western blot analysis were used to detect the efficiency of PSD-95 silencing in neurons. The results showed that PSD-95 expression was reduced in response to sh-PSD-95-1 and sh-PSD-95-2 (p < 0.05), of which sh-PSD-95-1 presented with a superior silencing efficiency (Fig. 6a,b). Thereby, sh-PSD-95-1 was selected for follow-up experiments. Western blot analysis revealed a downward trend in the expression of HDAC5 and MDM2 (p < 0.05) and an upward trend in the expression of NPAS4 and PSD-95 in DEX-treated cells (p < 0.05). In the presence of DEX, the expression of HDAC5, NPAS4, and MDM2 was not affected (p > 0.05), and PSD-95 expression was reduced following treatment with sh-PSD-95 (p < 0.05; Fig. 6c). In addition, treatment with DEX led to an enhancement in neuron viability (Fig. 6d) and a decline in LDH leakage rate (Fig. 6e) and apoptosis rate (Fig. 6f), but these trends were reversed following silencing of PSD-95 (all p < 0.05). Furthermore, the results of Western blot analysis displayed a decreased expression of cleaved caspase-3 in response to DEX, which was increased upon treatment with PSD-95 knockdown (p < 0.05), while no alterations were observed in caspase-3 expression upon any treatment (p > 0.05; Fig. 6g). These findings indicated that PSD-95 silencing neutralized the neuroprotective effect of DEX on OGD-treated neurons.

PSD-95 silencing abrogates the neuroprotective effect of DEX on OGD-treated neurons. a Silencing efficiency of PSD-95 in neurons evaluated by RT-qPCR. b Silencing efficiency of PSD-95 in neurons evaluated by Western blot analysis. *p < 0.05 vs. sh-NC-treated cells. OGD-induced cells were treated with DEX + sh-NC or DEX + sh-PSD-95. c Western blot analysis of HDAC5, NPAS4, MDM2, and PSD-95 proteins in cells. d Cell viability detected by CCK-8 assay. e LDH leakage rate in cells. f Cell apoptosis measured by flow cytometry. g Western blot analysis of cleaved caspase-3 and caspase-3 proteins in cells. *p < 0.05 vs. cells treated with normal saline + sh-NC. #p < 0.05 vs. cells treated with DEX + sh-NC. Data in (a)–(g) were analyzed by one-way ANOVA with Tukey’s tests
PSD-95 Knockdown Rescued the Neuroprotection of DEX on Cerebral Ischemic Injury in Mice
Finally, we aimed to validate whether DEX could promote the expression of PSD-95 through the HDAC5/NPAS4/MDM2 axis and protect cerebral ischemic injury in vivo. After intracerebroventricular injection of adenovirus, mice were subjected to MCAO. The results of Western blot analysis showed reduced expression of HDAC5 and MDM2 (p < 0.05) and elevated expression of NPAS4 and PSD-95 in MCAO mice treated with DEX (p < 0.05). Treatment with both DEX and sh-PSD-95 caused no changes in the expression of HDAC5, NPAS4, and MDM2 (p > 0.05) but decreased PSD-95 expression in MCAO mice (p < 0.05; Fig. 7a).

PSD-95 silencing abrogates the neuroprotective effect of DEX on cerebral ischemic injury in mice. MCAO mice were treated with DEX + sh-NC or DEX + sh-PSD-95. a Western blot analysis of HDAC5, NPAS4, MDM2, and PSD-95 proteins in mice. b Scatter diagram of mouse neurological score. c Scatter diagram of mouse foot faults. d Scatter diagram of mouse pole-climbing test. e HE staining analysis (× 400) of mouse cerebral tissues. f Cell apoptosis of mouse cerebral tissues measured by TUNEL staining. g TTC staining analysis of mouse cerebral infarction and quantitative analysis of infraction volume. *p < 0.05 vs. MCAO mice treated with normal saline + sh-NC. #p < 0.05 vs. MCAO mice treated with DEX + sh-NC. Data in (a)–(g) were analyzed by one-way ANOVA with Tukey’s tests. n = 10 for mice following each treatment
As shown in Fig. 7b, neurological score was increased in MCAO mice treated with DEX (p < 0.05), while MCAO mice treated with DEX + sh-PSD-95 had reduced neurological score (p < 0.05). In addition, DEX-treated MCAO mice exhibited a decreased number of foot faults (p < 0.05), which was negated by dual treatment with DEX and sh-PSD-95 (p < 0.05; Fig. 7c). Pole-climbing test results depicted in Fig. 7d clarified that Tturn and Ttotal of DEX-treated MCAO mice were shortened (p < 0.05); however, this trend was reversed in response to dual treatment with DEX and sh-PSD-95 (p < 0.05).
Moreover, histopathological analysis by HE staining revealed that DEX treatment could diminish the sparse cortex pyramidal cells, disordered arrangement, neuron loss, pyknosis and edema, deep staining, and unclear nucleolus in MCAO mice (p < 0.05), which was aggravated following further sh-PSD-95 treatment (p < 0.05; Fig. 7e).
TUNEL assay revealed diminished neuron apoptosis rate in DEX-treated MCAO mice (p < 0.05) yet enhanced apoptosis rate in MCAO mice co-treated with DEX and sh-PSD-95 (p < 0.05) (Fig. 7f). Additionally, the results of TTC staining demonstrated that the volume of cerebral infarction of DEX-treated MCAO mice was decreased (p < 0.05), while it was increased following additional treatment with sh-PSD-95 treatment (p < 0.05; Fig. 7g). The above-mentioned results supported the notion that the protective potential of DEX on cerebral ischemic injury of mice could be eliminated by PSD-95 knockdown.
Discussion
Cerebral ischemia is a major cause of neuronal injury or death that could subsequently result in severe neurological dysfunctions or even death in patients [17]. Polyphenol compounds, redox status, and the vitagene network have been highlighted to have correlations and biological relevance in neuroprotection. For instance, nutrition and dietary interventions support cognitive function and preserve brain health and could serve as potential protective methods against cognitive ageing [18]. A recent study also indicated the potential of plant polyphenols, or their molecular scaffolds, as nutraceuticals to combat aging and other related degenerative diseases [19]. Gammapyrone, compound of gamma-aminobutyric acid (GABA), can ameliorate spatial learning and memory impairment and prevent mitochondrial dysfunction and astroglial and microglial neuroinflammation [20]. In addition, anserine is a potent antioxidant that activates the intracellular Hsp70/HO-1 defense system under oxidative and glycative stress [21], thereby exerting neuroprotective effects against permanent focal ischemia [22]. Hormesis is the underlying cause for the aforementioned therapeutic effects [23, 24]; specifically, the Ginkgo biloba (a plant-derived agent) dose responses indicate the occurrence of hormetic dose responses which consistently appear independent of the biological model, inducing agent, endpoint, and/or mechanism [25]. Thus, ischemic insult is also a preconditioning signal leading to cellular protection. In this study, we found that DEX could potentially play a significant neuroprotective role in cerebral ischemic injury by impairing HDAC5-mediated NPAS4 inhibition and preventing MDM2-induced PSD-95 ubiquitination and degradation.
Some HDACs have been preferentially expressed in the brain and have been identified as regulators of neurodegeneration [26]. In several neurologic diseases, including Alzheimer’s disease and cerebral ischemic injury, inhibition of HDAC has neuroprotective properties, as indicated by findings obtained from animal studies [27]. The expression and activity of HDAC5 has been found to be upregulated in a rat cerebral I/R injury model induced by MCAO, and it can effectively inhibit MRTF-A-provoked anti-apoptotic effects on neurons [9]. This finding is in agreement with our current study suggesting that HDAC5 was extensively expressed in both MCAO-provoked mouse cerebral ischemic injury models and OGD-treated neurons and could be associated with neuron apoptosis.
Previous evidence revealed that DEX treatment can result in a decrease in the expression of HDAC5 and consequently relieves sepsis-induced acute kidney injury [28]. We also found that DEX could potentially downregulate the expression of HDAC5 in MCAO mice and OGD-treated neurons. The neuroprotective effect of DEX on cerebral I/R injury has been extensively reported. DEX can markedly decrease the number of apoptotic cells and suppress TLR4/NF-kB signaling pathway, thus preventing the cerebral I/R injury [16]. Postconditioning with DEX could protect against cerebral I/R injury by increasing the neuronal cell survival and reducing the production of autophagic vesicles [29]. Another recent study revealed that DEX can protect the neurons and astrocytes against Aβ cytotoxicity via brain-derived neurotrophic factor (BDNF) production [7]. Concurrently, pharmacological inhibition of HDAC5 has been shown to confer neuroprotective effects and prevent stroke-induced brain damage [30]. Based on the aforementioned findings, we can conclude that overexpression of HDAC5 might abolish the protective potential of DEX on OGD-treated neurons.
NPAS4 is a potent neuroprotective transcription factor against MCAO-induced brain damage [31]. Kumar et al. discovered that HDAC5 binds to the enhancer of the NPAS4 gene and negatively regulates NPAS4 expression [10]. NPAS4 deficiency leads to the upregulated expression of the E3 ubiquitin ligase MDM2 in olfactory bulb granule cells [12], which is indicative of the presence of a negative correlation between NPAS4 and MDM2. Both stroke tissues and OGD/R-treated cells presented with enhanced MDM2 expression, the loss of which augments cell proliferation and attenuates apoptosis, inducing a decrease in the infarct size in MCAO/R brains [32]. Therefore, HDAC5 could potentially promote MDM2 expression via NPAS4 inhibition in OGD-treated neurons.
Another key finding from the present study revealed that MDM2 ubiquitinated and degraded PSD-95 in OGD-treated neurons. Similarly, activation of MDM2 has been reported to trigger ubiquitination, degradation, and synapse elimination of PSD-95 in Fmr1 KO neurons [14]. The parietal cortex and hippocampus of the MCAO rats presented with a decreased expression of PSD-95 [33]. Furthermore, isoflurane can result in decreased PSD-95 expression, increased neuronal cell apoptosis, and reduced spatial learning and memory abilities, all of which can be abolished following treatment with DEX [34]. The administration of DEX (20 μg/kg) and its ability to significantly enhance of neurogenesis and astrogenesis in the hippocampus and upregulate PSD-95 expression has been previously highlighted [35]. Propofol can evidently result in neuronal injury in rat hippocampus, which, however, is ameliorated by DEX at different doses, manifested by reduced hippocampal neuronal cell apoptosis yet increased PSD-95 expression [36]. The aforementioned data led to us concluding that PSD-95 silencing could eliminate the protective effect of DEX on OGD-treated neurons and cerebral ischemic injury.
In summary, our present study provided evidence that DEX has a potential neuroprotective property against cerebral ischemic injury via the HDAC5/NPAS4/MDM2/PSD-95 axis (Fig. 8). These findings may provide novel insights in the development of new therapies for the prevention of cerebral ischemic injury. Nonetheless, further studies are required to verify the underlying target gene-based mechanisms with the use of advanced approaches, along with the clinical potentials and safety of DEX, particularly in neonatal medicine. In addition, the knockout and knockdown of HDAC5 and its potential protection of neurons against OGD damage require further experiments.

The mechanism graph of the regulatory network and function of DEX. DEX downregulates HDAC5 expression, increases NPAS4 expression, and reduces PSD-95 ubiquitination and degradation caused by MDM2, ultimately preventing cerebral ischemic injury
Data Availability
These data and materials of study were available.
References
Gao M, Wang R, Yu F, You J, Chen L (2018) Imaging and evaluation of sulfane sulfur in acute brain ischemia using a mitochondria-targeted near-infrared fluorescent probe. J Mater Chem B 6(17):2608–2619
Belov Kirdajova D, Kriska J, Tureckova J, Anderova M (2020) Ischemia-triggered glutamate excitotoxicity from the perspective of glial cells. Front Cell Neurosci 14:51
Zeng Q, Lian W, Wang G, Qiu M, Lin L, Zeng R (2020) Pterostilbene induces Nrf2/HO-1 and potentially regulates NF-kappaB and JNK-Akt/mTOR signaling in ischemic brain injury in neonatal rats. 3 Biotech 10(5):192
Zhang P, Zhang Y, Zhang J, Wu Y, Jia J, Wu J, Hu Y (2013) Early exercise protects against cerebral ischemic injury through inhibiting neuron apoptosis in cortex in rats. Int J Mol Sci 14(3):6074–6089
Peng M, Ling X, Song R, Gao X, Liang Z, Fang F, Cang J (2019) Upregulation of GLT-1 via PI3K/Akt pathway contributes to neuroprotection induced by dexmedetomidine. Front Neurol 10:1041
Gao Y, Yin H, Zhang Y, Dong Y, Yang F, Wu X, Liu H (2019) Dexmedetomidine protects hippocampal neurons against hypoxia/reoxygenation-induced apoptosis through activation HIF-1alpha/p53 signaling. Life Sci 232:116611
Wang Y, Jia A, Ma W (2019) Dexmedetomidine attenuates the toxicity of betaamyloid on neurons and astrocytes by increasing BDNF production under the regulation of HDAC2 and HDAC5. Mol Med Rep 19(1):533–540
Demyanenko S, Neginskaya M, Berezhnaya E (2018) Expression of class I histone deacetylases in ipsilateral and contralateral hemispheres after the focal photothrombotic infarction in the mouse brain. Transl Stroke Res 9(5):471–483
Li P, Chen H, Chen S, Mo X, Li T, Xiao B, Yu R, Guo J (2017) Circular RNA 0000096 affects cell growth and migration in gastric cancer. Br J Cancer 116(5):626–633
Taniguchi M, Carreira MB, Cooper YA, Bobadilla AC, Heinsbroek JA, Koike N, Larson EB, Balmuth EA et al (2017) HDAC5 and its target gene, Npas4, function in the nucleus accumbens to regulate cocaine-conditioned behaviors. Neuron 96(1):130–144 e136
Choy FC, Klaric TS, Koblar SA, Lewis MD (2015) The role of the neuroprotective factor Npas4 in cerebral ischemia. Int J Mol Sci 16(12):29011–29028
Yoshihara S, Takahashi H, Nishimura N, Kinoshita M, Asahina R, Kitsuki M, Tatsumi K, Furukawa-Hibi Y et al (2014) Npas4 regulates Mdm2 and thus Dcx in experience-dependent dendritic spine development of newborn olfactory bulb interneurons. Cell Rep 8(3):843–857
Vecino R, Burguete MC, Jover-Mengual T, Agulla J, Bobo-Jimenez V, Salom JB, Almeida A, Delgado-Esteban M (2018) The MDM2-p53 pathway is involved in preconditioning-induced neuronal tolerance to ischemia. Sci Rep 8(1):1610
Tsai NP, Wilkerson JR, Guo W, Huber KM (2017) FMRP-dependent Mdm2 dephosphorylation is required for MEF2-induced synapse elimination. Hum Mol Genet 26(2):293–304
Aguiar RP, Soares LM, Meyer E, da Silveira FC, Milani H, Newman-Tancredi A, Varney M, Prickaerts J et al (2020) Activation of 5-HT1A postsynaptic receptors by NLX-101 results in functional recovery and an increase in neuroplasticity in mice with brain ischemia. Prog Neuro-Psychopharmacol Biol Psychiatry 99:109832
Wang SL, Duan L, Xia B, Liu Z, Wang Y, Wang GM (2017) Dexmedetomidine preconditioning plays a neuroprotective role and suppresses TLR4/NF-kappaB pathways model of cerebral ischemia reperfusion. Biomed Pharmacother 93:1337–1342
Chen YM, He XZ, Wang SM, Xia Y (2020) Delta-opioid receptors, microRNAs, and neuroinflammation in cerebral ischemia/hypoxia. Front Immunol 11:421
Miquel S, Champ C, Day J, Aarts E, Bahr BA, Bakker M, Banati D, Calabrese V et al (2018) Poor cognitive ageing: vulnerabilities, mechanisms and the impact of nutritional interventions. Ageing Res Rev 42:40–55
Leri M, Scuto M, Ontario ML, Calabrese V, Calabrese EJ, Bucciantini M, Stefani M (2020) Healthy effects of plant polyphenols: molecular mechanisms. Int J Mol Sci 21:4
Pilipenko V, Narbute K, Amara I, Trovato A, Scuto M, Pupure J, Jansone B, Poikans J et al (2019) GABA-containing compound gammapyrone protects against brain impairments in Alzheimer's disease model male rats and prevents mitochondrial dysfunction in cell culture. J Neurosci Res 97(6):708–726
Peters V, Calabrese V, Forsberg E, Volk N, Fleming T, Baelde H, Weigand T, Thiel C et al (2018) Protective actions of anserine under diabetic conditions. Int J Mol Sci 19(9)
Min J, Senut MC, Rajanikant K, Greenberg E, Bandagi R, Zemke D, Mousa A, Kassab M et al (2008) Differential neuroprotective effects of carnosine, anserine, and N-acetyl carnosine against permanent focal ischemia. J Neurosci Res 86(13):2984–2991
Brunetti G, Di Rosa G, Scuto M, Leri M, Stefani M, Schmitz-Linneweber C, Calabrese V, Saul N (2020) Healthspan maintenance and prevention of Parkinson's-like phenotypes with hydroxytyrosol and oleuropein aglycone in C elegans. Int J Mol Sci 21:7
Calabrese V, Santoro A, Trovato Salinaro A, Modafferi S, Scuto M, Albouchi F, Monti D, Giordano J et al (2018) Hormetic approaches to the treatment of Parkinson's disease: perspectives and possibilities. J Neurosci Res 96(10):1641–1662
Calabrese EJ, Calabrese V, Tsatsakis A, Giordano JJ (2020) Hormesis and Ginkgo biloba (GB): Numerous biological effects of GB are mediated via hormesis. Ageing Res Rev:101019. https://doi.org/10.1016/j.arr.2020.101019101019
Kassis H, Shehadah A, Chopp M, Roberts C, Zhang ZG (2015) Stroke induces nuclear shuttling of histone deacetylase 4. Stroke 46(7):1909–1915
He M, Zhang B, Wei X, Wang Z, Fan B, Du P, Zhang Y, Jian W et al (2013) HDAC4/5-HMGB1 signalling mediated by NADPH oxidase activity contributes to cerebral ischaemia/reperfusion injury. J Cell Mol Med 17(4):531–542
Hsing CH, Lin CF, So E, Sun DP, Chen TC, Li CF, Yeh CH (2012) alpha2-Adrenoceptor agonist dexmedetomidine protects septic acute kidney injury through increasing BMP-7 and inhibiting HDAC2 and HDAC5. Am J Physiol Ren Physiol 303(10):F1443–F1453
Tang Y, Jia C, He J, Zhao Y, Chen H, Wang S (2019) The application and analytical pathway of dexmedetomidine in ischemia/reperfusion injury. J Anal Methods Chem 2019:7158142
Formisano L, Laudati G, Guida N, Mascolo L, Serani A, Cuomo O, Cantile M, Boscia F et al (2019) HDAC4 and HDAC5 form a complex with DREAM that epigenetically down-regulates NCX3 gene and its pharmacological inhibition reduces neuronal stroke damage. J Cereb Blood Flow Metab. https://doi.org/10.1177/0271678X19884742271678X19884742
Buchthal B, Weiss U, Bading H (2018) Post-injury nose-to-brain delivery of activin A and SerpinB2 reduces brain damage in a mouse stroke model. Mol Ther 26(10):2357–2365
Zhang T, Wang H, Li Q, Fu J, Huang J, Zhao Y (2018) MALAT1 activates the P53 signaling pathway by regulating MDM2 to promote ischemic stroke. Cell Physiol Biochem 50(6):2216–2228
Xu X, Ye L, Ruan Q (2009) Environmental enrichment induces synaptic structural modification after transient focal cerebral ischemia in rats. Exp Biol Med (Maywood) 234(3):296–305
Pang X, Zhang P, Zhou Y, Zhao J, Liu H (2020) Dexmedetomidine pretreatment attenuates isoflurane-induced neurotoxicity via inhibiting the TLR2/NF-kappaB signaling pathway in neonatal rats. Exp Mol Pathol 112:104328
Zhang Y, Gao Q, Wu Z, Xue H, Liu B, Zhao P (2019) Dexmedetomidine promotes hippocampal neurogenesis and improves spatial learning and memory in neonatal rats. Drug Des Devel Ther 13:4439–4449
Xing N, Xing F, Li Y, Li P, Zhang J, Wang D, Zhang W, Yang J (2020) Dexmedetomidine improves propofol-induced neuronal injury in rat hippocampus with the involvement of miR-34a and the PI3K/Akt signaling pathway. Life Sci 247:117359
Acknowledgments
We would like show sincere appreciation to the reviewers for critical comments on this article.
Funding
This project was supported by the Natural Science Foundation of Shanghai (No. 14ZR1407500).
Author information
Authors and Affiliations
Contributions
Hu Lv, Ying Li, and Qian Cheng designed the study, collated the data, and carried out data analyses. Wei Chen and Jiawei Chen contributed to drafting and polishing the manuscript. All authors have read and approved the final submitted manuscript.
Corresponding authors
Ethics declarations
This study was conducted with the approval of the ethics committee of Shanghai Medical College of Fudan University and in strict accordance with the Guide for the Care and Use of Laboratory animals published by the US National Institutes of Health, with extensive efforts made to minimize animal suffering.
Conflict of Interest
The authors declare that they have no conflict of interest.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Hu Lv, Ying Li, and Qian Cheng are regarded as co-first authors.
Rights and permissions
About this article

Cite this article
Lv, H., Li, Y., Cheng, Q. et al. Neuroprotective Effects Against Cerebral Ischemic Injury Exerted by Dexmedetomidine via the HDAC5/NPAS4/MDM2/PSD-95 Axis. Mol Neurobiol 58, 1990–2004 (2021). https://doi.org/10.1007/s12035-020-02223-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-020-02223-7