
Abstract
Treatment of antioxidants is necessary to protect ischemic stroke associated neuronal damage. Xanthohumol (XN), a natural flavonoid extracted from hops, has been reported to have potential function as an antioxidant and can be used for neuro protection. However, the role of XN in ischemic stroke remains unclear. Here, we studied the neuroprotective effects of XN through experimental stroke models. Middle cerebral artery occlusion (MCAO) and oxygen–glucose deprivation (OGD) was used as in vivo and in vitro model, respectively. We found that the treatment of XN improved MCAO-induced brain injury by reducing infarct size, improving neurological deficits, reversing neuronal damage, reducing oxidative stress injury and cell apoptosis. Further experimental studies showed that XN could revive neuronal apoptosis induced by OGD by preventing oxidative stress injury. In addition, our study suggested that these effects were related to the inhibition of phosphorylation of p38-MAPK and the mediation of nuclear Nrf2 activation. In conclusion, the neuroprotective effects of XN showed in this study make XN a promising supplement for ischemic stroke protection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ischemic stroke is still one of the major causes of disability and death worldwide. Neuronal damage caused by sudden decrease of cerebral blood flow after stroke is one of the main causes of cerebral ischemic injury [1, 2]. Re-establishment of cerebral blood supply can induce additional oxidative stress injury, which is the driving force of ischemic penumbra neuron injury after ischemia/reperfusion (I/R), and ultimately lead to neuronal apoptosis [3]. In addition to the treatment after the occurrence of cerebral infarction, the preventive protection of cerebral infarction is also important. Supplementary prophylactic drugs play important roles in reducing neuronal damage after cerebral infarction. Although progress has been made in the research of neuroprotective drugs, more attention should been paid to the pretreatment of cerebral infarction [4].
Many components extracted from natural plants are supplemented with exogenous antioxidants to prevent neurological diseases [5]. In the past few years, flavonoids have been found to be involved in neurogenesis and neuronal regeneration as potential neuroprotective agents [6]. XN, a flavonoid extracted from hops (Humulus lupulus; Fig. 1a), has attracted significant attention due to its antioxidant biological properties [7, 8]. As a food additive, XN can prevent neurodegenerative diseases by reducing oxidative damage [9]. In addition, XN has been found to reduce acute ethanol-induced brain oxidative damage by reducing ROS accumulation, as well as atherosclerotic plaque formation and hypercholesterolemia [10, 11]. The discovery of these effects provides a new direction for the preventive application of XN, which is the potential to become an effective supplement for the prevention of cerebral infarction. However, the effect and specific mechanism of XN in neuronal protection needs to be further studied.

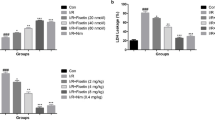
Neuroprotective effects of XN. a Structure of Xanthohumol. b Diagram of experimental design. c Neurobehavioral tests results of Bederson score, corner test, and survival rate. d HE staining and e quantitative analysis of infarct volume. NS no significance; *P < 0.05 XN versus MCAO group, **P < 0.01 XN versus MCAO group
Many studies have confirmed that inhibiting p38 mitogen-activated protein kinase (p38-MAPK) can activate the nuclear factor erythrocyte 2-related factor 2 (Nrf2)–heme oxygenase 1 (HO-1) which can reduce the excessive accumulation of ROS induced by ischemic stroke [5, 12, 13]. XN has been reported as an activator of Nrf2 and has the ability to inhibit the pathological accumulation of ROS [10, 11, 14]. Therefore, in this study, we investigated the neuroprotective effects of XN in middle cerebral artery occlusion (MCAO) and glucose and oxygen deprivation (OGD) models. We found that XN protected focal cerebral ischemia–reperfusion injury by inhibiting p38 MAPK phosphorylation and promoting Nrf2/HO-1 signaling pathway to inhibit oxidative stress injury and apoptosis.
Materials and methods
Animal model and treatment
All experiments were conducted in accordance with the guidelines for the care and use of animals in the National Institutes of Health (NIH, USA) Laboratory Animal Use Guidelines. For in vivo experiments, the male Sprague–Dawley (SD) rats (250–280 g body weights) were purchased from the experimental animal center of Harbin Medical University (ethical review under no. SYDW2019-8, Harbin, Heilongjiang Province, China). Rats were kept in standard cages under conventional laboratory conditions, and were fed with standard laboratory food at room temperature of 24 °C ± 2 °C for 12 h. After entering the room, rats were randomly divided into three groups: sham group (n = 12), MCAO group (n = 12) and XN group (n = 12). Rats in XN group were intraperitoneally (ip) injected with 0.4 mg/kg XN (Sigma, St. Louis, MO, USA) 10 min before MCAO. Rats in sham group and MCAO group were treated with saline as controls (Fig. 1b).
All rats were anesthetized by ip of chloral hydrate (350 mg/kg body weight; Beyotime, Beijing, China). As mentioned above, both MCAO and XN groups underwent MCAO surgery [15]. In brief, After 60 min, the embolus was gently removed and reperfused for 24 h. Sham-operated rats were operated the same way as the MCAO group and the XN group, but no embolus was inserted. During all the operations, the body temperature of the rats remained at 37 °C ± 0.3 °C. Observed the vital signs of rats since reperfusion and recorded the number of survivors and deaths. Survival rate = (survivors number/total number) * 100%. All rats were sacrificed 24 h after reperfusion for further study.
Neurobehavioral tests
Bederson score and corner test were measured and recorded to assess neurological impairment. Bederson score [16] was recorded before and 24 h after MCAO. A large enough angle of 30° was prepared for the corner test, and then the rats were allowed to enter the angle. Meanwhile, the direction of the rats turned in the angle [17] was recorded. All scores were recorded for further analysis. The Bederson score was repeated three times and the corner test was repeated ten times for analysis.
Infarct volume measurement
After 24 h of cerebral infarction reperfusion, rats were perfused with cold PBS (phosphate buffered saline, pH 7.4) and then perfused with cold 4% polyformaldehyde (PFA). Brain tissues were immersed in sucrose and finally cut into 20-µm thickness frozen coronal sections. To determine the infarct volume, brain sections were stained with hematoxylin and eosin (HE) and observed under optical microscope (ZEISS, Jena, Germany). Image J (National Institute of Health) was used to evaluate infarct volume (mm3) by counting pixels, and calculated according to the previous literature method [18].
Nissl staining
In order to observe the number and morphology of neurons, we performed Nissl staining. Briefly, frozen brain slices were stained with Nissl solution (Beyotime), then decolorized with alcohol, and finally immersed in xylene according to the manufacturer’s instructions. After sealing, neurons in the penumbra were observed by optical microscopy (ZEISS).
Immunohistochemical analysis
The frozen rat brain slices were sealed with 5% bovine serum albumin (BSA) at room temperature for 1 h, and then washed with cold PBS for three times. Sections were incubated with anti-4-hydroxy-2-nonaldehyde (4-HNE) antibody or anti-8-hydroxy-2′-deoxyguanosine (8-OHdG) antibody (JaICA, Shizuoka, Japan) at 4 °C for overnight. Sections were then incubated with Vector Laboratories (CA, USA) and finnaly treated with diaminobenzidine and then analyzed by optical microscopy (ZEISS).
Primary neuron culture and treatment
Primary cortical neuron culture was obtained from neonatal rats as descried previously [19]. In brief, cells were suspended in a nerve-based medium containing 2% B27 (Invitrogen, Carlsbad, CA, USA) and 10% fetal bovine serum and laid on a culture dish coated with polylysine (Sigma). Neurons were then preserved in a wet incubator containing 5% carbon dioxide at 37 °C, and the media were replaced every 3 days.
Neuron cultures were randomly divided into control group, OGD group and XN group. Cells in the XN group were incubated with 0.5 μg/mL XN solution for 10 min before OGD and maintained in a culture dish during OGD and reperfusion. In order to carry out the OGD procedure, cells were washed three times with PBS, and then cultured in glucose-free DMEM (Gibco, USA). Cells were transferred to the incubator filled with 95% nitrogen and 5% carbon dioxide at 37 °C. After 6 h of OGD, cell culture media were replaced with normal culture media and placed in 5% CO2 incubator for 24 h. At the same time, the control cells were kept in normal incubator until all cells were harvested.
Cell viability assay
Cell viability was assessed with CCK-8 (Boster Biological Technology, Wuhan, China) according to the manufacturer’s instructions. After removing the media and washing with PBS for three times, 100 μL DMEM (Hyclone) and 10 μL CCK-8 were added to each well and cultured in incubator at 37 °C for 1.5 h. A microplate reader was used to measure the OD450. OD values were recorded and analyzed.
Flow cytometry analysis
Flow cytometry was used to detect apoptotic rate, mitochondrial membrane potential and intracellular ROS expression with Annexin V-FITC/PI kit (Beyotime), JC-1 kit (Beyotime) and DCFH-DA (Sigma) according to the manufacturer’s instructions. Briefly, cells were harvested and then incubated in the mixture of Annexin V-FITC and PI at room temperature for 20 min for analyzing the extent of apoptosis. For detecting mitochondrial membrane potential and ROS, cells were dyed with JC-1 solution and DCFH-DA solution for 20 min at 37 °C in the dark, respectively. Cells were detected using flow cytometer (BD Biosciences). Flow data was analyzed using FlowJo LLC software.
Malondialdehyde (MDA), superoxide dismutase (SOD), catalase (CAT) kit and enzyme linked immunosorbent assay (ELISA)
Rat brain homogenates and cell supernatants were harvested to measure the expression levels of MDA, SOD, and CAT via assay kits (Nanjing Jiancheng Bioengineering Research Institute, Nanjing, China) according to the manufacturer’s instructions. The ratios of glutathione disulfide/glutathione GSSG/GSH ratios were mesured using assay kits (Abcam, Cambridge, MA, USA) according to the manufacturer’s instructions.
Western blot analysis
Western blot was carried out to quantitative protein expression level of brain tissue homogenates and primary neurons. Briefly, total proteins were extracted by using RIPA lysis buffer. Nuclear and cytoplasmic proteins were extracted via a nuclear and cytoplasmic protein extraction kit (Thermo Fisher, Walthman, MA, USA) according to the manufacturer’s instructions. Protein samples were separated by sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis and then transferred onto nitrocellulose membranes (Millipore Corporation, Billerica, MA, USA). Subsequently, membranes were blocked using 5% skimmed milk for 1 h at room temperature. After incubating with primary antibodies: anti-cleaved-caspase-3 antibody, anti-Bax antibody, anti-Bcl-2 antibody, anti-p38 antibody, anti-p-p38 antibody, anti-Nrf2 antibody and anti-HO-1 antibody (Abcam) overnight at 4 °C, the membranes were incubated with secondary antibodies at room temperature for 1 h. Blots were detected with the Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE, USA). The signal intensity was normalized to the control values and analyzed using Image J software (National Institute of Health).
Statistical analysis
After checking with Shapiro–Wilk test, all data were analyzed with the Mann–Whitney U test using SPSS 21.0 software. All test results with P < 0.05 were considered as statistically significant.
Results
XN had neuroprotective functions
To address the role of XN in protecting cerebral ischemia reperfusion-induced injury, rats were divided into three groups and treated as shown in Fig. 1b. XN group showed significance better Bederson score and corner test 24 h after treatment when comparing to MCAO group. In addition, we found that rats in the treatment group had slightly higher survival rate than those in the untreated group (Fig. 1c). We then used HE staining to detect the infarct volume. Massive cerebral infarction was observed in MCAO group. Importantly, the volume of cerebral infarction was significantly reduced when rats were pretreated with XN (Fig. 1d, e). These data indicated that pretreatment of XN had neuroprotective effects.
Pretreatment of XN protected neuron from damage and apoptosis
To further understand the mechanism of XN in protecting neuronal damage, we performed Nissl staining to observe the morphology change of ischemic neurons. As shown in Fig. 2a, neurons were intact in the sham group. Neurons became dark and triangular in shape in the MCAO group suggesting neuronal damage [20]. Neuron morphology in the XN group was more like the neuron shape that was observed in the sham group. We then performed flow cytometry to evaluate the effect of XN to the viability of neurons. As shown in Fig. 2b, CCK-8 cell viability analysis showed that neuron viability was significantly decreased after OGD. Importantly, XN treatment increased the viability as well as the survival rate of neurons after OGD injury. In addition, the apoptotic rate of neurons induced by OGD was significantly increased when comparing to the control group, while the apoptotic rate of neurons in the XN group was significantly lower (Fig. 2c, d). We further investigated the effect of XN on the expression level of apoptotic proteins by western blot. As shown in Fig. 2e–j, we found that the ratio of Bax/Bcl-2 was significantly increased after MCAO and OGD, but was significantly decreased with the pretreatment of XN. XN also showed inhibition of neuron apoptosis with the observation of decreased cleaved caspase-3 in XN group when comparing to MCAO group and OGD group. These data suggested that pretreatment of neuron with XN can reduce neuron damage and apoptosis.

XN protected against neurons injury and apoptosis. a Neuron morphology in sham, MCAO, and XN groups. b Cell viability. c Annexin V-FITC/PI and d quantitative analysis of apoptosis cells. Western blot bands and quantitative analysis in e–g MCAO and h–j OGD groups. *P < 0.05 XN versus OGD group, **P < 0.01 XN versus MCAO/OGD group, ##P < 0.01 XN versus sham/control group
XN improved oxidative stress injury induced by MCAO
Next, we evaluated the effects of XN on MCAO induced oxidative stress injury on neurons. Immunohistochemistry and ELISA were performed to determine the degree of oxidative stress injury. As shown in Fig. 3a–c, the numbers of 4-HNE and 8-OHdG positive cells were significantly increased in the MCAO group. However, these numbers were decreased in the XN group. In addition, the amount of MDA in the rat brain tissue was increased in the MCAO group. In comparison, the level of MDA was significantly lower in the XN group (Fig. 3d). Furthermore, the amounts of CAT and SOD were decreased in the MCAO group, but were significantly increased in the XN group (Fig. 3e, f). Moreover, XN treatment inhibited the ratio of GSSG/GSH that was induced by MCAO (Fig. 3g). Taken together, these results showed that pretreatment of XN could significantly inhibit oxidative stress injury induced by MCAO.

XN ameliorated oxidative stress injury caused by MCAO and OGD. Immunohistochemistry of a 4-HNE and 8-OHdG, and number of positive cells of b 4-HNE and c 8-OHdG. ELISA results of d, l MDA, e, m CAT, f, n SOD and g, o GSSG/GSH ratio in vivo/vitro. *P < 0.05 XN versus MCAO/OGD group, **P < 0.01 XN versus MCAO/OGD group, #P < 0.05 XN versus Sham/Contrl group, ##P < 0.01 XN versus Sham/Contrl group
XN alleviates oxidative stress injury induced by OGD
Next, we evaluated the effects of XN on OGD induced oxidative stress injury on neurons. Flow cytometry and ELISA were performed to detect the degree of oxidative stress injury. Pretreatment of XN improved mitochondrial membrane potential damage after OGD (Fig. 3h, i) as well as reduced excessive ROS accumulation induced by OGD (Fig. 3j, k). In addition, MDA expression level was increased after OGD, but was significantly decreased in the XN group (Fig. 3l). Moreover, with the pretreatment of XN, the amounts of CAT and SOD were increased when comparing to the OGD group (Fig. 3m, n). Furthermore, After OGD, the GSSG/GSH ratio was increased in the OGD group, but was significantly decreased in the XN group (Fig. 3o). These data indicated that XN pretreatment of XN could significantly inhibit oxidative stress injury induced by OGD.
XN triggered the expression of Nrf2/HO-1 after MCAO and OGD by inhibiting the expression of phosphorylated p38
Next, we investigated the potential mechanism of XN on ischemic injury in vivo and in vitro. Ischemia induced phosphorylation of p38-MAPK. We found that the XN group significantly inhibited the activation of p-p38 induced by MCAO and OGD (Fig. 4a, b). Further investigations showed that n-Nrf2 were up-regulated in the MCAO and OGD groups, while c-Nrf2 was down-regulated. The relative expression of n-Nrf2/c-Nrf2 was increased after MCAO and OGD. Treatment with XN enhanced the expression of n-Nrf2 as well as the relative expression level of n-Nrf2/c-Nrf2. In addition, HO-1, one of the downstream antioxidant proteins of Nrf2, showed an increased expression level after MCAO and OGD, which were further increased in the XN treatment group (Fig. 4c–f). These data indicated that XN induced the expression of Nrf2 and HO-1 after MCAO and OGD through by suppressing the expression of phosphorylated p38.

XN restrained the expression of phosphorylated p38 and triggered Nrf-2/HO-1 expression. Western blot results of a, b p38 MAPK in vivo and vitro. Western blot results of n-Nrf2, c-Nrf2, n/c (n-Nrf2/c-Nrf2) density ratio and HO-1 in c, d MCAO and e, f OGD. *P < 0.05 XN versus MCAO/OGD group, **P < 0.01 XN versus MCAO/OGD group, ##P < 0.01 XN versus sham/control group
Discussion
In ischemic brain cells, neuron death is irreversible and is particularly important in brain tissue damage [1, 4]. Give appropriate intervention before the occurrence of cerebral infarction could provide patients more benefit to alleviate cerebral ischemia injury. XN has many pharmacological effects and is considered as a potential natural beneficial substance. Although XN has been shown to have neuroprotective potential, the positive effects of XN on cerebral ischemia and stroke remain unclear [14]. In this study, we showed that XN as a natural plant extract has antioxidant, anti-apoptotic and therapeutic effects on cerebral ischemia-induced injury. This is supported with the observation that preventive treatment of XN can effectively reduce the volume of cerebral infarction and improve neurological deficits 24 h after MCAO. The reduction of infarct size attributes to the ability of XN to improve neurological function and the reversal of dead neurons [1, 3, 4]. In addition, we also observed the abnormal morphological changes of neurons 24 h after MCAO. XN showed positive effects in maintaining neuronal morphology close to normal morphology after cerebral ischemia injury.
Oxidative stress, especially excessive accumulation of ROS, is the most important factor for neuronal death induced by cerebral ischemia [21]. Increased ROS production in cells provides an oxidative environment within cells, which correspondingly leads to damage to basic cellular structural components such as lipids and DNA [22,23,24,25]. We observed that XN could reduce ROS accumulation, lipid peroxidation and DNA damage induced by MCAO and OGD. The structure and function of mitochondria were damaged when ROS accumulated in large quantities [26]. We confirmed that the preventive treatment of XN had protective effect on mitochondrial membrane potential damage. These results indicated that the neuroprotective effects of XN on MCAO and OGD injury were due to the antioxidant characteristic of XN.
Mitochondrial membrane damage affects the changes of apoptosis-related proteins. Under oxidative stress, the balance of Bcl-2 family protein expression, especially the Bax/Bcl-2 ratio, plays an important role in regulating apoptosis [27, 28]. We found that XN could reduce the Bax/Bcl-2 ratio, which can be induced by MCAO and OGD. This observation further supported the antioxidant effect of XN. To some extent, the increased ratio of Bax/Bcl-2 leads to the activation of caspase-3, which eventually leads to neuronal death after cerebral ischemia [29]. XN has been reported to inhibit cleaved-caspase-3 expression in ischemic region [30]. But there were no more evidences to prove the ability of XN to protect neurons. Our study also confirmed that XN reduced the protein level of cleaved-caspase-3 in ischemia neurons. We further studied the relationship between anti-apoptotic ability and neuroprotective function of XN. We found that XN reduced the number of apoptotic neurons after ischemic stroke. These results suggested that XN might inhibit neuronal apoptosis by inhibiting oxidative stress injury.
Apoptosis induced by intracellular accumulation of ROS can be eliminated by antioxidant enzymes, which convert peroxides into less toxic or harmless substances. We examined the effects of XN on the expression of antioxidant enzymes and provided evidence that XN may have antioxidant activity against oxidative stress. Under oxidative stress, Nrf2 is activated and transferred from cytoplasm to nucleus, which promotes the transcription of HO-1, SOD and CAT, and reduces the GSSG/GSH ratio after reperfusion [31]. The pretreatment of XN promoted this process in ischemic rats and primary neurons, suggesting that the activation of Nrf2 played an important role in the antioxidant capacity of XN.
Inhibiting the p38-MAPK pathway is one of the ways to reduce neuronal apoptosis induced by cerebral infarction [32]. Considering recent studies, the relationship among the activation of Nrf2 and the expression of enzymatic antioxidants and the inhibition of p38-MAPK in ischemic injury can not be ignored [33]. We detected the inhibition of XN preconditioning on p-p38 activation induced by cerebral ischemia. Therefore, we proposed that the mechanism of XN inhibiting neuronal apoptosis after cerebral ischemia is closely related to inhibiting the expression of p-p38 and activating Nrf2 pathway to promote the expression of enzymatic antioxidants. In conclusion, our study provided evidence for the neuroprotective effects of XN on cerebral ischemic stroke, and XN may have potential to be considered as a supplement for ischemic stroke protection (Fig. 5).

A proposed molecular mechanism underlies the neuroprotective effects of XN in experimental stroke model
References
Iadecola C, Anrather J (2011) The immunology of stroke: from mechanisms to translation. NatMed 17:796–808
Paspalj D, Nikic P, Savic M, Djuric D, Simanic I, Zivkovic V, Jeremic N, Srejovic I, Jakovljevic V (2015) Redox status in acute ischemic stroke: correlation with clinical outcome. Mol Cell Biochem 406:75–81
Ran YC, Zhu M, Li SJ, Zhang ZX, Wang X, Zhang Y, Cheng JL (2018) Related research and recent progress of ischemic penumbra. World Neurosurg 116:5–13
Puig B, Brenna S, Magnus T (2018) Molecular communication of a dying neuron in stroke. Int J Mol Sci 19:2834
Yang CH, Yen TL, Hsu CY, Thomas PA, Sheu JR, Jayakumar T (2017) Multi-targeting andrographolide, a novel NF-κB inhibitor, as a potential therapeutic agent for stroke. Int J Mol Sci 18:1638
Dias GP, Cavegn N, Nix A, do Nascimento Bevilaqua MC, Stangl D, Zainuddin MS, Nardi AE, Gardino PF, Thuret S (2012) The role of dietary polyphenols on adult hippocampal neurogenesis: molecular mechanisms and behavioural effects on depression and anxiety. Oxid Med Cell Longev 2012:541971
Dostálek P, Karabín M, Jelínek L (2017) Hop phytochemicals and their potential role in metabolic syndrome prevention and therapy. Molecules 22:1761
Liu M, Hansen PE, Wang G, Qiu L, Dong J, Yin H, Qian Z, Yang M, Miao J (2015) Pharmacological profile of xanthohumol, a prenylated flavonoid from hops (Humulus lupulus). Molecules 20:754–779
Yao J, Zhang B, Ge C, Peng S, Fang J (2015) Xanthohumol, a polyphenol chalcone present in hops, activating Nrf2 enzymes to confer protection against oxidative damage in PC12 cells. J Agric Food Chem 63:1521–1531
Doddapattar P, Radović B, Patankar JV, Obrowsky S, Jandl K, Nusshold C, Kolb D, Vujić N, Doshi L, Chandak PG, Goeritzer M, Ahammer H, Hoefler G, Sattler W, Kratky D (2013) Xanthohumol ameliorates atherosclerotic plaque formation, hypercholesterolemia, and hepatic steatosis in ApoE-deficientmice. Mol Nutr Food Res 57:1718–1728
Pinto C, Cestero JJ, Rodríguez-Galdón B (2014) Xanthohumol, a prenylated flavonoid from hops (Humulus lupulus L.), protects rat tissues against oxidative damage after acute ethanol administration. Toxicol Rep 1:726–733
Cheong SH, Lee DS (2017) Taurine chloramine prevents neuronal HT22 cell damage through Nrf2-related heme oxygenase-1. Adv Exp Med Biol 975:145–157
Yen TL, Chen RJ, Jayakumar T, Lu WJ, Hsieh CY, Hsu MJ, Yang CH, Chang CC, Lin YK, Lin KH, Sheu JR (2016) Andrographolide stimulates p38 mitogen-activated protein kinase-nuclear factor erythroid-2-related factor 2-heme oxygenase 1 signaling in primary cerebral endothelial cells for definite protection against ischemic stroke in rats. Transl Res 170:57–72
Li F, Yao Y, Huang H, Hao H, Ying M (2018) Xanthohumol attenuates cisplatin-induced nephrotoxicity through inhibiting NF-κB and activating Nrf2 signaling pathways. Int Immunopharmacol 61:277–282
Chen B, Zhang F, Li QY, Gong A, Lan Q (2016) Protective effect of Ad-VEGF-bone mesenchymal stem cells on cerebral infarction. Turk Neurosurg 26:8–15
Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H (1986) Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke 17:472–476
Li X, Blizzard KK, Zeng Z, DeVries AC, Hurn PD, McCullough LD (2004) Chronic behavioral testing after focal ischemia in the mouse: functional recovery and the effects of gender. Exp Neurol 187:94–104
Shang J, Liu N, Tanaka N, Abe K (2012) Expressions of hypoxic stress sensor proteins after transient cerebral ischemia in mice. J Neurosci Res 90:648–655
Qu Y, Mao M, Zhao F, Zhang L, Mu D (2009) Proapoptotic role of human growth and transformation-dependent protein in the developing rat brain after hypoxia-ischemia. Stroke 40:2843–2848
Oechmichen M, Meissner C (2006) Cerebral hypoxia and ischemia: the forensic point of view: a review. J Forensic Sci 51:880–887
Bakthavachalam P, Shanmugam PST (2017) Mitochondrial dysfunction-silent killer in cerebral ischemia. J Neurol Sci 375:417–423
Bai J, Mei Y (2011) Overexpression of aldehyde dehydrogenase-2 attenuates neurotoxicity induced by 4-hydroxynonenal in cultured primary hippocampal neurons. Neurotox Res 19:412–422
Gilgun-Sherki Y, Rosenbaum Z, Melamed E, Offen D (2002) Antioxidant therapy in acute central nervous system injury: current state. Pharmacol Rev 54:271–284
Sun K, Fan J, Han J (2015) Ameliorating effects of traditional Chinese medicine preparation, Chinese materia medica and active compounds on ischemia/reperfusion-induced cerebral microcirculatory disturbances and neuron damage. Acta Pharm Sin B 5:8–24
Yang JL, Mukda S, Chen SD (2018) Diverse roles of mitochondria in ischemic stroke. Redox Biol 16:263–275
Sanderson TH, Reynolds CA, Kumar R, Przyklenk K, Hüttemann M (2013) Molecular mechanisms of ischemia-reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol Neurobiol 47:9–23
D’Orsi B, Mateyka J, Prehn JHM (2017) Control of mitochondrial physiology and cell death by the Bcl-2 family proteins Bax and Bok. Neurochem Int 109:162–170
Rodrigo R, Fernández-Gajardo R, Gutiérrez R, Matamala JM, Carrasco R, Miranda-Merchak A, Feuerhake W (2013) Oxidative stress and pathophysiology of ischemic stroke: novel therapeutic opportunities. CNS Neurol Disord Drug Targets 12:698–714
Plesnila N, Zinkel S, Le DA, Amin-Hanjani S, Wu Y, Qiu J, Chiarugi A, Thomas SS, Kohane DS, Korsmeyer SJ (2001) BID mediates neuronal cell death after oxygen/glucose deprivation and focal cerebral ischemia. Proc Natl Acad Sci USA 98:15318–15323
Yen TL, Hsu CK, Lu WJ, Hsieh CY, Hsiao G, Chou DS, Sheu GJ, Wu JR (2012) Neuroprotective effects of xanthohumol, a prenylated flavonoid from hops (Humulus lupulus), in ischemic stroke of rats. J Agric Food Chem 60:1937–1944
Zhang R, Xu M, Wang Y, Xie F, Zhang G, Qin X (2017) Nrf2-a promising therapeutic target for defensing against oxidative stress in stroke. Mol Neurobiol 54:6006–6017
Sun J, Nan G (2016) The mitogen-activated protein kinase (MAPK) signaling pathway as a discovery target in stroke. J Mol Neurosci 59:90–98
Li C, Zhang WJ, Frei B (2016) Quercetin inhibits LPS-induced adhesion molecule expression and oxidant production in human aortic endothelial cells by p38-mediated Nrf2 activation and antioxidant enzyme induction. Redox Biol 9:104–113
Acknowledgements
This work was supported by the National Natural Science Foundation of China under Grant Nos. 81571166, 81771361 and 81820108014; Heilongjiang Provincial Postdoctoral Science Foundation under Grant No. LBH-Z17138; Health and Family Planning Commission of Heilongjiang Province under Grant No. 2016-072.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
All the authors have declared that no conflict interest exists.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Jiao, Y., Cao, Y., Lu, X. et al. Xanthohumol protects neuron from cerebral ischemia injury in experimental stroke. Mol Biol Rep 47, 2417–2425 (2020). https://doi.org/10.1007/s11033-019-05128-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-019-05128-4