
Abstract
The SAMP8 strain spontaneously develops learning and memory deficits with characteristics of aging, and is a good model for studying the mechanism of cognitive dysfunction with age. Oxidative stress occurs systemically in SAMP8 from early on in life and increases with aging. Neuropathological changes such as the deposition of Aβ, hyperphosphorylation of tau, impaired development of dendritic spines, and sponge formation, and neurochemical changes were found in the SAMP8 brain. These changes may be partially mediated by oxidative stress. Oxidative damage is a major factor in neurodegenerative disorders and aging. A decline in the respiratory control ratio suggesting mitochondrial dysfunction was found in the brain of SAMP8. The rise in oxidative stress following mitochondrial dysfunction may trigger neuropathological and neurochemical changes, disrupting the development of neural networks in the brain in SAMP8.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The senescence-accelerated mouse (SAM) was originally derived from AKR/J strain, litters of which showing characteristic of aging such as hair loss, lack of glossiness, skin coarseness, and a short life span were selected as the progenitors of the senescence-prone series (P series). Litters in which the aging process was normal were also selected as the progenitors of a senescence-resistant series (R series). Offspring of these progenitors, P series and R series, were mated by selective breeding to establish inbred strains as SAMP and SAMR strains, respectively. Thereafter, SAMP strains that spontaneously develop accelerated senescence and specific pathologic phenotypes and SAMR strains that show normal aging were established [1].
SAMP8, one of the prone strains of senescence-accelerated mice, is known as a model of senile dementia. These animals exhibit age-related deficits of learning and memory from an early age. Previous studies by ourselves and others revealed reduction in the amounts of neurotransmitters released from brain slices [2, 3], decreases in levels of muscarinic acetylcholine, and serotonine-1A and NMDA receptors [4, 5], and decreases in protein kinase C [5, 6] and synaptic spine density [7]. Genes related to Alzheimer’s disease (AD) were found in the SAMP8 brain [8], and β-amyloid (Aβ) deposits were found in the brains of aged SAMP8 mice [9, 10]. Abnormal granular structures, which stained positively with periodic acid-Schiff (PAS-positive granular structures; PGS), have also been observed in the SAMP8 brain [11]. In addition, spongiform degeneration due to the demyelinisation of neurons was found in the brainstem and arcuate nucleus of the hypothalamus [12]. These changes may be contributed to the decline of cognitive function in SAMP8 mice. However, the mechanism underlying the development of learning and memory deficits is not clear. Recently, genetic analysis to detect the genes responsible for the cognitive dysfunction in SAMP8 revealed that a small number of genes are related to the impaired development of learning and memory [13, 14] and that mitochondrial genes are abnormally expressed in the brain [15]. In this review, the current findings from neurochemical, neuropathological, and genetic studies are summarized.
Neuropathological Changes in Brain of SAMP8
SAMP8 mice spontaneously develop deficits of learning and memory at a young age, and the impairment worsens with age. Learning and memory in the SAMP8 strain has been assessed using several paradigms, including water-maze, T-maze, passive avoidance, and one-way active avoidance tests, with declines in cognitive function evident at 2 months of age, and significantly at 4 months as compared with normal aging SAMR1 mice [16]. In an immunoblot analysis, we found that amyloid precursor protein (APP)-like protein was increased with age in the brain of SAMP8, associated with an increase in gliosis and glial fibrillary acidic protein (GFAP) [9]. Recently, Takemura et al. [17] found that β/A4 protein-like immunoreactive granular structures (β-LIGS) increased with age using a polyclonal antibody to β/A4 protein fragments 10–42 in SAMP8 and SAMR1. β-LIGS occur throughout the brain, including in the medial septum, cerebral cortex, hippocampus, cerebellum, and cranial nerve nuclei, and significantly increase in number and density with age in SAMP8. Morley et al. [10] reported age-related increases in protein and mRNA levels of APP in the hippocampus of SAMP8 mice. However, in a study using immunocytochemistry, Aβ plaque was detected in the hippocampus of SAMP8 mice by 16 months of age, but no neuronal loss or apoptosis was detected in the hippocampus. No neuronal loss was seen in APP transgenic mice either [18, 19]. This suggests that there may be an apoptosis-resistant factor in the mouse or an additional pathological factor other than Aβ contributes to the massive neuronal loss in AD [19].
The deposition of Aβ is a major pathological hallmark of AD. Level of APP and its mRNA are increased in AD. The accumulation of Aβ (1–42) is considered to be a cause of AD. Therefore, a reduction in the amount of Aβ (1–42) accumulated might lead to a cure. When anti-sense oligonucleotide for Aβ was administrated intracerebroventricularly into the brain of SAMP8 mice, a reduction in the level of APP protein in the amygdale, septum, and hippocampus, a decrease in protein oxidation and lipid peroxidation, and an improvement of deficits in learning and memory were observed [20, 21]. Furthermore, immunotherapy with anti-Aβ antibody performed in APP transgenic and SAMP8 mice resulted in a reduction in the amount of Aβ deposited and a reversal of learning and memory deficits [22, 23].
Another hallmark of AD is the formation of neurofibrillary tangles (NFTs). NFTs develop as a result of the accumulation of hyperphosphorylated tau. Recently, Canudas et al. [24] reported that tau is hyperphosphorylated in the cortex, striatum, and hippocampus of 5-month-old SAMP8 compared with age-matched SAMR1. Glycogen synthase kinase-3β (GSK-3β) is one of the most implicated tau kinase involved in Alzheimer-like tau hyperphosphorylation [25–27]. However, the activity of GSK3β in the SAMP8 brain did not differ from that in the SAMR1, but the level of Cdk5 was higher in SAMP8 than SAMR1. Tau hyperphosphorylation was accompanied by an increase in Cdk5/p25 activity and an increase in Cdk5 expression, indicating a direct relationship between phosphorylation enzyme activities in the aging process in this senescence model [24]. A higher level of α-synuclein was also found in the brain of SAMP8 mice [28]. α-Synuclein is a potent inducer of tau hyperphosphorylation in neurodegenerative disorders [28, 29]. Oxidative stress is known to be directly associated with increases in the concentration of α-Ssynuclein [30]. Since the weak activity of antioxidant enzymes and the oxidative state are potentiated in SAMP8, abnormal phosphorylation of tau and increases in the α-synuclein level may be mediated in part by the production of reactive oxygen species (ROS) [28].
Neurochemical and Morphological Changes in the SAMP8 Brain
Neurochemical changes in the brains of SAMP8 mice have been reported by previously. We investigated the N-methyl-d-aspartic acid (NMDA)-evoked [3H] acetylcholine (ACh) and noradrenaline (NA) release from brain slices and age-related changes of the release in SAMP8 and SAMR1 mice. The NMDA (1 mM)-evoked [3H] ACh release did not change in SAMR1 between 2 and 14 months of age. However, it decreased markedly in SAMP8 at 6 and 14 months old [3]. Also, the NMDA (100 μM)-evoked [3H] NA release in SAMR1 was constant at 1–12 months of age, whereas, in SAMP8, it showed a sharp decline at 6–12 months old [2]. These neurochemical changes reflect age-related impairments of learning and memory in SAMP8 mice. Moreover, binding assays of muscarinic ACh (M1) receptors and NMDA receptors in the cerebral cortex, hippocampus, and cerebellum of SAMP8 mice demonstrated that numbers of M1 and NMDA receptors were decreased in the hippocampus at 2 and 12 months of age, and in the cerebral cortex at 12 months of age [7]. Amounts of protein kinase C (PKC) in the cytosol and membrane were also significantly reduced in the hippocampus of SAMP8 at age 12 months [7]. Armbrecht et al. [6] also reported that levels of PKC-gamma and calbindin in the hippocampus of SAMP8 decreased markedly with age, and the level of PKC-gamma was well correlated with a decline in learning ability assessed using the T-maze. These results suggest that the loss of calbindin decreases intracellular Ca2+ levels and changes in Ca2+ homeostasis, resulting in an alternation of Ca2+-dependent protein kinase activity. All these findings indicate a decrease in synaptic activity in the hippocampus of SAMP8.
In the hippocampal neurons, cyclic AMP-response element binding protein (CREB) is phosphorylated by protein kinase A (PKA), PKC, Ca2+/calmoduline-dependent kinase (CaMK) II, and CaMKIV following Ca2+ influx through the NMDA receptors [31–34]. CREB plays an important role for synaptic plasticity and long-term memory (LTM) formation. Phosphorylated CREB (p-CREB) is essential for lasting late-phase long-term potentiation (L-LTP), which is considered to be important for the promotion of gene expression and synaptic spine formation [31, 35–37]. We investigated CREB phosphorylation in the hippocampal CA1 neurons of SAMP8. Figure 1 shows the time-course changes of p-CREB in the hippocampal CA1 region of SAMP8 and SAMR1 at 2-month-old after electric shocks by a step-through passive avoidance apparatus. SAMR1 showed a biphasic peak of CREB phosphorylation at 3 and 12 h [38]. These two peaks were also observed in step-down avoidance and forced swimming tasks using normal rats [39, 40]. In contrast, one slight peak at 9 h was observed in the hippocampus of SAMP8 (Fig. 1). CREB phosphorylation produces a biphasic peak during hippocampal LTP [41]. Mutant mice, with defective NMDA receptor, CaMKII, and PKC genes, do not exhibit early-phase LTP (E-LTP) in the hippocampus [42–44]. In contrast, CREB knockout mice were deficient in the hippocampal L-LTP and LTM [45]. In the hippocampus of SAMP8, E-LTP was not occurred and L-LTP was decremented in short. This may be associated with a decrease in the NMDA receptors and PKC in the hippocampus of SAMP8.

Time-course of changes in the level of p-CREB in the hippocampal CA1 region after electric shock. Values represent means ± SEM *P < 0.05, **P < 0.01 versus control (C: non-shocked mice)
The remodeling of synaptic structures and the formation of new synaptic contacts are necessary for learning and memory. L-LTP induces dendritic spine formation [37]. Sugiyama et al. [7] reported a decrease in the number of dendritic spines in the hippocampal CA1 pyramidal cells of SAMP8. This may be due to a transient L-LTP by an impairment of CREB phosphorylation in the hippocampus of SAMP8. In addition, hippocampal synaptic spine density is affected by hormones [46]. Apical dendritic spine density of female rats was greater in proestrus than estrus in the hippocampal CA1 pyramidal cells. Synaptic spine density of ovariectomized (OVX) animals decreased with the depletion of estrogen [47]. OVX animals treated with estrogen showed an increase in synaptic spine density in the hippocampal CA1 neurons [46, 47]. In addition, estrogen induces new spines to form, but not dendritic lengthing or branching [47, 48]. Shors et al. [49] reported that there are sex differences in hippocampal spine density correlated with sex hormones, estrogen and testosterone. In the hippocampal CA1 pyramidal cells of SAMP8, the number of dendritic spines was smaller in females than males [7]. This might reflect the sex difference in the learning and memory ability of SAMP8: deficits in learning and memory are more severe in females than males (Fig. 2). It has been reported that the mRNAs of both estrogen receptor (ER) isoforms (ERα and ERβ) are expressed in the hippocampal neurons [50], and that levels of ERβ mRNA are higher than ERα mRNA in the cerebral cortex and hippocampus [51]. ERβ was also localized in the membrane of synapses in the hippocampal neurons [52]. Recently, the function of ERα and ERβ in the brain has been investigated using knockout mice. It has been demonstrated that ERβ is required for emotional behavior and spatial learning [53, 54], and that the phosphorylation of CREB in the hippocampal CA1 neurons is blocked completely in ERβ knockout mice [55]. Recently, Zhou et al. [56] reported age-related changes of ERα and ERβ gene expression in the hippocampus of SAMP8. The ERα gene in the hippocampus is constantly expressed at 3–15 months of age in both male and female SAMR1 and SAMP8, and no differences in the expression in both strains were observed. However, ERβ gene expression in the hippocampus decreased with age in both sexes in SAMR1 and SAMP8, and the level of ERβ mRNA in SAMP8 at 9–15 months was significantly lower than that in age-matched SAMR1. In our study, a significant reduction of ERβ gene expression was also observed in the hippocampal CA1 neurons of SAMP8 at 2 months of age as compared with age-matched SAMR1, but not in the cerebral cortex (data not shown). Also, the level of ERβ mRNA in the hippocampus was well correlated with learning and memory ability in SAMP8 (data not shown). These results suggest that an impairment of CREB phosphorylation occurred with the alteration by neurochemicals of the NMDA receptor, CaMKII, and PKC. These changes may be associated with ERβ, resulting in a deficiency in the formation of dendritic spines in hippocampal pyramidal cells.

Age-related changes in learning and memory abilities in SAMP8. Learning and memory abilities in SAMP8 mice were assessed using step-through passive avoidance apparatus. a Age-related changes in the mean retention time of SAMP8 mice. The values are mean ± SEM *significantly different from the mean value of females at 5 months of age (P < 0.05; t-test). b Severity of learning and memory deficits at different ages in male and female SAMP8 mice. Retention times were classified into the following three grades: severe impairment, 0–99 s; moderate impairment, 100–199 s; mild impairment, 200–299 s
Oxidative Stress and Dietary Interventions in SAMP8
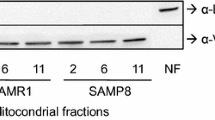
Oxidative stress is considered to be a causal factor of aging and the development of various diseases. A higher oxidative state has been detected in various organs of SAMP strains compared with normal aging SAMR1. It has been reported that the levels of lipid hydroperoxide and protein carbonyl increased with age and were higher at an early age in the brain, liver, and heart of SAMP8 compared with SAMR1 [57–61]. In contrast, the activities of antioxidant enzymes, superoxide dismutase (SOD), catalase, and glutathione peroxidase (GPx), are decreased in the cerebral cortex, hippocampus, and liver of SAMP8 [57, 62, 63]. These results suggest that the level of oxygen free radicals is systemically increased in SAMP8. In aged SAMP8, a decrease in the respiratory control ratio was found in the liver mitochondria, suggesting insufficient synthesis of ATP [64]. A higher redox state and greater activity for mitochondrial respiration with a lower respiration control ratio were found in the brain of SAMP8 at 2-month-old compared to age-matched SAMR1 [65]. Xu et al. [66] reported a significant decrease in platelet mitochondrial membrane potential (∆ψ m) in 2–9-month-old SAMP8 and hippocampal ATP in 6–9-month-old mice. In addition, mitochondrial DNA deletion was increased in the brain of SAMP8 at 4–8 weeks of age [67]. These results suggested that mitochondrial dysfunction occurred from a relatively early stage of life in SAMP8.
Free radical scavengers can be used to prevent oxidative damages to various organs. The administration of N-tert-butyl-α-phenylnitrone as a spin-trapping antioxidant reduced lipid peroxide and protein carbonyl levels in the cerebral cortex [68] and prolonged life span [69] of SAMP8. It has been reported that a reduction in spatial cognition in the water maze task was correlated with an increased amount of protein carbonyl in the cerebral cortex of aged mice [70], and that the administration of antioxidants improved memory impairment in the behavioral tests such as passive avoidance and radial-arm maze tests in old mice and gerbils [71–73]. The administration of α-lipoic acid and N-acetylcysteine reversed cognitive dysfunction in T-maze footshock avoidance and reduced oxidative stress in the brain of 12-month-old SAMP8 [60]. In addition, administration of di-n-propyl trisulfide, an organosulfur constituent of onion, reduced lipid hydroperoxide levels in the hippocampus and ameliorated impairments of learning and memory in aged SAMP8 [74]. Thus, these results suggest that antioxidant interventions are applicable for prevention of aging and cognitive dysfunction in SAMP8.
Caloric restriction (CR) prolongs life span in C. elegans and rodents [75–78] and reduces the risk factor of diseases in rodents and humans [79–82]. Recently, evidence supporting a potential neuroprotective role for CR was obtained using an aminal model of AD (Tg2576 mice). The development of AD-type amyloid plaques in the neocortex and hippocampus of Tg2576 mice was completely prevented by a low carbohydrate diet. The low carbohydrate diet increased in 30% ADAM10 (a disintegrin and metalloproteinase) concentration with the elevation of neocortical α-secretase activity [83]. CR is known to reduce oxidative stress [84]. It has been reported that CR decreased levels of ROS generated in mitochondria and oxidative damage to DNA, protein, and lipid [85–91]. Hyun et al. [92] reported that CR increased activities of plasma membrane redox system enzymes (NADH-ascorbate free radical reductase, NADH-quinone oxidoreductase 1, NADH-ferrocyanide reductase, NADH-coenzyme Q10 reductase, and NADH-cytochrome c reductase) and antioxidant levels (α-tocopherol and coenzyme Q10) in the brain of rats during aging. In SAMP8 mice, levels of ROS were significantly lower in the CR group than normal diet group. In contrast, levels of SOD activity were higher in the CR group. CR also markedly reduced the grading score of senescence and prolonged the life span of SAMP8 by about 30% [93]. Moreover, learning and memory impairments in SAMP8 were ameliorated by the administration of a CR diet in a passive avoidance apparatus. Interestingly, CR affects the metabolism of neurotransmitters in the brain of SAMP8 [94]. The levels of dopamine and serotonin were significantly increased in the cortex and hippocampus of the CR group. The acetylcholine level was also increased in the hippocampus of the CR group. This change was consistent with the increased ChAT activity in the CR group. The generation of ROS was reduced in the brain of SAMP8 given the CR diet. Thus, dietary restriction may be another intervention for the possible prevention of aging and neurodegenerative disorders.
Heredity and Gene Expression Profiling in SAMP8
Molecular genetic characterization of the SAM strains was performed using 27 biochemical and immunogenetic markers, and 74 endogenous murine leukemia virus (MuLV) proviral markers. A comparison of the panels of provirus loci revealed that all the SAM strains genetically resemble, but are distinguishable from one another, and that all strains are clearly different from the parental AKR/J strain. These findings indicate SAM strains to be a group of recombinant inbred or related inbred strains developed by some accidental out-breeding between AKR/J and one or more unknown strains [95, 96]. Furthermore, Xia et al. [97] performed genotyping of the SAM strains with 581 microsatellite markers. Genomic DNA was extracted from males of the SAM strains and AKR/J at 3 months of age, and amplified by PCR with microsatellite markers. Comparison of the distribution of the alleles of loci among the SAMP and SAMR series revealed a notable difference in D4Mit54, D14Mit92, D16Mit30, and D17Mit176. Estimation of genetic similarity between two inbred strains based on the percentage of loci with identical alleles revealed high similarity among the SAMR1, SAMR3A, and SAMR4 strains and between SAMP7 and SAMP9. Therefore, it is possible that some of these four chromosomal regions contain the genes responsible for accelerated senescence in the SAMP strains.
SAMP strains spontaneously develop accelerated senescence and specific pathological characteristics. To obtain information on heredity for SAMP strains, we investigated the inheritance of cognitive dysfunction in SAMP8 [14]. SAMP8 was crossed with Japanese fancy mouse 1 (JF1) normal mice to obtain F1, F2, and backcross generations. Memory retention times of the F1, F2, and backcross generations at 5 months of age were intermediate between those of the parent strains (Table 1). The incidence of learning and memory deficits (LMD) in the F2 generation was 70% in males and 76% in females (Table 2). This result is in good agreement with a 3:1 segregation. In the backcross generation, the incidence of LMD was 44% in males and 43% in females, and the segregation ratio of LMD to normal was 1:1. These results suggest the presence of a single major gene for LMD that may be an incomplete dominance. If LMD in SAMP8 is regulated by only one gene, the distribution of memory retention time in the F2 generation would show a bimodal distribution. However, the distribution was broad. This suggests that more than one gene is involved in the development of LMD. Also, the estimation of gene numbers controlling LMD was 1.64–3.58. Therefore, these results suggest that at least 2–4 genes are involved in the inheritance of LMD in SAMP8. In another study, the number of genes involved in LMD in SAMP8 was estimated by using successive backcross mating between SAMP8 and CD-1. The result showed that a few genes may play a major role in the pathogenesis of the cognitive deterioration of SAMP8 [13]. This number matched to our data. Thus, it is possible that relatively few genes are involved in the age-related cognitive dysfunction in SAMP8.
Moreover, to clarify the genes involved in age-related memory dysfunction in SAMP8, a quantitative trait loci (QTL) analysis was performed with SAMP8 and JF1 normal mice. The analysis demonstrated significant LOD scores for D1Mit178 on chromosome 1, D12Mit150 on chromosome 12, D13Mit 228 on chromosome 13, and D15Mit136 and D15Mit120 on chromosome 15 in SAMP8 [98]. These four chromosomes are different from the chromosomes found by Xia et al. [97]. Wei et al. [8] reported that AD-related genes such as those for βAPP, PS2, APOE, tau, IL-1β, IL-6, and TNF-α are abnormally expressed in the brain of SAMP8. However, these genes did not match our QTLs. Interestingly, the QTL on chromosome 1 had different effects on cognitive function depending on genotype and sex. Cognitive dysfunction derived from SAMP8 alleles in the QTL on chromosome 1 is more severe in female homozygous (P8/P8) than male homozygous (P8/P8) mice. This sex difference was seen only in homozygous (P8/P8) mice, not in the heterozygous (JF1/P8) and homozygous (JF1/JF1) mice [98]. Thus, the sex difference of LMD in SAMP8 may be due to hormonal effects on chromosome 1.
Recently, gene-expression profiling was performed on young and old SAMP8, SAMP10, SAMR1 and C57BL/6J [99]. Retina, hippocampus, and spleen were removed from the four strains, and total RNA was extracted. Analysis of the genes whose expression changed with aging in each strain revealed that one gene, encoding complement component 4 (C4), changed with age in the hippocampus of all four strains, and that seven genes increased in C57BL/J and at least one SAM. Also, the expression of two genes was downregulated with aging in all three SAM strains. The majority of the genes altered during aging (65%) were specific to one of four strains. In addition to the C4 gene, another gene changed during aging in all strains. The expression of phosphatidylserine decarboxylase (PSDC) was upregulated in the retina of all aged strains and in the hippocampus of aged SAMP8, SAMR1, and C57BL/J. This gene encodes an enzyme located in the inner mitochondrial membrane that is involved in the biosynthesis of phospholipids. Thus, it is possible that the upregulation of PSDC expression provides a mechanism to compensate for oxidative damage to membranes in the central nervous system [100]. A genome-wide evaluation of the genes differentially expressed between SAMP8 and the other strains revealed 61, 69, and 82% of the genes to be upregulated in the hippocampus of SAMR1, SAMP10, and C57BL/J, respectively, whereas, 46% of genes were upregulated with age in the hippocampus of SAMP8. These same trends were seen in the retina. The SAMP10, SAMR1, and C57BL/J strains showed upregulation of at least 80% of genes. However, only 24% of genes were upegulated in the retina of SAMP8. These results suggest SAMP8 to have an abnormal transcriptional response to the aging process. Thus, it is possible that SAMP10 mice exhibit an acceleration of certain normal molecular responses to aging, while SAMP8 mice exhibit a malfunction of normal transcriptional responses.
In another study, the differential gene expression related to cognitive deterioration with aging between SAMP8 and SAMR1 was profiled [15]. Of 91 differentially expressed genes, 50 were upregulated and 41 were downregulated in the hippocampus of SAMP8 at 12 months of age compared with age-matched SAMR1. Mitochondrial genes such as those for cytochrome c oxidase subunit I (MTCO1), MTCO3, and ubiquinol-cytochrome c reductase subunit (UQCRFS1), the genes involved in the cell skeleton such as those encoding dynein cytoplasmic heavy chain 1, kinectin, and neurofilament protein L, and also tyrosine familiy protein kinases such as Eph receptor B6 and neurotrophic tyrosine kinase receptor type 2 were significantly upregulated in the hippocampus of SAMP8 compared with SAMR1, whereas, CaMKIIA and Map41k6-pending genes involved in memory formation such as LTP and the growth of dendritic spines are downregulated in the hippocampus of SAMP8 compared with SAMR1 [15]. These results indicate the functions of mitochondria and cytoskeleton to be impaired in the brain of SAMP8. This data is consistent with the neurochemical data showing a high redox state and abnormal spine formation in the brain of SAMP8 [8, 38, 57–59]. Taken together, these results suggest that the genes abnormally expressed in SAMP8 are closely associated with the cognitive dysfunction of SAMP8 [15].
Conclusion
SAMP8 is a useful animal model of senile dementia. Neurochemical and morphological data show that the SAMP8 brain partially mimics the pathogenesis of AD. But, the mechanism underlying the development of LMD in SAMP8 is not fully understood. In this review, we discussed the morphological changes of neurons by neurochemical alterations, possible therapeutic interventions, and the heredity of SAMP8. Recent genetic analyses revealed that genes related to mitochondria and the cytoskeleton is abnormally expressed in the brain in SAMP8. This suggests that ROS may be generated early on in life, resulting in insufficient formation of the neural network due to oxidative damage in the brain. However, antioxidants helped to remedy the aging and neuronal damage from oxidative stress in SAMP8. Also, CR may be a good intervention for protecting neurons from oxidative damage. A possible mechanism underlying the development of cognitive dysfunction in SAMP8 is shown in Fig. 3. Abnormal gene expression may trigger the deposition of Aβ, mitochondrial dysfunction, and neurochemical changes. Then, an increase in oxidative stress with age due to Aβ and mitochondrial dysfunction also induces neurochemical changes. These changes may lead to shortcoming in the formation of dendritic spines and neural networks, resulting in cognitive dysfunction in SAMP8.

Scheme of the mechanism underlying the development of learning and memory deficits in SAMP8
References
Hosokawa M (2002) A higher oxidative status accelerates senescence and aggravates age-dependent disorders in SAMP strains of mice. Mech Ageing Dev 123:1553–1561. doi:10.1016/S0047-6374(02)00091-X
Zhao XH, Awaya A, Kobayashi H, Ohnuki T, Tokumitsu Y, Nomura Y (1990) Age-related changes in uptake and release on L-[3H] noradrenaline in brain slices of senescence accelerated mouse. Int J Dev Neurosci 8:267–272. doi:10.1016/0736-5748(90)90032-W
Zhao XH, Kitamura Y, Nomura Y (1992) Age-related changes in NMDA-induced [3H] acetylcholine release from brain slices of senescence accelerated mouse. Int J Dev Neurosci 10:121–129. doi:10.1016/0736-5748(92)90040-7
Kitamura Y, Zhao XH, Ohnuki T, Takei M, Nomura Y (1992) Age-related changes in transmitter glutamate and NMDA receptor/channels in the brain of senescence-accelerated mouse. Neurosci Lett 137:169–172. doi:10.1016/0304-3940(92)90396-O
Nomura Y, Kitamura Y, Ohnuki T, Arima Y, Yamanaka Y, Sasaki K et al (1997) Alterations in acetylcholine, NMDA, benzodiazepine receptors and protein kinase C in the brain of the senescence-accelerated mouse: an animal model useful for studies on cognitive enhances. Behav Brain Res 83:51–55. doi:10.1016/S0166-4328(97)86045-7
Armbrecht HJ, Boltz MA, Kumar VB, Flood JF, Morley JE (1999) Effect of age on calcium-dependent protein in hippocampus of senescence-accelerated mice. Brain Res 842:287–293. doi:10.1016/S0006-8993(99)01802-8
Sugiyama H, Akiyama H, Akiguchi I, Kameyama M, Takeda T (1987) Loss of dendritic spines in hippocampal CA1 pyramidal cells of senescence accelerated mouse (SAM)—A quantitative Golgi study. Clin Neurol 27:841–845
Wei X, Zhang Y, Zhou J (1999) Alzheimer’s disease-related gene expression in the brain of senescence accelerated mouse. Neurosci Lett 268:139–142. doi:10.1016/S0304-3940(99)00396-1
Nomura Y, Yamanaka Y, Kitamura Y, Arima T, Ohnuki T, Oomura Y et al (1996) Senescence-accelerated mouse. Neurochemical studies on aging. Ann N Y Acad Sci 786:410–418. doi:10.1111/j.1749-6632.1996.tb39080.x
Morley JE, Kumar VB, Bernardo AE, Farr SA, Uezu K, Tumosa N et al (2000) β-amyloid precursor polypeptide in SAMP8 mice affects learning and memory. Peptides 21:1761–1767. doi:10.1016/S0196-9781(00)00342-9
Akiyama H, Kameyama M, Akiguchi I, Sugiyama H, Kawamata T, Fukuyama H et al (1986) Periodic acid-Schiff (PAS)-positive, granular structures increase in the brain of senescence accelerated mouse (SAM). Acta Neuropathol 72(2):124–129. doi:10.1007/BF00685973
Yagi H, Irino M, Matsushita T, Katoh S, Umezawa M, Tsuboyama T et al (1989) Spontaneous spongy degeneration of the brain in SAM-P/8 mice, a newly developed memory-deficient strain. J Neuropathol Exp Neurol 48:577–590. doi:10.1097/00005072-198909000-00008
Flood JF, Morley PMK, Morley JE (1995) Age-related changes in learning, memory, and lipofuscin as a function of the percentage of SAMP8 genes. Physiol Behav 58(4):819–822
Tomobe K, Isobe M, Okuma Y, Kitamura K, Oketani Y, Nomura Y (2005) Genetic analysis of learning and memory deficits in senescence-accelerated mouse (SAM). Physiol Behav 84(4):505–510. doi:10.1016/j.physbeh.2004.12.012
Cheng XR, Zhou WX, Zhang YX, Zhou DS, Yang RF, Chen LF (2007) Differential gene expression profiles in the hippocampus of senescence-accelerated mouse. Neurobiol Aging 28(4):497–506. doi:10.1016/j.neurobiolaging.2006.02.004
Miyamoto M, Kiyota Y, Yamazaki N, Nagaoka A, Matsuo T, Nagawa Y et al (1986) Age-related changes in learning and memory in the senescence-accelerated muse (SAM). Physiol Behav 38:399–406. doi:10.1016/0031-9384(86)90112-5
Takemura M, Nakamura S, Akiguchi I, Ueno M, Oka N, Ishikawa S et al (1993) β/A4 proteinlike immunoreactive granular structure in the brain of senescence-accelerated mouse. Am J Pathol 142:1887–1897
Irizarry MC, Soriano F, McNamara M, Page KJ, Schenk D, Games D et al (1997) Abeta deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J Neurosci 17(18):7053–7059
Takeuchi A, Irizarry MC, Duff K, Saido TC, Hsiao Ashe K, Hasegawa M et al (2000) Age-related amyloid beta deposition in transgenic mice overexpressing both Alzheimer mutant presenilin 1 and amyloid beta precursor protein Swedish mutant is not associated with global neuronal loss. Am J Pathol 157(1):331–339
Kumar VB, Farr SA, Flood JF, Kamlesh V, Franko M, Banks WA et al (2000) Site-directed antisense oligonucleotide decreases the expression of amyloid precursor protein and reverses deficits in learning and memory in aged SAMP8 mice. Peptides 21(12):1769–1775. doi:10.1016/S0196-9781(00)00339-9
Poon HF, Joshi G, Sultana R, Farr SA, Banks WA, Morley JE et al (2004) Antisense directed at the Abeta region of APP decreases brain oxidative markers in aged senescence accelerated mice. Brain Res 1018(1):86–96. doi:10.1016/j.brainres.2004.05.048
Wilcock DM, Alamed J, Gottschall PE, Grimm J, Rosenthal A, Pons J et al (2006) Deglycosylated anti-amyloid-beta antibodies eliminate cognitive deficits and reduce parenchymal amyloid with minimal vascular consequences in aged amyloid precursor protein transgenic mice. J Neurosci 26(20):5340–5346. doi:10.1523/JNEUROSCI.0695-06.2006
Banks WA, Farr SA, Morley JE, Wolf KM, Geylis V, Steinitz M (2007) Anti-amyloid beta protein antibody passage across the blood-brain barrier in the SAMP8 mouse model of Alzheimer’s disease: an age-related selective uptake with reversal of learning impairment. Exp Neurol 206(2):248–256. doi:10.1016/j.expneurol.2007.05.005
Canudas AM, Gutierrez-Cuesta J, Rodríguez MI, Acuña-Castroviejo D, Sureda FX, Camins A et al (2005) Hyperphosphorylation of microtubule-associated protein tau in senescence-accelerated mouse (SAM). Mech Ageing Dev 126(12):1300–1304. doi:10.1016/j.mad.2005.07.008
Yamaguchi H, Ishiguro K, Uchida T, Takashima A, Lemere CA, Imahori K (1996) Preferential labeling of Alzheimer neurofibrillary tangles with antisera for tau protein kinase (TPK) I/glycogen synthase kinase-3 beta and cyclin-dependent kinase 5, a component of TPK II. Acta Neuropathol 92(3):232–241
Baum L, Seger R, Woodgett JR, Kawabata S, Maruyama K, Koyama M et al (1995) Overexpressed tau protein in cultured cells is phosphorylated without formation of PHF: implication of phosphoprotein phosphatase involvement. Brain Res Mol Brain Res 34(1):1–17. doi:10.1016/0169-328X(95)00111-5
Pei JJ, Tanaka T, Tung YC, Braak E, Iqbal K, Grundke-Iqbal I (1997) Distribution, levels, and activity of glycogen synthase kinase-3 in the Alzheimer disease brain. J Neuropathol Exp Neurol 56(1):70–78. doi:10.1097/00005072-199701000-00007
Alvarez-García O, Vega-Naredo I, Sierra V, Caballero B, Tomás-Zapico C, Camins A et al (2006) Elevated oxidative stress in the brain of senescence-accelerated mice at 5 months of age. Biogerontology 7(1):43–52. doi:10.1007/s10522-005-6041-2
Frasier M, Walzer M, McCarthy L, Magnuson D, Lee JM, Haas C et al (2005) Tau phosphorylation increases in symptomatic mice overexpressing A30P alpha-synuclein. Exp Neurol 192(2):274–287. doi:10.1016/j.expneurol.2004.07.016
Shendelman S, Jonason A, Martinat C, Leete T, Abeliovich A (2004) DJ-1 is a redox-dependent molecular chaperone that inhibits alpha-synuclein aggregate formation. PLoS Biol 2(11):e362. doi:10.1371/journal.pbio.0020362
Bito H, Deisseroth K, Tsien RW (1996) CREB phosphorylation and dephosphorylation: a Ca2+- and stimulus duration-dependent switch for hippocampal gene expression. Cell 87:1203–1214. doi:10.1016/S0092-8674(00)81816-4
Ho N, Liauw JA, Blaeser F, Wei F, Hanissian S, Muglia LM et al (2000) Impaired synaptic plasticity and cAMP response element-binding protein activation in Ca2+/calmodulin-dependent protein kinase type IV/Gr-deficient mice. J Neurosci 20:6459–6472
Walton M, Henderson C, Mason-Parker S, Lawlor P, Abraham WC, Bilkey D et al (1999) Immediate early gene transcription and synaptic modulation. J Neurosci Res 58:96–106. doi:10.1002/(SICI)1097-4547(19991001)58:1<96::AID-JNR10>3.0.CO;2-N
Xie H, Rothstein TL (1995) Protein kinase C mediates activation of nuclear cAMP response element-binding protein (CREB) in B lymphocytes stimulated through surface Ig. J Immunol 154:1717–1723
Impey S, Mark M, Poser S, Chavkin C, Storm DR (1996) Induction of CRE-mediated gene expression by stimuli that generate long-lasting LTP in area CA1 of the hippocampus. Neuron 16:973–982. doi:10.1016/S0896-6273(00)80120-8
Luscher C, Nicoll RA, Malenka RC, Muller D (2000) Synaptic plasticity and dynamic modulation of the postsynaptic membrane. Nat Neurosci 3:545–550. doi:10.1038/75714
Toni N, Buchs PA, Nikonenko I, Bron CR, Muller D (1999) LTP promotes formation of multiple spine synapses between a single axon terminal and a dendrite. Nature 402:421–425. doi:10.1038/46574
Tomobe K, Okuma Y, Nomura Y (2007) Impairment of CREB phosphorylation in the hippocampal CA1 region of the senescence-accelerated mouse (SAM) P8. Brain Res 1141:214–217. doi:10.1016/j.brainres.2006.08.026
Bernabeu R, Bevilaqua L, Ardenghi P, Bromberg E, Schmitz P, Bianchin M et al (1997) Involvement of hippocampal cAMP/cAMP-dependent protein kinase signaling pathways in a late memory consolidation phase of aversively motivated learning in rats. Proc Natl Acad Sci USA 94:7041–7046. doi:10.1073/pnas.94.13.7041
Bilang-Bleuel A, De Carli RS, Holsboer F, Reul HM (2002) Forced swimming evokes a biphasic response in CREB phosphorylation in extrahypothalamic limbic and neocortical brain structure in the rat. Eur J Neurosci 15:1048–1060. doi:10.1046/j.1460-9568.2002.01934.x
Schulz S, Siemer H, Krug M, Höllt V (1999) Direct evidence for biphasic cAMP responsive element-binding protein phosphorylation during long-term potentiation in the rat dentate gyrus in vivo. J Neurosci 19:5683–5692
Silva AJ, Stevens CF, Tonegawa S, Wang Y (1992) Deficient hippocampal long-term potentiation in a-calcium-calmodulin kinase II mutant mice. Science 257:201–206. doi:10.1126/science.1378648
Abeliovich A, Chen C, Goda Y, Silva AJ, Stevens CF, Tonegawa S (1993) Modified hippocampal long-term potentiation in PKC gamma-mutant mice. Cell 75:1253–1262. doi:10.1016/0092-8674(93)90613-U
Sakimura K, Kutsuwada T, Ito I, Manabe T, Takayama C, Kushiya E et al (1995) Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor 1 subunit. Nature 373:151–155. doi:10.1038/373151a0
Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva A (1994) Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell 79:59–68. doi:10.1016/0092-8674(94)90400-6
Leranth C, Shanabrough M, Horvath TL (2000) Hormonal regulation of hippocampal spine synapse density involves subcortical mediation. Neuroscience 101(2):349–356. doi:10.1016/S0306-4522(00)00369-9
Woolley CS, Weiland NG, McEwen BS, Schwartzkroin PA (1997) Estradiol increases the sensitivity of hippocampal CA1 pyramidal cells to NMDA receptor-mediated synaptic input: correlation with dendritic spine density. J Neurosci 17(5):1848–1859
Weiland NG, Orikasa C, Hayashi S, McEwen BS (1997) Distribution and hormone regulation of estrogen receptor immunoreactive cells in the hippocampus of male and female rats. J Comp Neurol 388(4):603–612. doi:10.1002/(SICI)1096-9861(19971201)388:4<603::AID-CNE8>3.0.CO;2-6
Shors TJ, Chua C, Falduto J (2001) Sex differences and opposite effects of stress on dendritic spine density in the male versus female hippocampus. J Neurosci 21(16):6292–6297
Prange-Kiel J, Wehrenberg U, Jarry H, Rune GM (2003) Para/autocrine regulation of estrogen receptors in hippocampal neurons. Hippocampus 13(2):226–234. doi:10.1002/hipo.10075
Shughrue PJ, Lane MV, Merchenthaler I (1997) Comparative distribution of estrogen receptor-α and -β mRNA in the rat central nervous system. J Comp Neurol 388:507–525. doi:10.1002/(SICI)1096-9861(19971201)388:4<507::AID-CNE1>3.0.CO;2-6
Nishio M, Kuroki Y, Watanabe Y (2004) Subcellular localization of estrogen receptor β in mouse hippocampus. Neurosci Lett 355:109–112. doi:10.1016/j.neulet.2003.10.064
Krezel W, Dupont S, Krust A, Chambon P, Chapman PF (2001) Increased anxiety and synaptic plasticity in estrogen receptor beta -deficient mice. Proc Natl Acad Sci USA 98(21):12278–12282. doi:10.1073/pnas.221451898
Rssman EF, Heck AL, Leonard JE, Shupnik MA, Gustafsson JA (2002) Disruption of estrogen receptor beta gene impairs spatial learning in female mice. Proc Natl Acad Sci USA 99(6):3996–4001. doi:10.1073/pnas.012032699
Ábrahám IM, Han S-K, Todman MG, Korach KS, Herbison AE (2003) Estrogen receptor β meditates rapid estrogen actions on gonadotropin-releasing hormone neurons in vivo. J Neurosci 23:5771–5777
Zhou W, An S, Fu Y, Zhang Y (2004) Age-related changes of the hippocampal estrogen receptor gene expression in senescence-accelerated mouse. In: Nomura Y, Takeda T, Okuma Y (eds) The senescence-accelerated mouse (SAM): an animal model of senescence. Elsevier, Amsterdam, pp 237–242
Nomura Y, Wang BX, Qi SB, Namba T, Kaneko S (1989) Biochemical changes related to aging in the senescence-accelerated mouse. Exp Gerontol 24:49–55. doi:10.1016/0531-5565(89)90034-X
Butterfield DA, Howard BJ, Yatin S, Allen KL, Carney JM (1997) Free radical oxidation of brain proteins in accelerated senescence and its modulation by N-tert-butyl-alpha-phenylnitrone. Proc Natl Acad Sci USA 94(2):674–678. doi:10.1073/pnas.94.2.674
Okatani Y, Wakatsuki A, Reiter RJ, Miyahara Y (2002) Melatonin reduces oxidative damage of neural lipids and proteins in senescence-accelerated mouse. Neurobiol Aging 23:639–644. doi:10.1016/S0197-4580(02)00005-2
Farr SA, Poon HF, Dogrukol-Ak D, Drake J, Banks WA, Eyerman E et al (2003) The antioxidants alpha-lipoic acid and N-acetylcysteine reverse memory impairment and brain oxidative stress in aged SAMP8 mice. J Neurochem 84(5):1173–1183. doi:10.1046/j.1471-4159.2003.01580.x
Yasui F, Ishibashi M, Matsugo S, Kojo S, Oomura Y, Sasaki K (2003) Brain lipid hydroperoxide level increases in senescence-accelerated mice at an early age. Neurosci Lett 350:66–68. doi:10.1016/S0304-3940(03)00827-9
Sato E, Kurokawa T, Oda N, Ishibashi S (1996) Early appearance of abnormality of microperoxisomal enzymes in the cerebral cortex of senescence-accelerated mouse. Mech Ageing Dev 92:175–184. doi:10.1016/S0047-6374(96)01832-5
Kurokawa T, Asada S, Nishitani S, Hazeki O (2001) Age-related changes in manganese superoxide dismutase activity in the cerebral cortex of senescence-accelerated prone and resistant mouse. Neurosci Lett 298:135–138. doi:10.1016/S0304-3940(00)01755-9
Nakahara H, Kanno T, Inai Y, Utsumi K, Hiramatsu M, Mori A et al (1998) Mitochondrial dysfunction in the senescence accelerated mouse (SAM). Free Radic Biol Med 24(1):85–92. doi:10.1016/S0891-5849(97)00164-0
Nishikawa T, Takahashi JA, Fujibayashi Y, Fujisawa H, Zhu B, Nishimura Y et al (1998) An early stage mechanism of the age-associated mitochondrial dysfunction in the brain of SAMP8 mice; an age-associated neurodegeneration animal model. Neurosci Lett 254(2):69–72. doi:10.1016/S0304-3940(98)00646-6
Xu J, Shi C, Li Q, Wu J, Forster EL, Yew DT (2007) Mitochondrial dysfunction in platelets and hippocampi of senescence-accelerated mice. J Bioenerg Biomembr 39(2):195–202. doi:10.1007/s10863-007-9077-y
Fujibayashi Y, Yamamoto S, Waki A, Konishi J, Yonekura Y (1998) Increased mitochondrial DNA deletion in the brain of SAMP8, a mouse model for spontaneous oxidative stress brain. Neurosci Lett 254:109–112. doi:10.1016/S0304-3940(98)00667-3
Butterfield DA, Koppal T, Howard B, Subramaniam R, Hall N, Hensley K et al (1998) Structural and functional changes in proteins induced by free radical-mediated oxidative stress and protective action of the antioxidants N-tert-butyl-alpha-phenylnitrone and vitamin E. Ann N Y Acad Sci 854:448–462. doi:10.1111/j.1749-6632.1998.tb09924.x
Edamatsu R, Mori A, Packer L (1995) The spin-trap N-tert-alpha-phenyl-butylnitrone prolongs the life span of the senescence accelerated mouse. Biochem Biophys Res Commun 211(3):847–849. doi:10.1006/bbrc.1995.1889
Forster MJ, Dubey A, Dawson KM, Stutts WA, Lal H, Sohal RS (1996) Age-related losses of cognitive function and motor skills in mice are associated with oxidative damage in the brain. Proc Natl Acad Sci USA 93:4765–4769. doi:10.1073/pnas.93.10.4765
Martinez M, Hernandez AI, Martinez N (2000) N-acetylcysteine delays age-associated memory impairment in mice: role in synaptic mitochondoria. Brain Res 855:100–106. doi:10.1016/S0006-8993(99)02349-5
Floyd RA (1991) Oxidative damage to behavior during aging. Science 254:1597. doi:10.1126/science.1684251
Smith CD, Carney JM, Starke-Reed PE, Oliver CN, Standtman ER, Floyd RA et al (1991) Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer’s disease. Proc Natl Acad Sci USA 88:10540–10543. doi:10.1073/pnas.88.23.10540
Nishimura H, Higuchi O, Tateshita K, Tomobe K, Okuma Y, Nomura Y (2006) Antioxidative activity and ameliorative effects of memory impairment of sulfur-containing compounds in Allium species. Biofactors 26(2):135–146
Bishop NA, Guarente L (2007) Two neurons mediate diet-restriction-induced longevity in C. elegans. Nature 447(7144):545–549. doi:10.1038/nature05904
Panowski SH, Wolff S, Aguilaniu H, Durieux J, Dillin A (2007) PHA-4/Foxa mediates diet-restriction-induced longevity of C. elegans. Nature 447(7144):550–555. doi:10.1038/nature05837
Pugh TD, Oberley TD, Weindruch R (1999) Dietary intervention at middle age: caloric restriction but not dehydroepiandrosterone sulfate increases lifespan and lifetime cancer incidence in mice. Cancer Res 59(7):1642–1648
Fernandes G, Yunis EJ, Good RA (1976) Influence of diet on survival of mice. Proc Natl Acad Sci USA 73(4):1279–1283. doi:10.1073/pnas.73.4.1279
Stern JS, Gades MD, Wheeldon CM, Borchers AT (2001) Calorie restriction in obesity: prevention of kidney disease in rodents. J Nutr 131(3):913S–917S
Jiang T, Liebman SE, Lucia MS, Phillips CL, Levi M (2005) Caloric restriction modulates renal expression of sterol regulatory element binding proteins, lipid accumulation, and age-related renal disease. J Am Soc Nephrol 16(8):2385–2394. doi:10.1681/ASN.2004080701
Varady KA, Hellerstein MK (2007) Alternate-day fasting and chronic disease prevention: a review of human and animal trials. Am J Clin Nutr 86(1):7–13
Heilbronn LK, Ravussin E (2003) Calorie restriction and aging: review of the literature and implications for studies in humans. Am J Clin Nutr 78(3):361–369
Wang J, Ho L, Qin W, Rocher AB, Seror I, Humala N et al (2005) Caloric restriction attenuates β-amyloid neuropathology in a mouse model of Alzheimer’s disease. FASEB J 19(6):659–661
Merry BJ (2004) Oxidative stress and mitochondrial function with aging—the effects of calorie restriction. Aging Cell 3(1):7–12. doi:10.1046/j.1474-9728.2003.00074.x
Gredilla R, Barja G, López-Torres M (2001) Effect of short-term caloric restriction on H2O2 production and oxidative DNA damage in rat liver mitochondria and location of the free radical source. J Bioenerg Biomembr 33(4):279–287. doi:10.1023/A:1010603206190
Gredilla R, Sanz A, Lopez-Torres M, Barja G (2001) Caloric restriction decreases mitochondrial free radical generation at complex I and lowers oxidative damage to mitochondrial DNA in the rat heart. FASEB J 15(9):1589–1591
Lambert AJ, Merry BJ (2004) Effect of caloric restriction on mitochondrial reactive oxygen species production and bioenergetics: reversal by insulin. Am J Physiol Regul Integr Comp Physiol 286(1):R71–R79. doi:10.1152/ajpregu.00341.2003
Lopez-Torres M, Gredilla R, Sanz A, Barja G (2002) Influence of aging and long-term caloric restriction on oxygen radical generation and oxidative DNA damage in rat liver mitochondria. Free Radic Biol Med 32:882–889. doi:10.1016/S0891-5849(02)00773-6
Pamplona R, Portero-Otín M, Requena J, Gredilla R, Barja G (2002) Oxidative, glycoxidative and lipoxidative damage to rat heart mitochondrial proteins is lower after 4 months of caloric restriction than in age-matched controls. Mech Ageing Dev 123(11):1437–1446. doi:10.1016/S0047-6374(02)00076-3
Sanz A, Caro P, Ibañez J, Gómez J, Gredilla R, Barja G (2005) Dietary restriction at old age lowers mitochondrial oxygen radical production and leak at complex I and oxidative DNA damage in rat brain. J Bioenerg Biomembr 37(2):83–90. doi:10.1007/s10863-005-4131-0
Stuart JA, Karahalil B, Hogue BA, Souza-Pinto NC, Bohr VA (2004) Mitochondrial and nuclear DNA base excision repair are affected differently by caloric restriction. FASEB J 18(3):595–597
Hyun DH, Emerson SS, Jo DG, Mattson MP, de Cabo R (2006) Calorie restriction up-regulates the plasma membrane redox system in brain cells and suppresses oxidative stress during aging. Proc Natl Acad Sci USA 103(52):19908–19912. doi:10.1073/pnas.0608008103
Choi JH, Kim D (2000) Effects of age and dietary restriction on lifespan and oxidative stress of SAMP8 mice with learning and memory impairments. J Nutr Health Aging 4(3):182–186
Kim DW, Choi JH (2000) Effects of age and dietary restriction on animal model SAMP8 mice with learning and memory impairments. J Nutr Health Aging 4(4):233–238
Higuchi K (1997) Genetic characterization of senescence-accelerated mouse (SAM). Exp Gerontol 32:129–138. doi:10.1016/S0531-5565(96)00060-5
Kitado H, Higuchi K, Takeda T (1994) Molecular genetic characterization of the senescence-accelerated mouse (SAM) strains. J Gerontol 49:B247–B254
Xia C, Higuchi K, Shimizu M, Matsushita T, Kogishi K, Wang J et al (1999) Genetic typing of the senescence-accelerated mouse (SAM) strains with microsatellite markers. Mamm Genome 10(3):235–238. doi:10.1007/s003359900979
Isobe M, Tomobe K, Sawada M, Kondo A, Kurokawa N, Nomura Y (2004) Quantitative trait loci for age-related memory dysfunction in SAMP8 and JF1 mice. In: Nomura Y, Takeda T, Okuma Y (eds) The senescence-accelerated mouse (SAM): an animal model of senescence. Elsevier, Amsterdam, pp 29–34
Carter TA, Greenhall JA, Yoshida S, Fuchs S, Helton R, Swaroop A et al (2005) Mechanisms of aging in senescence-accelerated mice. Genome Biol 6(6):R48. doi:10.1186/gb-2005-6-6-r48
Salvador GA, López FM, Giusto NM (2002) Age-related changes in central nervous system phosphatidylserine decarboxylase activity. J Neurosci Res 70(3):283–289. doi:10.1002/jnr.10385
Author information
Authors and Affiliations
Corresponding author
Additional information
Special issue article in Honour of Dr. Akitane Mori.
Rights and permissions
About this article
Cite this article
Tomobe, K., Nomura, Y. Neurochemistry, Neuropathology, and Heredity in SAMP8: A Mouse Model of Senescence. Neurochem Res 34, 660–669 (2009). https://doi.org/10.1007/s11064-009-9923-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-009-9923-x