
Abstract
Epidemiologic studies have shown that foods rich in polyphenols, such as flavonoids, can lower the risk of ischemic disease; however, the mechanism of protection has not been clearly investigated. In this study, we hypothesized that pretreatment effect of catechin hydrate (CH) on functional outcome, neuronal damage and on secondary injuries in the ischemic brain of rats. To test this hypothesis, male Wistar rats were pretreated with CH (20 mg/kg b.wt) for 21 days and then subjected to 2 h middle cerebral artery occlusion (MCAO) followed by 22 h of reperfusion. After 2 h MCAO/22 h reperfusion, neurological deficit, infarct sizes, activities of antioxidant enzymes and cytokines level were measured. Immunohistochemistry and western blot were used to analyse the expression of glial fibrillary acidic protein (GFAP), inducible nitric oxide (iNOS) and NF-kB in ischemic brain. The administration of CH showed marked reduction in infarct size, reduced the neurological deficits, suppressed neuronal loss and downregulate the iNOS, GFAP and NF-kB expression in MCAO rats. A significantly depleted activity of antioxidant enzymes and content of glutathione in MCAO group were protected significantly in MCAO group pretreated with CH. Conversely, the elevated level of thiobarbituric acid reactive species and cytokines in MCAO group was attenuated significantly in CH pretreated group when compared with MCAO group. The results indicated that CH protected the brain from damage caused by MCAO, and this effect may be through downregulation of NF-kB expression.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Stroke, a catastrophe which threatens life, is the third leading cause of death and the first major cause of long term disability in developing nation [1]. Approximately 80–85 % of stoke is ischemic in nature, characterized by the disruption/severe reduction of cerebral blood flow and lack of oxygen in cerebral arteries that leads to the ischemia–reperfusion (I/R) insult [2]. Ischemia reperfusion injury following ischemic stroke results from the extremely interconnected multiple pathobiological mechanisms includes excitotoxicity, oxidative stress, inflammation and apoptosis [3–5].
Oxidative stress plays a fundamental role in cerebral I/R injury because of large consumption of oxygen by brain. As brain is not well equipped with antioxidant defenses, and so free radicals/oxidants released by inflammatory cells threaten tissue viability in the vicinity of the ischemic core [6]. At the time of cerebral I/R, robust reactive oxygen species (ROS) are generated that promotes damage to cellular macromolecular such as lipids, proteins and DNA that leads to the irreversible neuronal injury and cell death [7].
In addition to oxidative stress, an inflammatory mechanism has been recently proposed in acute neuronal cell death following cerebral ischemia. This response is necessary to remove cell debris and tissue remodeling after injury. After I/R, energy depletion and necrotic neuron death in the local ischemic area start the inflammatory cascades. The reperfusion generated ROS induces the upregulation of NF-kB that leads to production of cytokines and chemokines which participate in the inflammatory process. Cytokines along with iNOS are thought to be the major mediators behind these complex inflammatory responses. Astrocyte involvement have also been linked to proinflammatory cytokines and iNOS upregulation in many pathologies of the CNS including cerebral ischemia [8] which markedly upregulates the expression of glial fibrillary acidic protein (GFAP).
Experimental models of cerebral ischemia have been developed to improve the understanding of deleterious mechanisms involved in the brain ischemic damage, and to identify the potential efficiency of therapeutic strategies. Animal model of MCAO closely mimic the changes that occur during and after human ischemic stroke because most human ischemic strokes are caused by occlusion of the middle cerebral artery (MCA) and so animal models were developed to induce ischemia in this arterial territory.
In the present era, much attention has been given to the use of naturally occurring compounds as the alternative therapeutic agents and their formulations for the treatment of different neurodegenerative diseases including stroke. These effects are attributed to their antioxidant as well as anti-inflammatory potential. Catechin hydrate (CH) is an abundant the major flavonoid present in the grape seed and green tea [9]. It exhibits biological and pharmacological properties and a number of studies have examined the antioxidant activity and anti-inflammatory properties of CH using a variety of assay systems [10]. Keeping this in view, the present study was primarily aimed to examine the neuroprotective effect of CH against MCAO induced neuronal damage in rats. Neuroprotective effects of CH were assessed by studying markers of oxidative damage and NF-kB mediated neuro inflammation in rat brain.
Experimental Procedure
Chemicals and Reagents
Oxidized glutathione (GSSG), reduced glutathione (GSH), glutathione reductase (GR), nicotinamide adenine dinucleotide phosphate (NADPH), 1-chloro-2,4-dinitrobenzene (CDNB), 5-5′-dithio-bis-2-nitrobenzoic acid (DTNB), thiobarbituric acid (TBA), trichloroacetic acid (TCA), 2,3,5-triphenyltetrazolium chloride (TTC), ethylene diamine tetra-acetic acid (EDTA), (−)epinephrine, paraformaldehyde, glycine, poly-l-lysine, cresyl violet and catechin hydrate (CH) were purchased from Sigma–Aldrich Chemicals Pvt. Ltd., India. TNF-α, and IL-1β Immuno Assay Kits were purchased from eBioscience & BD Bioscience, USA. Polyclonal NF-kB was purchased from Sigma–Aldrich Chemicals Pvt. Ltd., and anti-rabbit IgG was purchased from Jackson Immuno Research Laboratories Inc., West Groove, PA, USA.
Animals and Treatments
Male Wistar rats obtained from Central Animal House of Jamia Hamdard (Hamdard University), weighing 250 ± 10 g and age 16 weeks (approx.) at the start of the experiment, were used. Rats were housed in groups of four animals per cage at an ambient temperature of 25 ± 2 °C and relative humidity of 45–55 % with 12 h light/dark cycles and had free access to standard rodent pellet diet and water ad libitum. The food was withdrawn 12 h before the surgical procedure. Experiments were conducted in accordance with the Animal Ethics Committee of the University, approved by the Government of India, New Delhi, India.
Drug Administration
Catechin hydrate (20 mg/kg body weight in normal saline) was administered orally (p.o.) once daily for 21 days before MCAO. We examined the effects of different doses of catechin hydrate (CH) on cerebral ischemia reperfusion injury in pilot studies to determine the optimal dose of CH that provides the most neuroprotection against ischemia/reperfusion injury and also the CH dose (20 mg/kg) and treatment regimen used in this experiment was supported from previous studies showing that this amount provided the maximal protective effects in the treatment of different types of brain injury [52–54].
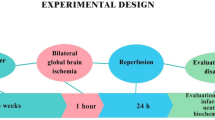
Experimental Design
To investigate the neuroprotective effects of catechin hydrate in an experimental model of cerebral ischemia, we used rat MCAO model [4]. Animals were divided into four groups each having eight animals. The first group served as sham (S) and saline was given orally, the second was middle cerebral artery occluded (MCAO), i.e., ischemia was induced for 2 h followed by reperfusion for 22 h, the third was pretreated for 21 days with Catechin hydrate (20 mg/kg, orally) followed by MCAO for 2 h and reperfusion for 22 h (i.e., CH + MCAO group) and the fourth was pretreated for 21 days with Catechin hydrate alone, i.e., Catechin hydrate group (20 mg/kg, orally). After the completion of the reperfusion period, animals were assessed for neurobehavioral activities and then sacrificed. The brains were taken out to dissect the frontal cortex and striatum for biochemical estimations.
Post-Operative Care
Recovery of anaesthesia took approximately 4–5 h after surgery. The rats were kept in a well-ventilated room at 25 ± 3 °C in individual cages till they gained full consciousness and then housed one animal per cage. Food and water were kept inside the cages for the rest of 22 h so that animals could easily access it without any physical trauma due to overhead surgery.
Behavioral Studies
Adhesive-Removal Test
Adhesive removal test was performed by the method of [11]. For the adhesive-removal somatosensory test animals were tested 24 h after MCAO [12]. Two small adhesive-back paper dots each 8-mm diameter were placed on each forepaws to cover the hairless part of the forepaws. The animal was placed in a box (40 cm × 30 cm) and the time the animal took to remove the pieces of tape from the ipsilateral and contralateral paws was recorded. The animals were given a maximum of 180 s to sense the tapes and remove them and were scored as 180 s if they did not succeed.
Rota Rod (Muscular Coordination)
Omni Rotor (Omnitech Electronics, Inc., Columbus, OH, USA) was used to evaluate the muscular coordination skill after 2 h MCAO/22 h reperfusion as reported by [13]. It consisted of a rotating rod, 75 mm diameter, which was divided into four parts by compartmentalization to permit the testing of four rats at a time. The apparatus automatically recorded the time in 0.1 s, when the rat falls of the rotating shaft. The speed was set at 10 cycles per minute and cut of time was 180 s. The rats were trained 5 days prior to surgery for 5 times at 1 h interval per day. Rats that performed on the rota rod for more than 10 s were used. After the 22 h of MCAO the performance time was measured.
Grip Test
Grip test was performed by the method of [14]. The apparatus consist of a string of 50 cm length, pulled tight between two vertical supports and elevated 40 cm from a flat surface was used. The rat was placed on the string at a point midway between supports and evaluated according to the following scale, 0—fall off, 1—hangs onto string by two forepaws, 2—as for 1 but attempts to climb on string, 3—hangs onto string by two forepaws plus one or both hind paws, 4—hangs onto string by all fore paws plus tail wrapped around string, 5—escape.
Evaluation of Ischemic Damage
Extent of ischemic damage was assessed using TTC and cresyl violet staining.
Infarct Volume Analysis
The animals were sacrificed after 2 h of occlusion followed by 22 h of reperfusion. The brains were dissected out and kept in a brain matrix. A 1.5 mm coronal sections of the brains was cut down with the help of sharp blades and stained with 0.1 % triphenyltetrazolium chloride (TTC) prepared into normal saline at 37 °C for 15 min. TTC acts as a proton acceptor for many pyridine nucleotide-linked dehydrogenase along with the cytochromes, form an integral part of the inner mitochondrial membrane and make up the electron transport chain. The tetrazolium salt is reduced by the enzymes into a red lipid soluble formazan. Viable tissues stain deep red while the infarcts remain unstained. For imaging, the sections were scanned by a high-resolution scanner. The infarct volume was calculated by taking the average of infarct area on both sides of the slice and multiplying it by section thickness. Infarct volumes from each section were then summed to determine total brain infarct volume and adjusted for edema [4].
Assessment of Neuronal Damage
After 2 h MCAO/22 h reperfusion, animals were anesthetized with chloral hydrate (400 mg/kg, i.p.) and perfused transcardially with 0.9 % sodium chloride at 4 °C, followed by 4 % paraformaldehyde in 0.1 M phosphate-buffer (PB, pH 7.4).The brain was removed, kept in the same fixative for overnight at 4 °C and immersed in 0.1 M PBS containing 20 %, 30 % sucrose overnight at 4 °C, respectively. Sections were incubated in 0.5 % cresyl violet solution for 3–5 min. The sections were rinsed in distilled water and differentiated in 95 % alcohol, followed by dehydration in 100 % alcohol. The sections were cleared in xylene and mounted in DPX mounting medium. Images were acquired using the light microscopy (BX51, Olympus, Japan).
Biochemical Studies
Tissue Preparation for Antioxidant Enzymes and Glutathione Assays
After behavioral study, the animals were sacrificed and their brains were taken out to dissect striatum and frontal cortex to give 5 % (w/v) homogenate (10 mM PB, pH 7.0 having 10 μl/ml protease inhibitors, 5 mM leupeptin, 1.5 mM aprotinin, 2 mM phenylmethylsulphonylfluoride (PMSF), 3 mM peptastatin A, 0.1 mM EGTA, 1 mM benzamidine and 0.04 % butylated hydroxytoluene) and centrifuged at 800g for 5 min at 4 °C. This supernatant (S1) was used for the assay of TBARS and remaining S1 was recentrifuged at 10,500×g for 15 min at 4 °C (S2) to separate post mitochondrial supernatant (PMS) which was used for the estimation of antioxidant enzymes and GSH.
Assay for TBARS
LPO was estimated by measuring the TBARS level in accordance with the method as described previously [15] with some modification. Briefly, 0.2 ml S1 was pipetted in 2.0 ml flat bottom eppendorf tube and incubated at 37 °C in a metabolic water bath shaker at 120 strokes up and down; another 0.2 ml of the same S1 was pipetted in an eppendorf tube and placed at 0 °C incubation. After 1 h of incubation, 0.4 ml of 5 % TCA and 0.4 ml of 0.67 % TBA was added in both samples (i.e., 0 and 37 °C). The reaction mixture was centrifuged at 3,000g for 15 min. The supernatant was transferred to another test tube and placed in a boiling water bath for 10 min. Thereafter, the test tubes were cooled and the absorbance of the color was read at 535 nm. The rate of LPO was expressed as nmol of TBARS formed/h/mg protein using molar extinction coefficient of 1.56 × 105 M−1 cm−1.
Estimation of Protein Carbonyl
Protein oxidation was estimated by determination of protein carbonyl content as described previously [4]. The tissue homogenate (0.25 ml) was mixed with an equal volume of 20 % TCA. Thereafter, 0.25 ml of 10 mM 2, 4-dinitrophenylhydrazine (DNPH) in 2.0 M HCl was added and allowed to standing at room temperature for 1 h, with vortexing every 10–15 min. Then 0.5 ml of 20 % TCA was added and centrifuged at 11,000g for 5 min. The supernatant was discarded and pellet was washed 3 times with 1 ml of ethanol-ethyl acetate (1:1) to remove free reagent. The sample was allowed to stand for 10 min before centrifugation and the supernatant was discarded each time. Precipitated protein was redissolved in 0.6 ml guanidine hydrochloride solution within 15 min at 37–50 °C and then centrifuged at 11,000g for 5 min to remove any insoluble material. The carbonyl content of the supernatant was measured spectrophotometrically at 370 nm. The results were expressed as nmol of DNPH incorporated/mg protein using molar extinction coefficient of 22 × 103 M−1 cm−1.
Assay for GSH Level
GSH was determined as described previously [16]. PMS 0.1 ml was precipitated with 0.1 ml of sulfosalicylic acid (4 %). The samples were kept at 4 °C for 1 h and then subjected to centrifugation at 1,200g for 15 min at 4 °C. The assay mixture contained 0.1 ml of filtered aliquot, 1.7 ml PB (0.1 M, pH 7.4) and 0.2 ml DTNB (4 mg/ml, 0.1 M PB, pH 7.4) in a total volume of 2.0 ml. The yellow color developed was read immediately at 412 nm. The GSH level was calculated as nmol GSH mg − 1 protein, using molar extinction coefficient of 13.6 × 103 M−1 cm−1.
Determination of GR Activity
GR activity was assayed by the method [17] as describe by Mohandas et al. (1984). The assay system consisted of 0.1 M PB pH 7.6, 0.1 mM NADPH, 0.5 mM EDTA, 1.0 mM GSSG and 0.1 ml PMS in a total volume of 2.0 ml. The enzyme activity was quantitated at room temperature by measuring the disappearance of NADPH at 340 nm and was calculated as nmol NADPH oxidized/min/mg protein using molar extinction coefficient of 6.22 × 103 M−1 cm−1.
Determination of Glutathione Peroxidase (GPx) Activity
GPx activity was measured according to the procedure of [18]. The reaction mixture consisted of 0.05 M PB pH 7.0, 1.0 mM EDTA, 1.0 mM sodium azide, 1.4 U of 0.1 ml GR, 1.0 mM glutathione, 0.2 mM NADPH, 0.25 mM hydrogen peroxide and 0.1 ml of PMS in a final volume of 2.0 ml. The disappearance of NADPH at 340 nm was recorded at room temperature. The enzyme activity was calculated as nmol NADPH oxidized/min/mg protein using molar extinction coefficient of 6.22 × 103 M−1 cm−1.
Determination of Superoxide Dismutase (SOD) Activity
SOD activity was measured spectrophotometrically, according to the method of [19] by monitoring the auto oxidation of (−)-epinephrine at pH 10.4 for 3 min at 480 nm. The reaction mixture contained glycine buffer (50 mM, pH, 10.4) and 0.2 ml of PMS. The reaction was initiated by the addition of (−)-epinephrine. The enzyme activity was calculated in terms of nmol (−)-epinephrine protected from oxidation/min/mg protein using molar extinction coefficient of 4.02 × 103 M−1 cm−1.
Determination of Catalase Activity
Catalase activity was assayed by the method as described previously [20]. Briefly, the assay mixture consisted of 0.05 M PB pH 7.0, 0.019 M hydrogen peroxide, and 0.05 ml PMS in a total volume of 3.0 ml. Changes in absorbance were recorded at 240 nm. Catalase activity was calculated in terms of nmol H2O2 consumed/min/mg protein.
Preparation of Nuclear Extracts and Western Blot Analysis
Brain cortical tissue was collected from the infarcted and surrounding areas. Nuclear extracts was prepared as described [39]. Samples of nuclear fractions containing equal amounts of protein (35 μg) were separated in 14 % SDS–polyacrylamide gel electrophoresis. Proteins were transferred onto PVDF membrane and incubated overnight at 4 °C with specific primary rabbit polyclonal antibody against NF-κB p65 (dilution, 1:1,000), followed by appropriate secondary antibodies conjugated to horseradish peroxidase. Proteins recognized by the antibody were visualized by enhanced chemiluminescence Femto kit (Thermo Scientific) according to manufacturer instructions. All blots were stripped and re-incubated with primary antibody specific to β-actin (Sigma) as a loading control. Intensity of the bands was measured by densitometry and quantified using NIH-Image J software.
Measurement of Cytokines
The brains of MCAO, CH + MCAO and sham animals were removed after cervical dislocation 24 h after MCAO. A 3-mm coronal section was cut from the parietal cortex and stored at −70 °C until use. Brain samples were homogenized in a buffer consisting of 20 mM Tris–HCl with pH 7.6, 100 mM KCl, 5 mM NaCl, 2 mM EDTA, 1 mM EGTA, 0.25 M sucrose, 2 mM DTT, 2 mM PMSF; pH 7.2. Homogenates were centrifuged at 4 °C and 12,000g for 15 min. Supernatants were removed and assayed in duplicate using TNF-α and IL-1β assay kits (eBioscience and BD Bioscience, USA) according to the manufacturer’s guidelines. Tissue cytokine concentrations were expressed as picograms of antigen per mg of protein.
Immunohistochemistry for GFAP and iNOS
After 22 h of reperfusion the animals were anesthetized with chloral hydrate (400 mg/kg, i.p.) and perfused transcardially with 0.9 % sodium chloride at 4 °C, followed by 4 % paraformaldehyde in 0.1 M phosphate-buffer (PB, pH 7.4). The brain was removed, kept in the same fixative for overnight at 4 °C and immersed in 0.1 M PBS containing 20 %, 30 % sucrose overnight at 4 °C, respectively. The tissues were kept in final sucrose solution till sectioning. The fixed tissues were embedded in OCT compound (polyvinyl glycol, polyvinyl alcohol, and water) and frozen at −20 °C. Coronal sections of 12 μm thicknesses were cut on a cryostat (Leica, Germany) and collected on gelatincoated slides and immersed in wash buffer (sodium phosphate.
0.1 M, sodium chloride 0.5 M, Triton X-100, sodium azide) pH 7.4 for 20 min. After a pre-incubation for 1 h in blocking solution (10 % normal goat serum, 0.3 % Triton X-100 in PBS), sections were incubated overnight at 4 °C with primary antibodies; rabbit anti-GFAP (anti-glial fibrillary acidic protein; 1:200 dilution; Chemicon International, Temecula, CA) nd mouse anti-iNOS (1:200 dilution; Sigma-Aldrich Chemicals Pvt. Ltd., India) diluted in a solution of 0.3 % triton X-100 in PBS. The slides were washed with PBS to remove the unbound antibodies and sections were incubated with 1:500 dilutions of goat anti-rabbit IgG and 1:500 dilutions of goat anti-mouse IgG secondary antibodies respectively for 1 h at room temperature. Finally, the sections were mounted on gelatin-coated slides, air dried and cover slipped. Omission of primary or secondary antibody served as controls.
Determination of Protein
Protein was determined by the method of [21] using Bovine Serum Albumin as standard.
Statistical Analysis
Results are expressed as mean ± SEM. Statistical analysis of the data was done by applying the analysis of variance (ANOVA), followed by Tukey’s Kramer. The p value <0.05 was considered statistically significant.
Results
Effect of Catechin Hydrate on Behavioral Output
Tape removal test is a technique that assesses sensory and motor impairments in forepaw function. After 2 h MCAO/22 h reperfusion, an increase in the time needed to remove adhesive tape from the contralateral and ipsilateral forepaws was observed in MCAO group as compared with sham group rats (Fig. 1a). Interestingly, catechin hydrate treated MCAO group (CH + MCAO) significantly shortened the time to remove the adhesive tapes from the forepaws compared with MCAO group (p < 0.01). Catechin hydrate alone pre-treated group (CH + S) showed no significant changes as compared to sham group. A significant decrease in muscular coordination skill was observed in MCAO group as compared to the sham group animals (p < 0.01) (Fig. 1b). Animals treated with catechin hydrate in CH + MCAO group afforded a significant protection in muscular coordination skill, as compared to MCAO group animals (p < 0.01). No significant difference was observed between sham and catechin hydrate treated sham group (CH + S).The grip strength was found to be significantly decreased (p < 0.01) in MCAO group as compared to sham group. Whereas catechin hydrate pretreatment significantly (p < 0.05) improved the grip strength in CH + MCAO group as compared to MCAO group. However, no significant alteration was observed in catechin hydrate pretreated sham group (CH + S) as compared to sham group (Fig. 1c).

Catechin hydrate (CH) pretreatment for 21 days improves performances in the tapes removal test, muscular coordination skill and grip strength after stroke. a Time needed to remove tapes from contralateral and ipsilateral forepaws was recorded on sham, MCAO and CH + MCAO group. A significant shortened time to remove the adhesive tapes from the forepaws was observed in CH + MCAO group as compared to MCAO group, indicating a profound improvement in sensory motor performance. Values are expressed as mean ± SEM of eight animals. *p < 0.01, MCAO versus sham; # p < 0.01, CH + MCAO versus MCAO. b A significant decrease in motor coordination in MCAO group as compared to sham group was observed and it was significantly protected in CH + MCAO group as compared to MCAO group. Values are expressed as mean ± SEM for six animals. *p < 0.01, MCAO versus sham; # p < 0.01, CH + MCAO versus MCAO. c The grip strength was decreased significantly in MCAO group animals as compared to sham group animals. Pretreating the animals with Catechin hydrate followed by MCAO has protected motor deficit as compared to MCAO group. Values are expressed as mean ± SEM of six animals. *p < 0.01, MCAO versus sham; # p < 0.05, CH + MCAO versus MCAO
Effect of Catechin Hydrate on TTC Stain and Infarct Volume
TTC staining of MCAO brain sections showed reproducible and readily detectable lesions in the areas that are supplied by the MCA after 22 h of reperfusion (Fig. 2a). The lesions were present in striatum and the overlying cortex. We hypothesized that catechin hydrate play a protective role in stroke. Indeed, MCAO group rats have shown a significantly increased infarct volume as compared with sham. Catechin hydrate pretreatment has reduced the infarct volume significantly (p < 0.01) as compared to MCAO group.

Effect of catechin hydrate pretreatment for 21 days on brain infarct size by TTC stain after middle cerebral artery occlusion for 2 h and reperfusion of 22 h. a Representative photographs of brain sections stained with 0.1 % TTC, and measurement of infarct volumes of MCAO and CH + MCAO group are presented. MCAO group produced a significant lesion over sham group (figure not shown). However, CH + MCAO group showed a significant (*p < 0.01) reduction in tissue damage as compared to MCAO group. b Cresyl violet staining shows the neuronal alterations in the ipsilateral brain after 2 h MCAO/22 h reperfusion. Normal morphologic features of neurons are present in sham. Characteristic features of hyperchromia, shrinkage of nucleus and cytoplasm in neurons present in MCAO groups. Administration of catechin hydrate clearly ameliorates ischemia-induced neuronal damaged in CH + MCAO. Magnification is 20×
Effect of Catechin Hydrate on Cresyl Violet Staining
To access the neuronal damage induced by ischemic-reperfusion, we evaluated MCAO group against CH + MCAO group animals. Sections of the MCAO group showed significant neuronal loss, neuronal shrinkage and marked vascular changes throughout the cortex and striatum. Intact neurons were almost absent in that area. The corresponding area in the sections of CH + MCAO group showed partial loss of neuronal staining with presence of intact neurons in between the vacuolated spaces. The section of the sham group showed normal neuronal staining with no pathological change (Fig. 2b).
Catechin hydrate Pretreatment Decreased the TBARS Level in Striatum and Frontal Cortex
The effect of catechin hydrate on TBARS level was measured to demonstrate the oxidative damage by lipid peroxidation (LPO) in frontal cortex and striatum of MCAO group rats. A significant increased (p < 0.001) TBARS level was observed in MCAO group animals as compared to sham group. Rats of CH + MCAO group has exhibited significant attenuation in TBARS level in frontal cortex (p < 0.05) and striatum (p < 0.01) as compared to MCAO group rats (Fig. 3). Catechin hydrate alone pre-treated sham group (CH + S) showed no significant changes in TBARS level as compared to the sham group.

Effect of catechin hydrate pretreatment on TBARS (a) and protein carbonyl (b) contents in the frontal cortex and striatum. TBARS and protein carbonyl contents were significantly increased in MCAO group as compared to sham group. Catechin hydrate pretreatment has decreased the TBARS and protein carbonyl contents significantly in frontal cortex and striatum. Values are expressed as mean ± SEM. (n = 8). *p < 0.001 MCAO versus S, # p < 0.05; ## p < 0.01 CH + MCAO versus MCAO
Effect of Catechin Hydrate on the Indices of Protein Oxidation
Protein oxidation was assessed by the determination of protein carbonyl content in the samples of frontal cortex and striatum. MCAO induced a significant (p < 0.01) increase in protein carbonyl content. Pre-treatment with CH, however reduced the MCAO-induced increase in protein carbonyl content significantly (p < 0.05) in the frontal cortex and striatum. There was no statistical significant reduction in protein carbonyl content in rat pretreated with CH alone in CH + Sham group (Fig. 3b).
Effect of Catechin Hydrate on Endogenous Antioxidant System
Protective effect of catechin hydrate on endogenous antioxidant defense system in frontal cortex and striatum was observed. The level of GSH or total free SH group was depleted significantly in frontal cortex and striatum in MCAO group as compared to sham group. Catechin hydrate pretreatment has protected its level significantly (p < 0.05) in CH + MCAO group as compared to MCAO group. Catechin hydrate alone pre-treated group (CH + S) exhibited no significant changes in GSH level total free SH group as compared to sham group (Fig. 4). The activities of antioxidant enzymes (GPx, GR, SOD and catalase) were decreased significantly in frontal cortex and striatum of MCAO group animals as compared to sham group animals and their activities were protected significantly in frontal cortex as well as in striatum of catechin hydrate pretreated MCAO group (CH + MCAO) animals as compared to MCAO group animals. No significant change in the activity of these enzymes was observed in CH + S group as compared to sham group (Tables 1, 2).

Effect of catechin hydrate pre-treatment on GSH level in frontal cortex and striatum. GSH level was significantly decreased in frontal cortex and striatum in MCAO group rats as compared to sham group. Catechin hydrate pretreatment has significantly increased the level of GSH in CH + MCAO group as compared to MCAO group. Values are expressed as mean ± SEM. (n = 8). *p < 0.01 MCAO versus S, #p < 0.05; ## p < 0.01 CH + MCAO versus MCAO
Effect of Catechin Hydrate on NF-kB Expression
A significant increased expression level of NF-kB was observed in MCAO group animals as compared to sham group. Catechin hydrate supplementation has significantly decreased the expression levels of NF-kB in catechin hydrate pretreated MCAO group (CH + MCAO) when compared with MCAO group (Figs. 5a). However no significant alteration was observed in catechin hydrate treated sham group (CH + S) as compared to sham group (Data not shown).

a Expression level of NF-kB and its quantification. b Quantification of interleukin-1β (IL-1 β) and TNF-α by ELISA in the frontal cortex and striatum of sham, MCAO and CH + MCAO groups are shown. Concentration of IL-1β and TNF-α were significantly increased in MCAO group as compared to sham group. Catechin hydrate pre-treatment significantly reduced IL-1β and TNF-α concentration in CH + MCAO as compared to MCAO group. Values are expressed as mean ± SEM. (n = 8). *p < 0.01; **p < 0.001 MCAO versus sham, # p < 0.01; CH + MCAO versus MCAO
Effect of Catechin Hydrate on IL-1β and TNF-α
Quantification of IL-1β and TNF-α in frontal cortex and striatum by ELISA showed a significant increased level of IL-1β (p < 0.01) and TNF-α (p < 0.001) in MCAO group as compared to sham group. Interestingly, catechin hydrate supplementation has significantly decreased the levels of IL-1β (p < 0.01) and TNF-α (p < 0.01) in catechin hydrate pretreated MCAO group (CH + MCAO) when compared with MCAO group (Fig. 5b). However, no significant alteration was observed in catechin hydrate treated sham group (CH + S) as compared to the sham group.
Effect of Catechin Hydrate Pretreatment on GFAP and iNOS Expression in MCAO Rats
The activation of astrocyte up regulation is associated with neuronal cell death in cerebral ischemia. GFAP expression was found to be remarkably high (p < 0.001) in ischemic hemisphere of MCAO group. A noticeable reduction (p < 0.01) in GFAP expression was observed in catechin hydrate pretreated group as compared to MCAO group. Expression of GFAP was seen to be very scarce in sham group animals (Fig. 6a). Catechin hydrate pre-treatment did not show any remarkable effects in the CH + S group as compared to sham group (data not shown).

a The left panel shows representative coronal brain sections of sham, MCAO and CH + MCAO stained for iNOS. b The right panel shows representative coronal brain sections of sham, MCAO and CH + MCAO stained for GFAP. n = 4. Magnification is 20×
Numbers of iNOS positive cells are remarkably high (p < 0.001) in ischemic hemisphere of MCAO group, which was significantly (p < 0.01) attenuated with the pretreatment of catechin hydrate. However, iNOS expression was found to be almost negligible in sham group (Fig. 6b). Catechin hydrate pre-treatment did not show any remarkable effects in the CH + S group as compared to sham group (data not shown).
Discussion
The present study provides the compelling evidence that catechin hydrate (CH) protects the rat cortex and striatum from ischemia reperfusion (I/R) injury. I/R tissue damage may be a consequence of oxidative stress and could be partially ameliorated by herbal supplementation. In this context, we have evaluated middle cerebral artery occlusion (MCAO) in rats, a model widely used to study neuroprotective effect of drugs because of its recapitulation of biochemical and pathological features of stroke in humans [4, 8].
Oxyradical-induced damage is considered an important factor in the functional deficit in ischemia reperfusion. The behavioral deficits are the common neurological sequel in patients of cerebral ischemic [22] and its assessment is a more powerful endpoint in evaluating neuroprotection. The behavioral testing data in the current study provides a sensitive evaluation of the ability of CH to provide protection in this MCAO model. The present study suggested that CH, a well know free radical scavenger has reduced neurobehavioral deficits by scavenging free radicals, which are thought to cause behavioral deficits in experimental animals [23]. The poor neurobehavioral outcome in MCAO group rats might be attributed to I/R induce necrosis in the frontal sensorimotor cortices and caudate–putamen, which comprise a wide range of motor and sensorimotor deficits including partial paralysis, poor locomotor activity and lack of coordination [24, 25]. We report here an appreciable increase in muscular coordination skill by rotarod and motor strength by grip and tape adhesive remove test following pretreatment with CH. The restoration of CH induce behavioral deficits confirm its protective role against I/R injury, which was in corroboration with the previous similar findings where exogenous antioxidants supplementation have been used to treat cerebral ischemia [4, 5, 26].
Overproduction of free radicals cause an imbalance in the redox environment of cells, and react with proteins and nucleic acids to alter their functions, or induce lipid peroxidation (LPO) and protein oxidation, leading to eventual cell death. Therefore, scavenging free radicals and preventing lipid and protein peroxidation, which are the main effects of CH, can directly suppress oxidative damage and inflammatory response. In the line of previous findings, we observed different oxidative damage parameters, the critical determinant in I/R brain injury [27] causes a significant increase in LPO and protein oxidation accompanied by significant depletion in brain GSH [28]. The above state can be reverted back by nullifying the overproduction of ROS by endogenous antioxidants, especially by GSH [29]. GSH is the major endogenous antioxidant in the brain [31], which scavenges free radicals, reduces peroxides and can be conjugated with electrophilic compounds, thereby providing cells with multiple defenses against both ROS and their by-products [32]. GSH level was found to be low in MCAO group thus often increase the susceptibility of plasma membranes towards peroxide and other free radicals attacks. The main cause of GSH loss during oxidative stress in brain ischemia is the formation of protein glutathione mixed disulphide (PrSSG) and loss of thiol proteins resulting in various membrane defunctioning [33]. We have observed an elevated level of TBARS accompanied by depleted GSH level in MCAO rat brain and pre-treatment with CH partially attenuates the elevated level of TBARS and depleted level of GSH which is in concomitant with the previous observations where antioxidants were used as a remedy in experimental stroke models [4, 30]. GSH along with GPx forms glutathione disulphide in a hydroperoxides reaction. GPx uses GSH as a proton donor, converts H2O2 to water and molecular oxygen; in this process GSH is oxidized to GSSG, which is reconverted to GSH by enzyme GR, thus maintaining the pool. A significant decrease in GPx and GR activity could suggest inactivation by ROS [34], which are increased in I/R injury. Another critical enzyme which provides the first line of defenses is SOD, which with GPx and catalase is thought to be the critical enzyme acting as free radical scavengers that could prevent tissue damage caused by peroxidase reactions. A decrease in the activity of the above antioxidant enzymes can lead to an excess availability of superoxide anion (O ∙−2 ) and hydrogen peroxide (H2O2), which in turn generate hydroxyl radicals (∙OH), resulting in initiation and propagation of lipid peroxidation. SOD can catalyze dismutation of O ∙−2 into H2O2, which is then deactivated to H2O by GPx or catalase [35, 36]. In the present study, pretreatment of CH significantly elevated the GSH and its related enzymes activities along with an increase in activities of SOD and catalase, which is in corroboration with the former reports where antioxidants have been used in experimental I/R models [1, 4, 5, 30].
In the brain ischemia, NF-κB is involved in excitotoxic, oxidative and inflammatory events associated with neurodegeneration [37–39, 51]. Activation of NF-κB also induces pro-inflammatory genes encoding enzymes, cytokines, and other adhesion molecules, all of which are known to promote inflammatory tissue injury [38, 50]. Consisting with this mechanism, we also found enhanced level of TNF-α and IL-1β, along with increased expression of proinflammatory enzyme iNOS in MCAO group animals. CH treatment downregulated the NF-kB expression and inhibited the upregulation of cytokines level and iNOS expression.
In the present study, we found that two important markers involved in the ischemic inflammatory cascade, GFAP and iNOS, were up-regulated. Increasing evidence points to a correlation between cerebral ischemia and GFAP over activation [40]. On the other hand flavonoids have been reported to act on glial cells [41]. A putative flavonoid binding sites for flavonoid have been described, such as adenosine, glutamate and GABA receptors present in post-mitotic neurons. Astrocytes are believed to be responsible for most glutamate uptake in synaptic areas and consequently are the major regulators of glutamate homeostasis [42]. An over expression of GFAP in MCAO rats was observed as compared to CH pre-treated rats where GFAP expression has been significantly reduced. The above protection offered by CH may be attributed to its regulatory mechanism of glutamate upregulation and thus reducing glial mediated ischemic injury.
Further astrocytes elaborate iNOS in response to a series of proinflammatory mediators, including cytokines such as IL-1β and TNF-α [43]. Nitric oxide (NO) derived from iNOS in astrocytes and its oxidative by-product peroxynitrite are thought to contribute to neuronal death due to oxidation of structural neuronal proteins during ischemia [44]. It has been demonstrated that flavonoids inhibit the expression of isoforms of inducible nitric oxide synthase which are responsible for the production of nitric oxide, as well as inflammatory mediators such as cytokines, chemokines or adhesion molecules [45]. Our finding shows that iNOS overexpression in MCAO rats were significantly attenuated with the supplementation of CH. These findings are in harmony with the earlier studies carried out by others [46, 47].
To conclude our findings, we sought to observe the changes in the levels of IL-1β, and TNF-α with reference to cerebral ischemia. Together all represents crucial mediator of neurodegeneration induced by ischemic brain injury [48]. The increased content of IL-1β and TNF-α in MCAO group was significantly protected in CH + MCAO group rats suggesting the neuroprotective effect of CH following cerebral ischemia may be in part, mediated through modulation of the injury caused by proinflammatory cascades. Pretreatment of CH significantly counteracted the NF-kB associated upregulation of proinflammatory cytokines; a piece of evidence attributed to its anti-inflammatory property of CH which was consistent with the previous finding [39, 49]. Taken together, the present study demonstrates that CH has neuroprotective effects against MCAO insult and its effect might be associated with inhibition of oxidative stress and NF-kB mediated downregulation of inflammatory response.
References
Cherubini A, Ruggiero C, Morand C et al (2008) Dietary antioxidants as potential pharmacological agents for ischemic stroke. Curr Med Chem 15:1236–1248
Allen CL, Bayraktutan U (2009) Oxidative stress and its role in the pathogenesis of ischemic stroke. Stroke 4:461–470
Doyle KP, Simon RP, Stenzel-Poore MP (2008) Mechanisms of ischemic brain damage. Neuropharmacology 55:310–318
Khan MM, Ahmad A, Ishrat T et al (2009) Rutin protects the neural damage induced by transient focal ischemia in rats. Brain Res 1292:123–135
Raza SS, Khan MM, Ashfaq M, Ahmad A et al (2011) Silymarin protects neurons from oxidative stress associated damages in focal cerebral ischemia: a behavioral, biochemical and Immunohistological study in Wistar rats. J Neurol Sci 309:45–54
Lakhan SE, Kirchgessner A, Hofer M (2009) Inflammatory mechanisms in ischemic stroke: therapeutic approaches. J Transl Med 17:7–97
Chan PH (2001) Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab 21:2–14
Hu SX, Sheng WS, Peterson PK et al (1995) Differential regulation by cytokines of human astrocyte nitric oxide production. Glia 15:491–494
Yilmaz Y, Toledo RT (2004) Major flavonoids in grape seeds and skins: antioxidant capacity of catechin, epicatechin, and gallic acid. J Agric Food Chem 52:255–260
Li Q, Zhao HF, Zhang ZF, Liu ZG, Pei XR, Wang JB, Li Y (2009) Long-term green tea catechin administration prevents spatial learning and memory impairment in senescence-accelerated mouse prone-8 mice by decreasing Abeta1-42 oligomers and upregulating synaptic plasticity-related proteins in the hippocampus. Neuroscience 163:741–749
Schallert T, Upchurch M, Lobaugh N et al (1982) Tactile extinction, distinguishing between sensorimotor and motor asymmetries in rats with unilateral nigrostraital damage. Pharmacol Biochem Behav 16:455–462
Bouet V, Freret T, Toutain J et al (2007) Sensorimotor and cognitive deficits after transient middle cerebral artery occlusion in the mouse. Exp Neurol 203:555–567
Kelly MA, Rubinstein M, Phillips TJ et al (1998) Locomotor activity in D2 dopamine receptor deficient mice in determined by gene dosage, genetic background and developmental adaptations. J Neurosci 18:7470–7479
Moran PM, Higgins LS, Cordel B et al (1995) Age related learning deficits in transgenic mice expressing the 721-amino acid isoform of human beta- amyloid precursor protein. Proc Natl Acad Sci USA 92:5341–5345
Utley HC, Bernheim F, Hochslein P (1967) Effect of sulfhydryl reagent on peroxidation in microsome. Arch Biochem Biophys 260:521–531
Jollow DJ, Mitchell JR, Zampaglione N et al (1974) Bromobenzene-induced liver necrosis. Protective role of glutathione and evidence for 3,4-bromobenzene oxide as the hepatotoxic metabolite. Pharmacology 11:151–169
Carlberg I, Mannervik B (1975) Glutathione reductase levels in rat brain. J Biol Chem 250:5475–5480
Mohandas J, Marshall JJ, Duggin GG et al (1984) Differential distribution of glutathione and glutathione related enzymes in rabbit kidneys, possible implication in analgesic neuropathy. Cancer Res 44:5086–5091
Stevens M, Obrosova I, Cao X et al (2000) Effects of DL-alpha-lipoic acid on peripheral nerve conduction, blood flow, energy metabolism and oxidative stress in experimental diabetic neuropathy. Diabetes 49:1006–1015
Claiborne A (1984) Catalase activity. In: Green Wald RA (ed) CRC hand book of methods for oxygen radical research. CRC Press, Boca Raton, pp 283–284
Lowry OH, Rosenbrough NJ, Farr AL et al (1951) Protein measurement with the folin phenol reagent. J Biol Chem 193:265–275
Yousuf S, Atif F, Ahmad M et al (2009) Resveratrol exerts its neuroprotective effect by modulating mitochondrial dysfunctions and associated cell death during cerebral ischemia. Brain Res 1250:242–253
Hunter AJ, Mackay KB, Rogers DC (1998) To what extent have functional studies of ischaemia in animals been useful in the assessment of potential neuroprotective agents. Trends Pharmacol Sci 19:59–66
Yousuf S, Salim S, Ahmad M et al (2005) Protective effect of Khamira Abreesham Uood Mastagiwala against free radical induced damage in focal ischemia. J Ethnopharmacol 99:179–184
Khan MM, Ishrat T, Ahmad A et al (2010) Sesamin attenuates behavioral, biochemical and histological alterations induced by reversible middle cerebral artery occlusion in the rats. Chem Biol Interact 183:255–263
Chen SF, Hsu CW, Huang WH et al (2008) Post-injury baicalein improves histological and functional outcomes and reduces inflammatory cytokines after experimental traumatic brain injury. Br J Pharmacol 155:1279–1296
Saleem S, Ahmad M, Ahmad A et al (2006) Effect of Saffron (Crocus sativus) on neurobehavioral and neurochemical changes in cerebral ischemia in rats. J Med Food 9:246–253
Candelario-Jalil E (2009) Injury and repair mechanisms in ischemic stroke: considerations for the development of novel neurotherapeutics. Curr Opin Investig Drugs 10:644–654
Verbunt RJ, van Dockum WG, Bastiaanse EM et al (1995) Glutathione disulfide as an index of oxidative stress during postischemic reperfusion in isolated rat hearts. Mol Cell Biochem 144:85–93
Wang Z, Liu T, Gan L, Wang T, Yuan X, Zhang B et al (2010) Shikonin protects mouse brain against cerebral ischemia/reperfusion injury through its antioxidant activity. Eur J Pharmacol 643:211–217
Keelan J, Allen NJ, Antcliffe D et al (2001) Quantitative imaging of glutathione in hippocampal neurons and glia in culture using monochlorobimane. J Neurosci Res 66:873–884
Shukla D, Saxena S, Jayamurthy P et al (2009) Hypoxic preconditioning with cobalt attenuates hypobaric hypoxia-induced oxidative damage in rat lungs. High Alt Med Biol Spring 10:57–69
Reed DJ (1990) Status of calcium and thiols in hepatocellular injury by oxidative stress. Semin Liver Dis 4:285–292
Haung J, Philbert MA (1996) Cellular responses of cultured cerebellar astrocytes toethacrynic acid-induced perturbation of subcellular glutathione homeostasis. Brain Res 711:184–192
Aebi H (1984) Catalase in vitro. Methods Enzymol 105:121–126
Kumuhekar MH, Katyane SS (1992) Altered kinetic attributes of Na+–K+ ATPase activity in kidney, brain and erythrocyte membrane in alloxan diabetic rats. Indian J Exp Biol 30:26–32
Zhang W, Potrovita I, Tarabin V, Herrmann O, Beer V, Weih F, Schneider A, Schwaninger M (2005) Neuronal activation of NF-kappa B contributes to cell death in cerebral ischemia. J Cereb Blood Flow Metab 25:30–40
Schwaninger M, Inta I, Herrmann O (2006) NF-kappaB signalling in cerebral ischaemia. Biochem Soc Trans 34:1291–1294
Zhang HL, Xu M, Wei C, Qin AP, Liu CF et al (2011) Neuroprotective effects of pioglitazone in a rat model of permanent focal cerebral ischemia are associated with peroxisome proliferator-activated receptor gamma-mediated suppression of nuclear factor-κB signaling pathway. Neuroscience 176:381–395
Karin N, Ludwig Z, Csajbok MO et al (2007) Serum glial fibrillary acidic protein is related to focal brain injury and outcome after aneurysmal subarachnoid hemorrhage. Stroke 38:1489–1490
Vafeiadou K, Vauzour D, Yi LH et al (2009) The citrus flavanone naringenin inhibits inflammatory signalling in glial cells and protects against neuroinflammatory injury. Arch Biochem Biophys 484:100–109
Matute C, Domercq M, Sanchez-Gomez MV (2006) Glutamate-mediated glial injury: mechanisms and clinical importance. Glia 53:212–224
Vaughan CJ, Delanty N (1999) Neuroprotective properties of Statins in cerebral ischemia and stroke. Stroke 30:1969–1973
Tunon MJ, Garcia-Mediavilla MV, Sanchez-Campos S et al (2009) Potential of flavonoids as anti- inflammatory agents: modulation of proinflammatory gene expression and signal transduction pathways. Curr Drug Metab 10:256–271
SunM Zhao Y, Gu Y et al (2009) Inhibition of nNOS reduces ischemic cell death through down- regulating calpain and caspase-3 after experimental stroke. Neurochem Int 54:339–346
Zhang N, Komine-Kobayashi M, Tanaka R et al (2005) Edaravone reduces early accumulation of oxidative products and sequential inflammatory responses after transient focal ischemia in mice brain. Stroke 36:2220–2225
Shohami E, Bass R, Wallach D et al (1996) Inhibition of tumor necrosis factor alpha (TNF alpha) activity in rat brain is associated with cerebroprotection after closed head injury. J Cereb Blood Flow Metab 16:378–384
Touzani O, Boutin H, Chuquet J (1999) Potential mechanisms of IL-1 involvement in cerebral ischemia. J Neuroimmunol 100:203–215
Tu XK, Yang WZ, Wang CH, Shi SS, Zhang YL et al (2010) Zileuton reduces inflammatory reaction and brain damage following permanent cerebral ischemia in rats. Inflammation 33:344–352
Harari OA, Liao JK (2010) NF-κB and innate immunity in ischemic stroke. Ann NY Acad Sci 1207:32–40
Chen TY, Lin MH, Lee WT et al (2012) Nicotinamide inhibits nuclear factor-kappa B translocation after transient focal cerebral ischemia. Crit Care Med 40:532–537
Kalender Y, Kaya S, Durak D, Uzun FG, Demir F (2012) Protective effects of catechin and quercetin on antioxidant status, lipid peroxidation and testis-histoarchitecture induced by chlorpyrifos in male rats. Environ Toxicol Pharmacol 33:141–148
Daisy P, Balasubramanian K, Rajalakshmi M, Eliza J, Selvaraj J (2010) Insulin mimetic impact of Catechin isolated from Cassia fistula on the glucose oxidation and molecular mechanisms of glucose uptake on Streptozotocin-induced diabetic Wistar rats. Phytomedicine 17:28–36
Uzun FG, Demir F, Kalender S, Bas H, Kalender Y (2010) Protective effect of catechin and quercetin on chlorpyrifos-induced lung toxicity in male rats. Food Chem Toxicol 48:1714–1720
Acknowledgments
Authors are thankful to the Department of Ayurveda, Yoga and Naturalpathy, Unani, Siddha and Homeopath (AYUSH), Ministry of Health and Family Welfare, Government of India, New Delhi for financial assistance. The authors wish to thanks Mr. Dharamvir singh for his assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
All the authors (except Mohammed M. Safhi): Fund for the Improvement of Science and Technology sponsored by DST and Special Assistance Programme sponsored by UGC.
Rights and permissions
About this article
Cite this article
Ashafaq, M., Raza, S.S., Khan, M.M. et al. Catechin Hydrate Ameliorates Redox Imbalance and Limits Inflammatory Response in Focal Cerebral Ischemia. Neurochem Res 37, 1747–1760 (2012). https://doi.org/10.1007/s11064-012-0786-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-012-0786-1