
Abstract
Transient receptor potential V1 (TRPV1) is specifically expressed in the nociceptive receptors and can detect a variety of noxious stimuli, thus potentiating pain sensitization. While peripheral delivery of capsaicin causes the desensitization of sensory neurons, thus alleviating pain. Therefore capsaicin is used in the clinical treatment of various types of pain; however, these treatments will bring many side effects, such as a strong burning pain in the early stages of treatment which hampers the further use of capsaicin. Thus, the studies of the functional regulation of TRPV1 are mainly focused on two aspects: to develop more potent analogues of capsaicin with less side effects; or to elucidate the mechanisms of TRPV1 in pain sensitivity, especially of that TRPV1 as a target of various protein kinases such as PKD1 and Cdk5 is involved pain hypersensitivity. Thus we would summarize the progress of these two aspects in this mini review.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pain is not only one of the most common signs in the clinic, the chronic pain is also considered to be a type of mental disease. The lack of adequate and effective treatment makes the chronic pain as a serious problem affecting the daily work and life of the people. Therefore, exploring the intrinsic molecular mechanism of chronic pain to provide new targets for analgesic pharmacology is becoming the key step in the clinical treatment for pain. It is also important to study how to improve the effects of the existing analgesics and how to reduce the side effects of these drugs such as the burning pain caused by capsaicin and the tolerance of morphine.
Pivotal Role of TRPV1 in Pain and Pain Modulation
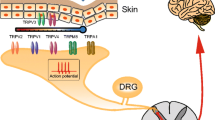
Among the mechanisms of pain, the most important mechanism is thought to be peripheral (dorsal root ganglion, DRG) and central (spinal dorsal horn) sensitization. Many factors contribute to the development and maintenance of pain sensitivity, such as neurotrophic factors, protein kinases and ion channels; these factors ultimately lead to the strengthening of synaptic transmissions and the development of sensitization [17, 45]. The nociceptive sensors in DRG neurons are the first station in the transmission of pain; therefore, the DRG has become an important target for pain treatment. At the same time, the specific nociceptive receptors in DRG have become the focus of pain research.
The transient receptor potential V1 (TRPV1) channel is one of the nociceptive receptors in DRG neurons. TRPV1, also known as vanilloid receptor 1 or capsaicin receptor was cloned in 1997 [4], and it is a member of a large family of calcium-permeable nonselective cation channels which are gated by heat, low pH, or endogenous ligands termed “endovanilloids’’ including anandamide, lipoxygenase derivatives of arachidonic acid, and long-chain, linear fatty acid dopamines such as N-arachidonyldopamine [4–6, 12, 24, 31, 38, 50]. This channel is specifically expressed in the nociceptive receptors which can detect a variety of noxious stimuli such as thermal, chemical stimuli like capsaicin and resiniferatoxin (RTX) causing pain, inflammation and hyperalgesia [11]. Studies with TRPV1-deficient mice demonstrate that the TRPV1 channel is essential for selective modalities of thermal hyperalgesia induced by tissue injury and inflammation, supporting the hypothesis that TRPV1 is a molecular integrator of painful stimuli [4]. On the other hand, peripheral delivery of capsaicin causes the desensitization of sensory neurons with C fibers and Aδ fibers, thus alleviating pain. Based on this characteristic, capsaicin is used in the clinical treatment of various types of pain, including diabetes mellitus neuropathic pain, joint inflammatory pain and post-herpetic neuropathic pain [29, 30]. However, these treatments bring many side effects, such as a strong burning pain in the early stages of treatment that can prevent the further use of capsaicin. Therefore, understanding the mechanism of the functional regulation of TRPV1, or developing potent capsaicin analogues with minor side effects are two major focuses in pain research.
TRPV1 Acts as a Target for a Variety of Protein Kinases and Plays a Pivotal Role in Pain Sensitization
Pain sensitization can occur on two levels, that is, post-transcriptional modification and changes in the level of transcription, which mediate early and long-term pain sensitization, respectively [17, 45]. To date, however, there is no factor that can fully explain the development of pain sensitivity, which undoubtedly has more complex molecular mechanisms. Among these mechanisms, the most attractive is the signal transmission network formed by the close link and the interaction among the intracellular signal molecules. In this network, both the post-transcriptional modification and the changes in the level of transcription act through second messengers and specific protein kinases. At present, the involvement of protein kinases in pain and in pain modulation is relatively clear. It is well established that the ERK protein kinase participates in both the peripheral and central mechanisms of pain sensitization, and other protein kinases such PKA, PKC, CaMK and PI3K are also involved in the regulation of pain [14, 16]. For example, a recent study indicated that peripheral nerve injury could increase the phosphorylation of ERK, p38 and JNK in DRG neurons. The phosphorylation of p38 in DRG neurons was upregulated for as long as three weeks after spinal nerve ligation. Nerve injury can cause the activation of MAPK kinases in non-DRG neurons such as satellite cells, and stimulate the synthesis of inflammatory mediators. Inhibitors of ERK, JNK and p38 can alleviate neuropathic pain [14]. The latest research showed that ERK activation in the amygdala could enhance peripheral sensitization caused by inflammation [3], indicating that inflammatory injury could activate ERK signaling not only in the peripheral nociceptive receptors, but also in the central nervous system, which further strengthened the hyperalgesia.
We mentioned above that TRPV1 plays an important role in the transmission of pain [4, 5, 38]. At the early stages of inflammation and injury, locally-released inflammatory mediators, such as prostaglandin (mainly prostaglandin-2), bradykinin, substance P, NGF and protease, are the primary contributors to pain sensitization. These inflammatory mediators cannot activate pain receptors directly but can lower the nociceptive receptor threshold. They often activate protein kinases such as ERK, p38 and PKC through the corresponding receptors. Activated protein kinases can phosphorylate TRPV1, leading to rapid and dynamic changes in pain sensitivity [1, 15, 25, 34, 47, 49]. Our pharmacological study indicates an interaction between a novel protein kinase, PKD1 and TRPV1. PKD1 was successfully cloned by two laboratories from the United Kingdom and Germany in 1994. The human homologue is protein kinase C μ (protein kinase C μ, PKC μ), and the mouse homologue is called PKD1. This protein was first considered to be a new member in protein kinase C family and was classified as an atypical PKC subfamily [18, 40]. However, the enzymatic properties and molecular structure of PKD1 are very different from that of PKC. Based on this, PKD1 is now classified to a new protein kinase family that is distinct from PKC.
The protein kinase D family has a variety of important biological functions such as regulation of nuclear transcription factor NFκ B-mediated gene expression [32, 33], participation in the Na+/H+ transport [9], the promotion of Golgi vesicle transport and release [8, 13], and Golgi-to-membrane trafficking [23]. To date, however, almost of the research on PKD is concentrated on non-neural cells, and most of these studies are based on the overexpression of exogenous PKD. Very little is known about the function of PKD in the nervous system.
We first proved that PKD1 can directly phosphorylate rat TRPV1 (rTRPV1) at Serine 116 which is located in the N-terminal of rTRPV1 in vitro, and we identified that PKD1 can bind to the N-terminal but not C-terminal of rTRPV1. Furthermore, mutation of Ser116A in rTRPV1 both blocked the phosphorylation of rTRPV1 by PKD1 as well as the enhancement by PKD1 of the rTRPV1 response to capsaicin. We conclude that PKD1 functions as a direct modulator of rTRPV1 [42]. Next, we detected an interaction between PKD1 and TRPV1 in animal model of inflammatory hyperalgesia caused by CFA. Through the study of PKD1 phosphorylation and the use of intrathecal gene delivery and electrophysiological techniques, we found that PKD1 mediated the effect of heat hyperalgesia but not of mechanic hyperalgesia. We concluded that PKD1 in DRG, through interaction with TRPV1, is involved in the developing and maintaining inflammatory heat hypersensitivity [48]. The phosphorylation site identified in TRPV1 and the involvement of PKD1 in inflammatory hyperalgesia will provide a new target for the design of novel analgesics thus having great theoretical and practical significance.
In addition, we confirmed cell cycle-dependent protein kinase 5 (Cdk5), involved in pain hypersensitivity. Cdk5 was cloned in 1992 by several laboratories [10, 21]. The studies of Cdk5 for two decades indicate that Cdk5 is a unique member which is functionally different from the other members of the Cdk family. Cdk5 is widely distributed in the nervous system with high kinase activity which participates in a variety of the functions of the nervous system, including neuronal migration during development, regulation of cytoskeletal dynamics and changes in synaptic plasticity [7, 22]. Similar to other Cdks [26], Cdk5 activity requires the presence of its activators, p35 and p39 [20, 37, 39] or their truncated forms, p25 and p30 [19, 20, 37].
The Pareek group and us proved almost simultaneously that Cdk5 activation was involved in [7] pain transmission and heat hyperalgesia [28, 46]. However, there are discrepancies of the results between these two groups. First, we used different animal models: Pareek group indicated that Cdk5 participated in carrageenan-induced acute pain and pain transmission, while our group showed that Cdk5 was involved in CFA-induced chronic pain and peripheral and central pain sensitization. Second, the results from Pareek group were mainly based on the p35 knock-in and knockout mice, whereas the conclusions from our group were acquired largely through intrathecal gene delivery. Since p35 was also distributed in motor neurons with abundant content, thus gene delivery into the spinal cord may be more specific toward elucidating the role of Cdk5 in pain modulation. Third, the results from Pareek group showed that the upregulation of Cdk5 activity in DRG and in the spinal cord was due to the increased levels of p25. However, we found that p25 was not detected in the DRG and that the increase in Cdk5 activity in DRG was due to the upregulation of p35, but in the dorsal horn, the increase in Cdk5 activity was due to the increased levels of p25. Lastly, they found that Cdk5 was involved in the basic state of pain conduction, but we found that Cdk5 did not affect the pain threshold under basic conditions. In summary, the results of these two groups are complementary which illustrate the participation of Cdk5 in pain transmission, providing a new target for the treatment of pain.
The further studies indicate that one of the mechanisms underlying the role of Cdk5 in pain sensitization is through direct phosphorylation of TRPV1. The evidences are as follows: First, Cdk5 colocalizes with TRPV1 in DRG neurons. Second, inhibition of Cdk5 activity attenuates TRPV1 mediated calcium influx in cultured DRG neurons. Third, Cdk5 directly phosphorylates TRPV1 at Threonine 407. Fourth, conditional depletion of Cdk5 in C-fiber specific neurons abrogated phosphorylation of TRPV1 at Threonine 407 and induced hypoalgesia [27, 28]. In our ongoing studies, we also found that Cdk5 could regulate the membrane trafficking of TRPV1; this observation needs to be further investigated.
Development of the Agonist and Antagonist of TRPV1
The Blumberg laboratory at the NIH found a potent capsaicin analogue, RTX. Compared with capsaicin, RTX causes a stronger desensitization of pain sensory neurons but only caused a moderate degree of burning pain, thus this compound is more suitable for clinical use [35]. In addition to using capsaicin and its analogues to desensitize pain sensory neurons, capsaicin receptor antagonists also can be used as a supplementary treatment. As mentioned above, capsaicin first activates the TRPV1 in C fiber sensory neurons and then causes receptor desensitization. Receptor desensitization is a lasting process, and the early and brief activation of the receptor causes burning pain. Thus, if a capsaicin receptor antagonist is combined with capsaicin, we may be able to alleviate the sensation of burning pain by blocking the early activation of TRPV1 but still retain the desensitized effect of capsaicin. Unfortunately, at present, the only commonly-used TRPV1 antagonist is capsazepine. Capsazepine is a TRPV1 antagonist with moderate activity, but to some extent, capsazepine also acts in other channels including voltage-dependent calcium channels and the ATP receptor [2, 36]. Therefore, it is necessary to develop more potent and more specific TRPV1 antagonists. Several laboratories have initiated the development of new TRPV1 antagonists. The Thomsen lab has identified a highly efficient TRPV1 antagonist, 5-iodo-RTX. This TRPV1 antagonist is 40 times potent than capsazepine [41] and can block the burning pain caused by capsaicin in animal experiments. However, 5-iodo-RTX is a non-competitive antagonist of TRPV1, and it may weaken the analgesic effect of capsaicin; this may limit clinical utility.
RTX, which was designed by changing a few chemical radicals in the trunk structure of capsaicin, can desensitize pain sensory neurons more effectively than capsaicin while reducing the burning sensation [35]. This observation suggests that different chemical radicals in capsaicin may contribute to the distinctive biological effects. Based on this principle, we analyzed the structure of capsaicin and capsazepine and changed certain chemical radicals, thereby designing and synthesizing a series of analogues of capsaicin and capsazepine. We then identified two specific and efficient TRPV1 antagonists through pharmacological and cell biological experiments. These newly-discoveredTRPV1 antagonists have the following advantages: (1) potent antagonism, 25 and 60 times potent as compared to 5-iodo-RTX and capsazepine, respectively; (2) high specificity, including no activation of the ATP receptor; (3) effective inhibition of TRPV1 that includes blocking the activation of TRPV1 by capsaicin, H+ and temperature; (4) being competitive antagonists. Our results further indicate that we can change the pharmacological properties of capsaicin and its analogues by varying certain chemical radicals in their structures. Therefore, we can acquire capsaicin analogs, which provide hints for the development of new capsaicin analogs with efficient analgesia and minor side effects, thereby expanding the options for the treatment of pain [43].
During the search for new types of capsaicin analogues, we found that the same capsaicin analogue can present distinctive pharmacological properties under different physiological and pathological conditions. The partial agonists had some properties of weak antagonists and thus could be mistakenly considered to be antagonists under certain conditions. Furthermore, these partial agonists could be converted to full agonists depending on the pH, temperature, TRPV1 receptor density and the degree of PKC activation. Discovery of this phenomenon has two implications: it reveals the complexity of the role of capsaicin analogs and exerts a warning for the future screening of TRPV1 antagonists with respect to the importance of the strict control of drug screening conditions; and these findings suggest that partial agonists may also have potential clinical applications. The effects of weak partial agonists might be greatly enhanced under inflammatory or other pathological conditions. Since the weak effect of partial agonists remains unchanged in non-pathological tissues, they may reduce the systemic side effects of capsaicin. In summary, this study explored a new area for the synthesis of novel TRPV1 agonists and provided new evidence for the clinical treatment of a number of diseases [44].
In summary, TRPV1 is a multi-signal detector and integrates a variety of pain stimuli. It is necessary to investigate how TRPV1 can detect these different stimuli as a multi-signal pain receptor in physiological and pathological conditions. It is also important to elucidate how the structure and function of TRPV1 vary with changes in the external environment. All of these results will help us to understand the transfer and integration of pain signaling by DRG and the dorsal spinal horn, and to find new analgesic drugs with strong positive effects and minor side effects. At present, little is known about the molecular mechanisms that underlie the involvement of TRPV1 in pain sensitization, although numerous studies have focused on structural and functional analysis of TRPV1. In addition, the mechanisms of TRPV1 desensitization are far from being elucidated. Thus, the structure and function of TRPV1, as well as its role in pain sensitivity, are still an important area in pain research.
References
Amadesi S, Cottrell GS, Divino L, Chapman K, Grady EF, Bautista F, Karanjia R, Barajas-Lopez C, Vanner S, Vergnolle N, Bunnett NW (2006) Protease-activated receptor 2 sensitizes TRPV1 by protein kinase Cepsilon- and A-dependent mechanisms in rats and mice. J Physiol 575:555–571
Bevan S, Szolcsanyi J (1990) Sensory neuron-specific actions of capsaicin: mechanisms and applications. Trends Pharmacol Sci 11:330–333
Carrasquillo Y, Gereau RW (2007) Activation of the extracellular signal-regulated kinase in the amygdala modulates pain perception. J Neurosci 27:1543–1551
Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D (1997) The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389:816–824
Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, Harries MH, Latcham J, Clapham C, Atkinson K, Hughes SA, Rance K, Grau E, Harper AJ, Pugh PL, Rogers DC, Bingham S, Randall A, Sheardown SA (2000) Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature 405:183–187
De Petrocellis L, Di Marzo V (2005) Lipids as regulators of the activity of transient receptor potential type V1 (TRPV1) channels. Life Sci 77:1651–1666
Dhavan R, Tsai LH (2001) A decade of CDK5. Nat Rev Mol Cell Biol 2:749–759
Diaz Anel AM, Malhotra V (2005) PKCeta is required for beta1gamma2/beta3gamma2- and PKD-mediated transport to the cell surface and the organization of the Golgi apparatus. J Cell Biol 169:83–91
Haworth RS, Sinnett-Smith J, Rozengurt E, Avkiran M (1999) Protein kinase D inhibits plasma membrane Na(+)/H(+) exchanger activity. Am J Physiol 277:C1202–C1209
Hellmich MR, Pant HC, Wada E, Battey JF (1992) Neuronal cdc2-like kinase: a cdc2-related protein kinase with predominantly neuronal expression. Proc Natl Acad Sci USA 89:10867–10871
Holzer P (1988) Local effector functions of capsaicin-sensitive sensory nerve endings: involvement of tachykinins, calcitonin gene-related peptide and other neuropeptides. Neuroscience 24:739–768
Hwang SW, Cho H, Kwak J, Lee SY, Kang CJ, Jung J, Cho S, Min KH, Suh YG, Kim D, Oh U (2000) Direct activation of capsaicin receptors by products of lipoxygenases: endogenous capsaicin-like substances. Proc Natl Acad Sci USA 97:6155–6160
Jamora C, Yamanouye N, Van Lint J, Laudenslager J, Vandenheede JR, Faulkner DJ, Malhotra V (1999) Gbetagamma-mediated regulation of Golgi organization is through the direct activation of protein kinase D. Cell 98:59–68
Ji RR, Kawasaki Y, Zhuang ZY, Wen YR, Zhang YQ (2007) Protein kinases as potential targets for the treatment of pathological pain. Handb Exp Pharmacol 359–389
Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ (2002) p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron 36:57–68
Ji RR, Strichartz G (2004) Cell signaling and the genesis of neuropathic pain. Sci STKE 2004:reE14
Ji RR, Woolf CJ (2001) Neuronal plasticity and signal transduction in nociceptive neurons: implications for the initiation and maintenance of pathological pain. Neurobiol Dis 8:1–10
Johannes FJ, Prestle J, Eis S, Oberhagemann P, Pfizenmaier K (1994) PKCu is a novel, atypical member of the protein kinase C family. J Biol Chem 269:6140–6148
Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH (2000) Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 405:360–364
Lew J, Huang QQ, Qi Z, Winkfein RJ, Aebersold R, Hunt T, Wang JH (1994) A brain-specific activator of cyclin-dependent kinase 5. Nature 371:423–426
Lew J, Winkfein RJ, Paudel HK, Wang JH (1992) Brain proline-directed protein kinase is a neurofilament kinase which displays high sequence homology to p34cdc2. J Biol Chem 267:25922–25926
Li BS, Sun MK, Zhang L, Takahashi S, Ma W, Vinade L, Kulkarni AB, Brady RO, Pant HC (2001) Regulation of NMDA receptors by cyclin-dependent kinase-5. Proc Natl Acad Sci USA 98:12742–12747
Liljedahl M, Maeda Y, Colanzi A, Ayala I, Van Lint J, Malhotra V (2001) Protein kinase D regulates the fission of cell surface destined transport carriers from the trans-Golgi network. Cell 104:409–420
Matta JA, Miyares RL, Ahern GP (2007) TRPV1 is a novel target for omega-3 polyunsaturated fatty acids. J Physiol 57:397–411
Morenilla-Palao C, Planells-Cases R, Garcia-Sanz N, Ferrer-Montiel A (2004) Regulated exocytosis contributes to protein kinase C potentiation of vanilloid receptor activity. J Biol Chem 279:25665–25672
Nurse P (1990) Univerasl control mechanism regultion onset of M-phase. Nature 344:503–508
Pareek TK, Keller J, Kesavapany S, Agarwal N, Kuner R, Pant HC, Iadarola MJ, Brady RO, Kulkarni AB (2007) Cyclin-dependent kinase 5 modulates nociceptive signaling through direct phosphorylation of transient receptor potential vanilloid 1. Proc Natl Acad Sci USA 104:660–665
Pareek TK, Keller J, Kesavapany S, Pant HC, Iadarola MJ, Brady RO, Kulkarni AB (2006) Cyclin-dependent kinase 5 activity regulates pain signaling. Proc Natl Acad Sci USA 103:791–796
Rains C, Bryson HM (1995) Topical capsaicin. A review of its pharmacological properties and therapeutic potential in post-herpetic neuralgia, diabetic neuropathy and osteoarthritis. Drugs Aging 7:317–328
Robbins W (2000) Clinical applications of capsaicinoids. Clin J Pain 16:S86–S89
Shin J, Cho H, Hwang SW, Jung J, Shin CY, Lee SY, Kim SH, Lee MG, Choi YH, Kim J, Haber NA, Reichling DB, Khasar S, Levine JD, Oh U (2002) Bradykinin-12-lipoxygenase-VR1 signaling pathway for inflammatory hyperalgesia. Proc Natl Acad Sci USA 99:10150–10155
Storz P, Doppler H, Toker A (2004) Protein kinase Cdelta selectively regulates protein kinase D-dependent activation of NF-kappaB in oxidative stress signaling. Mol Cell Biol 24:2614–2626
Storz P, Toker A (2003) Protein kinase D mediates a stress-induced NF-kappaB activation and survival pathway. EMBO J 22:109–120
Sugiura T, Tominaga M, Katsuya H, Mizumura K (2002) Bradykinin lowers the threshold temperature for heat activation of vanilloid receptor 1. J Neurophysiol 88:544–548
Szallasi A, Blumberg PM (1989) Resiniferatoxin, a phorbol-related diterpene, acts as an ultrapotent analog of capsaicin, the irritant constituent in red pepper. Neuroscience 30:515–520
Szallasi A, Blumberg PM (1993) Mechanisms and therapeutic potential of vanilloids (capsaicin-like molecules). Adv Pharmacol 24:123–155
Tang D, Yeung J, Lee KY, Matsushita M, Matsui H, Tomizawa K, Hatase O, Wang JH (1995) An isoform of the neuronal cyclin-dependent kinase 5 (Cdk5) activator. J Biol Chem 270:26897–26903
Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D (1998) The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 21:531–543
Tsai LH, Delalle I, Caviness VS Jr, Chae T, Harlow E (1994) p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature 371:419–423
Valverde AM, Sinnett-Smith J, Van Lint J, Rozengurt E (1994) Molecular cloning and characterization of protein kinase D: a target for diacylglycerol and phorbol esters with a distinctive catalytic domain. Proc Natl Acad Sci USA 91:8572–8576
Wahl P, Foged C, Tullin S, Thomsen C (2001) Iodo-resiniferatoxin, a new potent vanilloid receptor antagonist. Mol Pharmacol 59:9–15
Wang Y, Kedei N, Wang M, Wang QJ, Huppler AR, Toth A, Tran R, Blumberg PM (2004) Interaction between protein kinase Cmu and the vanilloid receptor type 1. J Biol Chem 279:53674–53682
Wang Y, Szabo T, Welter JD, Toth A, Tran R, Lee J, Kang SU, Suh YG, Blumberg PM, Lee J (2002) High affinity antagonists of the vanilloid receptor. Mol Pharmacol 62:947–956
Wang Y, Toth A, Tran R, Szabo T, Welter JD, Blumberg PM, Lee J, Kang SU, Lim JO, Lee J (2003) High-affinity partial agonists of the vanilloid receptor. Mol Pharmacol 64:325–333
Woolf CJ, Salter MW (2000) Neuronal plasticity: increasing the gain in pain. Science 288:1765–1769
Yang YR, He Y, Zhang Y, Li Y, Li Y, Han Y, Zhu H, Wang Y (2007) Activation of cyclin-dependent kinase 5 (Cdk5) in primary sensory and dorsal horn neurons by peripheral inflammation contributes to heat hyperalgesia. Pain 127:109–120
Zhang H, Cang CL, Kawasaki Y, Liang LL, Zhang YQ, Ji RR, Zhao ZQ (2007) Neurokinin-1 receptor enhances TRPV1 activity in primary sensory neurons via PKCepsilon: a novel pathway for heat hyperalgesia. J Neurosci 27:12067–12077
Zhu H, Yang Y, Zhang H, Han Y, Li Y, Zhang Y, Yin D, He Q, Zhao Z, Blumberg PM, Han J, Wang Y (2007) Interaction between protein kinase D1 and transient receptor potential V1 in primary sensory neurons is involved in heat hypersensitivity. Pain
Zhuang ZY, Xu H, Clapham DE, Ji RR (2004) Phosphatidylinositol 3-kinase activates ERK in primary sensory neurons and mediates inflammatory heat hyperalgesia through TRPV1 sensitization. J Neurosci 24:8300–8309
Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, Di Marzo V, Julius D, Hogestatt ED (1999) Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 400:452–457
Acknowledgments
All the works were supported by grants from the National Natural Science Foundation of China (30371635, 303330230 and 30770703), a grant from the Beijing Natural Science Foundation (7072040), and a grant from the specialized Research Fund for Doctoral Program of Higher Education (20060001121).
Author information
Authors and Affiliations
Corresponding author
Additional information
Special issue article in honor of Dr. Ji-Sheng Han.
Rights and permissions
About this article
Cite this article
Wang, Y. The Functional Regulation of TRPV1 and Its Role in Pain Sensitization. Neurochem Res 33, 2008–2012 (2008). https://doi.org/10.1007/s11064-008-9750-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-008-9750-5