
Abstract
Transient receptor potential vanilloid type 1 (TRPV1) is a nonselective cation channel that is intensively expressed in the peripheral nerve system and involved in a variety of physiological and pathophysiological processes in mammals. Its activity is of great significance in transmitting pain or itch signals from peripheral sensory neurons to the central nervous system. The alteration or hypersensitivity of TRPV1 channel is well evidenced under various pathological conditions. Moreover, accumulative studies have revealed that TRPV1-expressing (TRPV1+) sensory neurons mediate the neuroimmune crosstalk by releasing neuropeptides to innervated tissues as well as immune cells. In the central projection, TRPV1+ terminals synapse with the secondary neurons for the transmission of pain and itch signalling. The intense involvement of TRPV1 and TRPV1+ neurons in pain and itch makes it a potential pharmaceutical target. Over decades, the basis of TRPV1 channel structure, the nature of its activity, and its modulation in pathological processes have been broadly studied and well documented. Herein, we highlight the role of TRPV1 and its associated neurons in sensing pain and itch. The fundamental understandings of TRPV1-involved nociception, pruriception, neurogenic inflammation, and cell-specific modulation will help bring out more effective strategies of TRPV1 modulation in treating pain- and itch-related diseases.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
The transient receptor potential (TRP) channels are a superfamily that is made up of 28 members in mice and 27 members in humans [1]. As nonselective cation-permeable channels, the TRP family is capable to respond to multiple external and internal stimuli, including changes in thermal, pH, chemical irritants, as well as mechanical and osmotic cues. Therefore, they are implied in numerous physiological and pathological conditions and have become an increasing focus in basic science, translational research, and drug development.
Generally, the TRP superfamily is divided into six subfamilies: canonical (TRPC), vanilloid (TRPV), ankyrin (TRPA), melastatin (TRPM), polycystin (TRPP), and mucolipin (TRPML). Among them, the TRPV subfamily, which includes four members TRPV1 to TRPV4, are broadly involved in nature responses like pain and itch across mammals. The TPRV1 channel is also known as vanilloid receptor 1 and was cloned and identified as the receptor for heat and capsaicin in 1997 by David Julius lab [2]. It consists of six transmembrane domains with a pore formed by transmembrane segments 5–6 (S5–S6) and the intervening pore loop, which is flanked by S1–S4 voltage-sensor-like domains [3]. Various stimuli such as noxious heat (>43 °C), capsaicin, and pH acting on TRPV1 can directly lead to the channel opening and the consequent cation entry [2, 4]. TRPV1 is highly expressed in the peripheral nerve system (PNS) particularly C-fiber [5]. Ever since its discovery, TRPV1 has attracted broad attention in multiple studies that focus on pain or itch [6,7,8,9,10,11]. In this chapter, we will start with the introduction of basic biology in neuroscience and then interpret how TRPV1 participates in pain and itch perception. Furthermore, based on its biology, we will discuss the potential of TRPV1 modulation as drug interventions in pathologic sensory conditions.
2 TRPV1 Biology in Pain and Itch
2.1 The Basics of Pain and Itch
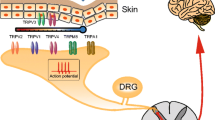
In mammals, the skin, mucous membrane, and muscles are highly innervated by primary somatosensory fibers. Their cell bodies are located in trigeminal ganglia which innervate the head and neck and the dorsal root ganglia (DRG) which receive signals from the rest of the body. These afferent neurons are pseudo-unipolar neurons, which project long axons to the skin and deeper body structures, and transmit impulse signaling through the other axonal branches synapse with neurons in the brain stem nuclei or spinal cord dorsal horn. The information is then relayed by higher order neurons towards the cortex where various senses such as warmth, coldness, touch, pressure, pain, and itch are ultimately perceived. While pain is defined as “an unpleasant sensory and emotional experience associated with actual or potential tissue damage” [12], the itch was introduced as “An unpleasant sensation that provokes the desire to scratch” [13]. Generally, sensory neurons that are specialized to sense pain and itch are referred to as nociceptors and pruriceptors, respectively. Due to the protective behaviors subsequentially aroused by pain and itch, hosts can be aware of or protected from environmental dangers.
Historically, sensory neurons have been classified according to the amount of myelination which dictates conduction velocity (fast or slow) and diameter (large or small). There are three main types of sensory neurons: large diameter (heavily and moderately myelinated Aβ fibers), medium diameter (thinly myelinated Aδ fibers), and small diameter (unmyelinated C fibers). While various mechanosensations such as touch and pressure are mainly elicited by Aδ and Aβ fibers, the vast majority of pain and itch is mediated by Aδ fibers and unmyelinated C fibers with free nerve endings.
2.2 TRPV1 and TRPV1+ Sensory Neurons
TRPV1 is the most well-characterized TRP channel and is the specific receptor of capsaicin (the spicy ingredient of chili peppers). However, a wide range of endogenous and exogenous stimuli such as noxious temperature (~43 °C), acidic or basic pH, vanilloid compounds, can also activate this ion channel [2, 4].
It is well established that TRPV1 activation and consequential cation ion flow are required for transduction of a number of pain or itch signaling. Meanwhile, nociceptors rely on molecular sensors like TRPV1 to detect noxious stimuli. It has been demonstrated in numerous studies that ~40–50% of sensory neurons express TRPV1 [14, 15]. More recently, emerging studies using unbiased single-cell RNA sequencing (scRNA seq) of murine DRG neurons updated the sensory neuron classification system according to their gene expression [16,17,18,19]. In 2015, Usoskin et al. identified that a subpopulation of Aδ fibers expresses the gene of calcitonin-gene related peptide (CGRP, Calca) and nerve growth factor receptor (TrkA; Ntrk1). They classified this population as peptidergic (PEP) 2. Those C fibers that express the above two genes plus neuropeptides substance P (SP; Tac1), are classified into PEP1. Other C fibers that were previously classified as non-peptidergic (NP) neurons (P2rx3 expression and predicted IB4 binding), were classified into three subclusters NP1, NP2, and NP3. Interestingly, some NP neurons like NP2 also contain markers for peptidergic neurons (i.e., CGRP gene Calca) [18, 19]. Therefore, NP neurons still have the capability to release neuropeptides. It has been revealed by sc-RNA seq data that Trpv1 is abundantly expressed in the PEP1, NP2, and NP3 [18, 19], suggesting the critical role played by these three subpopulations in transmitting noxious and pruriceptive stimuli (Figs. 12.1 and 12.2).

Maximum likelihood estimated expression of selected genes in mouse dorsal root ganglia neuron subpopulations. Nociceptors are considered to primarily be in PEP1 and PEP2 populations, while pruriceptor populations are considered to be NP1, NP2, and NP3. (Full database is available in Usoskin et al. [18])

Pain- and itch-sensing receptors are broadly expressed in TRPV1+ subpopulations. (a) Peptidergic TRPV1+ neurons express the majority body of pain-sensing receptors and release the corresponding neuropeptides such as SP (coded by gene Tac1), CGRP (coded by Calca), and NMB (coded by Nmb). (b, c) Itch-sensing neurons are mainly expressed in NP2 and NP3 TRPV1+ subpopulations. Neuropeptides releasing from these two subpopulations including NMB, CGRP, and SST (coded by gene Sst), Nppb (coded by gene Nppb). Of note, MrgprA3 and Nppb are the two well-accepted makers for itch populations
2.3 TRPV1 in Pain Sensation
2.3.1 Pain Classification
Pain can be simply divided into acute pain and chronic pain. Acute pain refers to the new onset of tissue damage or injuries, usually featured as self-limited healing and relief within hours or days. From this direction, acute pain provides warning signals and provokes avoidance from dangers. However, when the pain pathway is altered or pain sensation tends to last longer or recurs, chronic pain can be defined. In order to better identify chronic pain, given the first priority to pain etiology, the Task Force group of IASP (International Association for the Study of Pain) has defined chronic pain as “pain that lasts or recurs for longer than 3 months” in the year 2015 [20], regardless the specific pain phenotypes. Chronic pain is further classified as: (1) chronic primary pain; (2) chronic cancer-related pain; (3) chronic postsurgical or posttraumatic pain; (4) chronic neuropathic pain; (5) chronic secondary headache or orofacial pain; (6) chronic secondary visceral pain; and (7) chronic secondary musculoskeletal pain. More details regarding ICD-11 (the 11th version of International Classification of Diseases) codes for chronic pain were also extensively defined in the year 2019, which benefits to identify patients with chronic pain [21, 22].
The clear-cut definition and ICD-11 codes for chronic pain are attributed to the up-to-date efforts of the majority body of clinical and basic science research. Chronic pain has several debilitating features in which hypersensitivity is the most dominant one. Hypersensitivity refers to the “tissue or nerve damage elicits hyperactivity to promote guarding of the injury area” [23]. As a result, innocuous stimuli such as light touch could be perceived as pain (allodynia), and painful stimuli will induce greater intensity (hyperalgesia). This phenomenon is driven by various sensitization pathways and corresponding mechanisms. The mechanism for sensing and transmitting pain sensation is complex and involves multiple factors, leading to difficulties in pain treatment [23]. As the predominated nociceptor in the peripheral sensory system, TRPV1+ neurons possess numerous receptors for pain mediators such as adenosine triphosphate (ATP), serotonin, and bradykinin. Of the utmost importance, the TRPV1 channel is also a potent sensor for noxious stimuli and can be sensitized in a broad spectrum of chronic pain environments.
2.3.2 TRPV1 Serves as the Sensor for Pain Sensation
In general, direct activation of the TRPV1 channel by capsaicin and noxious heat (above 43 °C) can induce a burning sensation in the applied region. Capsaicin is known as the pain mediator long before its receptor was identified in 1997 [2]. By binding to the residue of Y511 located at the transmembrane spanning segment 4 (S4), capsaicin induces the opening of the TRPV1 channel and introduces calcium influx, the first sign for pain sensation singling in the peripheral. Similarly, noxious heat enables TRPV1 channel opening and triggers intracellular calcium increase. The intracellular calcium enhancement due to the receptor potential of TRPV1 activation could further trigger the voltage-gated ion channels to generate action potentials and result in the peripheral transmission of burning pain sensation. The property of sensing heat and capsaicin to produce pain sensation makes TRPV1 the sensor for these two fundamental noxious as well as thermal and chemical stimuli in the sensory nervous system.
Acidosis is common in the context of inflammation, tissue damage, and ischemia, in which protons are the important pain mediators. Proton (also described as hydrogen ion, acid, or low pH in different research papers) can directly activate the TRPV1 channel when the pH value hits lower than 6 at room temperature, by binding the E648 site of the extracellular loop and triggering cation entry for pain transmission [24]. Interestingly, in addition to protons, TRPV1 can also detect basic deviations from homeostatic pH. Alkaline pH such as ammonia can also induce irritant and pain sensation, which is common in the environment of artificial fertilizers and industrial pollutants. Via exposure to the external NH4Cl solutions to get the diffusion-free NH3 (ammonia), cultured DRG neurons and TRPV1-coexpressing human embryonic kidney 293 (HEK293) cells showed robust calcium responses [4]. In TRPV1-expressing HEK293 cells, the cell activity induced by intracellular pH of 9.5 was nearly abolished when TRPV1-antagonist was introduced. These results indicated the responsible role of TRPV1 in sensing alkaline chemicals and intracellular basic pH. Distinct from the proton binding site of E648, the base-sensing residue is detected at the site of H378 located in TRPV1 N-terminals [4]. Although TRPA1 (transient receptor potential ankyrin 1) can also respond to ammonia and intracellular base, so far, TRPV1 is the only identified ion channel that senses both acid and alkaline pH [4].
Some toxin peptides like venoms (which contains three types of inhibitor cysteine knot peptides) from spiders, snakes, cone snails, or scorpions, functioning as vanillotoxins, can also directly activate TRPV1 and induce inflammatory pain [25]. One such classical peptide toxin named as double-knot toxin (DkTx) from the Earth Tiger tarantula, can specifically bind to the residues within the S5-P-S6 pore region, and exhibit antibody-like bivalency to produce pain sensation in a virtually irreversible manner [26].
While sensing the noxious stimuli, TRPV1 activity is also magnified via sensitization in the context of inflammatory pain. Studies have revealed that TRPV1 responses to pain-producing chemicals and thermal stimuli, subsequently triggers heat-evoked pain or hypersensitivity in injured tissues [27]. Genetic TRPV1 knockout mice lack the thermal hypersensitivity in the context of inflammation, indicating that TRPV1 serves as the pain sensor upon activation by internal and external stimuli [11].
2.3.3 TRPV1+ Sensory Neuron in Pain Sensation
Besides the direct activation by heat and chemical irritants, TRPV1 protein actually labels the nociceptive PEP1 population based on the classification of sensory neuron types by the unbiased large-scale scRNA-seq [18]. Among the aforementioned types of pain classified by IASP, TRPV1 plays an extremely important role in inflammatory pain. Injuries or damaged tissues can release the classical “inflammatory soup”, which comprises the major pain-inducing chemicals such as ATP, bradykinin, prostaglandins, 5-HT (5-hydro-xytryptamine, also known as serotonin), and endothelin-1. From experimental data and the unbiased sequencing analysis, TRPV1+ neurons possess the dominant nociceptive populations sensing the above pain mediators. That is, TRPV1+ PEP1 neurons express purinergic receptor P2rX3, bradykinin B2 receptor, protease-activated receptor 2 (PAR2), and adenosine 2 receptor and so on for sensing pain mediators (Summarized in Fig. 12.2a) [1, 18]. In the field of acute pain, postoperative pain is the most common one that requires acute anti-pain treatment otherwise would potentiate chronic pain. Postoperative pain also attributes to inflammation in wounds. TRPV1 is necessary for heat (but not mechanical) hyperalgesia after incision, which has been evidenced by genetic knockout and pharmacologic antagonism in tested mice [28]. Genetic ablation of TRPV1-lineage neurons results in the overall deficit to either thermal hyperalgesia or neurogenic inflammation, although this is partially due to other thermal TRPs co-expressed in the TRPV1-lineage population [9]. Also, ablation of TRPV1+ neurons and fibers by the potent agonist resiniferatoxin (RTX) treatment has been shown to impair thermal nociception without affecting mechanical nociception in adult rats. Furthermore, during the projection from DRG to the dorsal horn, the loss of TRPV1+ neurons significantly eliminates the expression of presynaptic mu opioid receptors in DRG, and also potentiates the analgesic effect of opioid, indicating the inhibition effect of the intact mechano-nociception afferents [29].
Most of the pain mediator receptors are not only located in the TRPV1+ neurons but also employ TRPV1 as the downstream ion channel for nociception sensitization. Activation of the bradykinin B2 receptor by bradykinin during tissue inflammation mainly elicits pain sensation by sensitizing TRPV1 via phospholipase C (PLC) and cyclooxygenase-1 activation [30]. Meanwhile, TRPV1 is also required for a number of G-protein coupled receptors (GPCRs), such as the 5-HT2 receptor and PAR2 receptor when sensing pain mediators [23]. Notably, TRPV1 may have some alterations upon chemical stimulation to augment the pain sensation. For example, during pain sensitization, multiple intracellular signals like protein kinase A (PKA) and protein kinase C (PKC) pathways can phosphorylate the TRPV1 channel and produce pain sensitization, which provides a unique strategy to modulate TRPV1 activity for analgesia [31].
2.3.4 TRPV1-TRPA1 Complex in Pain Sensation
TRPV1+ neurons co-express several TRP channels that are also nociception sensors. TRPA1 is the sole member of TRPA subfamily and serves a broad spectrum of biophysiological functions in different species. TRPA1 is largely co-expressed with TRPV1 in sensory neurons, where they act closely in modulating pain [11, 32]. TRPA1 senses exogenous irritant chemicals such as pungent products isothiocyanates (AITC) [33, 34] as well as endogenous metabolites such as reactive carbonyl species like 4-hydroxynonenal (4-HNE) and 4-oxoonenal (4-ONE) [35, 36]. Hence TRPA1 is viewed as the “gate-keeper” for inflammation within sensory nervous system [37]. The role of TRPA1 involved in pain sensation has been well-documented [38, 39]. The most intriguing inhibition effect of TRPV1 on TRPA1 has been observed in pain conditions [40]. Until recently, the finding of Tmen100 protein as the modulator for TRPA1-mediated hyperalgesia in a TRPV1-dependent manner [41], has shed light on a new strategy for pain treatment. In general, Tmem100 is a 134-amino-acid transmembrane protein highly conserved in vertebrates [42]. In the tested rodent DRGs, Tmem100 is exclusively expressed in the peptidergic population. Importantly, Tmem100 has the capacity to release the inhibition of TRPA1 by TRPV1, resulting in inflammatory-mediated hyperalgesia. Mutation of Tmem100 with Q-Q-Q in the charged K-R-R sequence in the C terminal enhances the binding with TRPV1, which causes disconnection or low affinity with TRPA1. The consequence of this structure change contributes to the relief of TRPA1-mediated hyperalgesia [41]. Therefore, the regulation effect of TRPV1-TRPA1 correlation is also considered as a new target in neurogenic inflammation [43].
2.4 TRPV1 in Itch Sensation
2.4.1 Itch Is a Distinct Neural Process from Pain
Although it has been long considered that an itch is a mild form of pain, recent innovations in neuroscience and neuroimmunology have identified how itch pathways are distinct from pain.
In 2007, Sun and Chen discovered the first itch-specific pathway in the nervous system defined by gastrin-releasing peptide (GRP) and its receptor (GRPR), indicating that itch is a trackable and distinct sensory process from pain [44]. Later in 2009, Liu et al. identified that murine mas-related G-protein-coupled receptor (Mrgpr) A3 is the receptor of chloroquine (CQ), an anti-malaria drug that can induce serve itch sensation [45]. Mrgpr family are GPCRs and comprise ~8 genes and pseudogenes in humans and 27 in murine, most of which are expressed by primary sensory neurons [46]. CQ can cause robust itch in both humans and mice [45]. Interestingly, deletion of a cluster of Mrgpr genes significantly reduced mice scratching behavior induced by CQ but not histamine. They also identified that human MRGPRX1 has a similar primary sequence to MrgprA3 and can be specifically activated by CQ [45]. In the year 2013, the same group further found that MrgprA3-expressing neurons were the itch selective population. To demonstrate this, they adopted a unique approach by inserting the TRPV1 cation channel only in MrgprA3+ sensory neurons in TRPV1-deficient mice. Following capsaicin intradermal injection, they observed itch-induced scratching behavior rather than pain-related behaviors in those animals [47]. Other Mrgpr family expressing in neurons mediates itch sensation include MrgprC11 and MrgprD, which can be activated by bovine adrenal medulla peptide 8–22 [45] and β-alanine [48], respectively. Collectively, the discoveries of specific Mrgprs on sensory neurons and their itch function provide landmark advances in itch biology and neurology.
2.4.2 TRPV1+ Sensory Neuron in Itch Sensation
The scRNA-seq studies classified MrgprA3 (overlapped with MrgprC11)-expressing neurons into NP2 itch subpopulation (Figs. 12.1 and 12.2b). In addition to the NP2 subpopulation, NP3 is the other itch sensory neuron with significant TRPV1 expression (Figs. 12.1 and 12.2c). However, during the sensory nervous system development, TRPV1-lineage neurons in mice differentiate into DRG neuron subsets with diverse TRP markers that include TRPV1, TRPA1, and TRPM8. Therefore, in addition to TRPV1+ neurons, the animal tool of TRPV1-linage deficiency may also have impaired TRPA1-, TRPM8-, and even Mrg-expressing neurons [9]. However, administration of RTX, a potent agonist of TRPV1, provides an effective study approach to ablate only TRPV1+ neurons in the fully developed sensory nervous system [49].
NP3 is characterized by high enrichment of gene Nppb for brain natriuretic peptide (BNP), a neurotransmitter that activates itch in the spinal cord, along with genes that code the pruritogen receptors: interleukin (IL)-31RA (Il-31ra) and CysLTR2 (Cysltr2) (Fig. 12.2c). Stimuli that act on sensory neurons and directly induce itch are defined as pruritogens. IL-31 is a type 2 immune cytokine and is predominantly released by T helper (Th) 2 cells in the context of atopic dermatitis (AD), a common itchy and inflammatory skin disease. IL-31 is the first cytokine that has been identified as a pruritogen. In 2014, Cevikbas et al. identified that IL31RA is expressed by both human and mouse DRG neurons [50]. Either intradermal or intrathecal injection of IL-31 was sufficient to evoke robust itch behavior in naive wild-type (WT) mice. Notably, those IL-31-responsive sensory neurons also largely co-express with TRPV1. Later, the connection between IL-31 and Nppb+ neurons was further confirmed by the findings in which IL-31 is sufficient to upregulate Nppb in both skin and DRG [51]. More recently, we identified that the potent pruritogen leukotriene (LT) C4 and its receptor CysLTR2 neural pathway is the key mechanism in AD-associated itch flares [52]. However, although CysLTR2 siRNA knockdown in trigeminal ganglia was sufficient to alleviate acute itch flares in this context, it remains undefined whether this phenotype is specifically dependent on TRPV1+ neurons. Notwithstanding this, the Hoon group directly demonstrated that Nppb+ neurons are sensors of mast cell-derived LTC4 and TRPV1-linage neurons are required for N-methyl LTC4 (N-met LTC4)-induced itch. Moreover, they also revealed that itch elicited by serotonin and sphingosine-1 phosphate (S1p) is dependent on the Nppb+ subpopulation and these itch pathways further rely on the canonical GRPR-spinal cord circuit. Taken together, these results highlight that the Nppb-labeled sensory neurons are specifically responsible for IL-31-, LTC4-, serotonin-, and S1p-evoked itch. However, whether the cellular sources of these pruritogens play different roles towards itch neuron populations is still an interesting question for further research.
Histamine is a classical pruritogen that can be released from mast cells and basophils. So far, four distinct GPCRs (H1R, H2R, H3R, and H4R) for histamine have been found. Three of these receptors are expressed by neurons. However, only H1R and H4R are expressed by pruriceptive DRG and mediate histaminergic itch [53, 54]. Both NP2 and NP3 are found to have histamine receptor RNA expressed in sc-RNA seq studies (Fig. 12.2). Consistent with this, TRPV1-DTA animals exhibited a remarkable reduction of scratch responses to histamine intradermal injection [55].
IL-4 and IL-13 are the other two canonical types 2 effector cytokines. They have been shown to directly activate sensory neurons and promote itch in the context of AD-like disease [56, 57]. Compared with the receptors for IL-31 (Il31ra and Osmrβ), the gene of IL-4 and IL-13 shared receptor subunit (Il4ra) is broadly expressed across NP1, NP2, and NP3 itch sensory neurons [18, 57]. More importantly, the humanized anti-IL-4Rα mAb (dupilumab) has demonstrated outstanding anti-itch effects in patients with AD or other chronic pruritic diseases such as chronic pruritus of unknown origin [58,59,60,61,62,63,64]. Taken together, these advances highlight how cytokines dramatically promote itch. However, whether these cytokine itch pathways will be inhibited by targeting TRPV1+ neurons is unclear. Further studies may unveil the role and mechanisms of specific sensory subpopulations in cytokine-induced itch.
2.4.3 Role of TRPV1 Ion Channel for Itch Signaling
In addition to investigating the function of TRPV1+ sensory neurons in itch signaling, numerous basic studies have explored the molecular mechanisms of itch sensation.
It is well established that histaminergic itch is TRPV1-dependent. TRPV1-deficient mice exhibited less histamine-induced scratch compared with WT controls, whereas α-5HT- or endothelin 1-elicited itch was unaffected. The TRPV1 channel opening is critically required for histamine to activate sensory neurons via H1R [2, 65]. TRPV1 opening consequently causes increases in intracellular calcium (Ca2+) concentrations that prepare for neuronal action potentials. This process likely involves phospholipase (PL) A2 or lipoxygenases (LO) as a PLA2 inhibitor or a LO inhibitor is sufficient to block the histamine-induced Ca2+ influx in sensory neurons [66]. In addition, other analyses indicated that phospholipase (PL) Cbeta3 has overlapped expression with H1R in a subpopulation of C fibers and PLCbeta3 specifically mediates histamine-induced calcium responses through the H1R in cultured sensory neurons. The PLCbeta3-deficient murine strain showed significant defects in histamine-induced scratching behavior [67]. Notwithstanding this, the precise molecular mechanisms underlying histamine receptor-TRPV1 pathways will be an exciting area for further investigation. For the newly discovered H4R, recent studies suggest that TRPV1 and PLC are also required for H4R-mediated itch signaling in vitro [68].
In addition to histaminergic itch, multiple non-histaminergic itch mechanisms that involve TRPV1 include IL-31, IL-4/IL-13, LTB4, and LTC4. To explore which TRP channels are involved in IL-31-mediated itch, Cevikbas et al. employed TRPV1- and TRPA1-deficient mouse strain, respectively, and found that either TRPV1 or TRPA1 deletion can significantly decrease IL-31-evoked itch [50]. Using calcium imaging in vitro, they confirmed that the percentage of IL-31-responsive neurons was significantly reduced in TRPV1-or TRPA1-deficient DRG. Taken together, these results indicate that IL-31 employs both TRPV1 and TRPA1 to fully mediate itch. To investigate the downstream of TRP channels in the IL-31 itch pathway, in the same study, authors first observed that IL-31-stimulated murine DRG neurons induced ERK1/2 phosphorylation in vitro. Then they demonstrated that administration of U0126 which can completely prevent ERK1/2 phosphorylation was sufficient to reduce scratching bouts in WT mice that received IL-31 intradermal injections. Thus, ERK1/2 might provide a future direction to study the TRP downstream phosphorylation in the IL-31-induced itch pathway.
Although it appears that cultured DRG neurons from either Trpa1−/− or Trpv1−/− mice exhibited fewer responses to both IL-4 and IL-13, whether TRPV1 or TRPA1 is required for IL-4 and IL-13 enhanced scratching behavior remains undefined. However, Oetjen et al. demonstrated that IL-4/IL-13 needs Janus kinase (JAK) to transmit their signals into neurons [57]. The JAK family has four members: JAK1, JAK2, JAK3, and TYK2 [69]. The conditional deletion of JAK1 in sensory neurons led to a marked reduction in scratching behavior in the setting of AD-like disease. Moreover, in humans, JAK inhibitors are shown to significantly reduce itch severity in patients suffering from chronic pruritus [70]. Therefore, whether there is any connection between TRPV1 and JAKs would be interesting for future investigation.
LTs are a family of eicosanoid inflammatory mediators and are produced in leukocytes, particularly mast cells and basophils [71, 72]. LTs are generated by the oxidation of arachidonic acid and the essential fatty acid eicosapentaenoic acid via the 5-LO pathway. LTs are divided into two classes: the chemoattractant LTB4 and the cysteinyl LTs (CysLTs: LTC4, LTD4, and LTE4) [73]. It has been shown that intradermal injections of LTB4 resulted in scratching behavior in mice [74, 75]. Similar to IL-31-induced itch, either TRPV1 or TRPA1 antagonists are sufficient to inhibit itch behavior proved by LTB4 [75]. For CysLTs, while the LTC4-CysLTR2 itch pathway has been confirmed by multiple research groups [52, 76, 77], the role of LTD4 and LTE4 as pruritogens remains controversial. Interestingly, although LTC4-activated sensory neurons showed large coexpression with TRPV1 and TRPA1, either TRPV1 or TRPA1 knockout could not reduce the percentage of LTC4-responsive DRG neurons. Only TRPV1 and TRPA1 compound deletion is capable to ameliorate the neuronal responsiveness towards LTC4. In line with these in vitro results, mice lacking either TRPV1 or TRPA1 exhibited unaltered scratching behavior following N-met LTC4 injection compared with their littermate controls. However, TRPV1 and TRPA1 double knockout attenuated LTC4-induced scratching responses. Taken together, we can conclude that CysLTR2 is equally reliant on either TRPV1 or TRPA1 to fully mediate LTC4-elicited itch. In other words, both canonical TRPV1 and TRPA1 signaling must be impaired to limit LTC4-mediated itch.
3 TRPV1 Activity Modulation in Pain and Itch
3.1 TRPV1 Upregulation in the Context of Pain
In many pathological conditions of chronic pain, the functional upregulation of TRPV1 is likely induced by inflammatory mediators such as nerve growth factors released from adjacent non-neural cells. The elevation of TRPV1 protein, but not mRNA levels, indicates the cytoplasm assembly upon activation, rather than transcription-independent overexpression [78]. Interestingly, the increased TRPV1 protein then is transported to the peripheral membrane of a cell body, but not the central C-fiber terminals for synapse transmission. The TRPV1 upregulation is implied in tissue inflammation and thermal hypersensitivity. Further studies revealed that p38 MAPK activation is required for TRPV1 overexpression [78], indicating MAPK pathways in induction and maintenance of peripheral sensitization and persistent chronic pain [79].
Under pathological situations, TRPV1 has been detected in previous TRPV1-absent sensory neurons. To a large extent, hyperalgesia could be ascribed to this mechanism. Take TRPM8-dependent cold allodynia as an example, we have demonstrated that pleasant cool temperature that only causes cool sensation to the skin surprisingly induces cold allodynia to the cornea [80]. The significance of this phenomenon is that such cold allodynia becomes more severe in multiple pathological conditions such as dry eyes. We further identified that TRPV1 is highly expressed in TRPM8+ neuron-innervated cornea rather than other tissues. The expression of TRPV1 in TRPM8+ neurons was further elevated under dry eye conditions. Via electrophysiological approaches, we found that TRVP1 enables TRPM8+ cold-sensing neurons to depolarize and fire action potentials more easily. In line with in vitro results, TRPV1 deficiency and pharmacological antagonist significantly alleviated corneal cold allodynia. Meanwhile, TRPV1 upregulation is sufficient to cause cold allodynia as genetical overexpression of TRPV1 in TRPM8-expressing sensory neurons led to cold allodynia in both cornea and skin even without any pathological changes [80]. Taken together, it is reasonable that the upregulated TRPV1 ion channel initiates neural hypersensitivity in the TRPV1+ neuron population and results in phenomena of hyperalgesia.
3.2 TRPV1 Upregulation in the Context of Chronic Itch
TRPV1 upregulation has also been seen in itchy conditions. In a pure chronic itch model, dry skin conditioning was performed by the acetone–ether–water procedure for sequential days to induce robust itch behavior, known as dry skin model. In the mouse model, we evidenced that the TRPV1+ fiber largely expanded their territory in the skin compared with the control treatment. This is confirmed by histochemistry staining of placental alkaline phosphatase in the genetic TRPV1-reporter mouse line [81]. In this study, the upregulation of TRPV1 was confirmed on both gene and protein levels and the upregulated TRPV1 was sufficient to provoke enhanced calcium responses to the low dose of capsaicin, indicating its intact function. Importantly, in this dry skin model, a low dose of capsaicin injection strikingly induced robust scratching behavior rather than pain behavior, suggesting that TRPV1 might have broader functions in some specific pathologic conditions.
3.3 TRPV1 Structure Modulation
TRPV1 channel possesses highly modulable pockets for cation of calcium, magnesium, and sodium ion. A three-dimensional study revealed that TRPV1 has an ion permeation pathway via S5 and S6 and an intervening pore loop region (S5-P-S6) [3], which serves as dominant modulation sites. Based on the high allosteric coupling between upper and lower gates during activation, inflammatory agents are capable to modulate TRPV1 and such modulation is found to contribute to acute and persistent pain [23]. TRPV1 has two activated structures that have been detected by pharmacological probes (a peptide toxin, DkTx, and a small vanilloid agonist, RTX). Such diversity highlights the modulable property of TRPV1 and implies its potentials in regulating neuron activities [82]. In 2007, the high-resolution structure of the TRPV1 domain was described as cytosolic ankyrin repeat domain (ARD). This advanced finding helps better understand protein interactions folding in TRPV1 and indicates that the calcium-dependent regulation may involve competitive interactions between ATP and calmodulin at the TRPV1-ARD-binding site [83].
3.4 TRPV1 Phosphorylation
3.4.1 PKC Pathway
As the dominant downstream TRP channels for GPCRs-mediated pain and itch, the TRPV1 channel can be modulated by three major pathways. The process of TRPV1 phosphorylation is the most important pathway for the transmission of pain and itch signaling. The PKC-dependent pathway is the most common way for TRPV1 sensitization. Generally, TRPV1 phosphorylation mediated by PKC involves the Gαq/11-PLC-phosphatidylinositol (4,5) bisphosphate (PIP2)-diacylglycerol (DAG)-PKC intracellular pathway [84]. This pathway is crucial for various GPCRs such as bradykinin B2 receptor, 5-HT receptors, purinergic receptors, protease-activated receptors, and Mrgprs. Many chemokines can also directly sensitize TRPV1 and contribute to hyperalgesia during inflammation. Pro-inflammatory chemokines such as CCL3 can activate CCR1 (co-expressing with TRPV1 in more than 85% of small-diameter neurons) and induce calcium influx and PKC activation, hence are responsible for sensitization in a receptor crosstalk manner [85].
PIP2 is one of the first noticeable intracellular molecules for phosphorylation in sensory neurons. PLC is capable to hydrolyze PIP2 to produce DAG and isositol triphosphate (IP3). IP3 further promotes the release of calcium ions in cells and acts synergistically with DAG in the process of activating PKC. PKC activation phosphorylates the TRPV1 channel, hence triggers the sensitization effect from the upstream signaling. In fact, TRPV1 interaction with PIP2 and calmodulin is the classical pathway for the activity modulation within neurons. Cation permeability induced by TRPV1 agonists is dynamic and could be intensively modulated in a PKC-dependent pathway. The permeability of cation led by TRPV1 opening is time- and agonist concentration-dependent for large cations and calcium entry. This phenomenon has been evidenced in native or recombinant TRPV1 channels in rats and attributes to the cation selectivity led by different agonists that phosphorylate site Ser800 or Ser502 in a PKC-dependent manner. This property may further change the TRPV1 activity in pain sensation, as well as in neurotransmitter release or agonist-related cytotoxicity [86]. The putative binding sites for TRPV1 activity have been indicated to be Lys571 and Arg575 in the linker between the S4 and S5 of one TRPV1 subunit and Lys694, and the predicted PIP2–TRPV1 interaction region has relied on Leu777-Ser820 [87].
However, controversial results have revealed the negative regulatory role of PIP2 on the TRPV1 channel [88]. Researchers reconstituted purified TRPV1 into artificial liposomes and tested the precise effect of various phosphoinositides by introducing them into the TRPV1-expression system, respectively. Through this way, they found that membrane lipids including PIP2, PI4P, and phosphatidylinositol, can actually inhibit TRPV1-mediated sensitivity [88]. They also claimed that TRPV1 is fully functional without phosphoinositides. The ongoing exploration found that phospholipids, specifically phosphoinositides are important for heat-induced channel open status [89]. With regard to the controversial results for PIP2 involvement in TRPV1 sensitization, as the TPRV1 channel possesses multiple binding sites for activity modulation, further evidence could be applied to confirm the role of phosphoinositide in TRPV1 modulation based on the binding site regulation [90].
3.4.2 PKA Pathway
The sensitization and phosphorylation of TRPV1 via Gαs-activation are relied on Gαs- adenylyl cyclase (AC)-cAMP-PKA pathway. Briefly, activation of Gαs by pain and itch mediators could activate AC to generate cAMP, leading to the activation of PKA. PKA activation further sensitizes TRPV1 channels and sequentially triggers the hyperactivity of cation entry. Hence, any disturbance of this pathway will impair the overall sensitivity of TRPV1 phosphorylation. The GPCR-related Gαs-activation is very important for pain and itch mediator transmission. While Gαs-coupled receptors (such as 5-HT4 and 5-HT7) have positive regulation effects on the cAMP-dependent modulation of TRPV1, the Gαi/o-coupled receptors (e.g. μ-opioid receptor, cannabinoid receptors 1 and 2) play a negative regulation role [84].
Prostaglandin E2 is a classical TRPV1-mediated noxious stimulus that could act on Gαs. In this pathway, the downstream desensitization of TRPV1 can be achieved by altering Ser116 and Thr370 at the PKA binding site. On the contrary, pretreatment with forskolin to activate AC is ineffective to reduce TRPV1 desensitization. These results indicate the involvement of PKA-dependent reduction of desensitization of capsaicin-activated currents [91]. Nevertheless, other binding sites for PKA-dependent phosphorylation at the site of Thr144 or Ser502 have also been discovered [87].
Either opioids or cannabis is effective to reduce nociception provoked by noxious stimuli. Interestingly, both of their receptors, particularly at the postsynaptic membrane, are largely co-expressed with TRPV1 in peripheral sensory neurons. They both negatively regulate TRPV1 activity by acting on Gαi/o-coupled receptors. μ-opioid receptor (MOR)-mediated activation of Gαi/o-coupled receptors is sufficient to reduce AC activity and cAMP levels, leading to the decreased neural activity and thus anti-pain effects [92]. However, opioid withdrawal can significantly increase the cAMP level and increase the PKA activity. It has been demonstrated in transfected HEK293 cells and dissociated DRG neurons that capsaicin-induced TRPV1 activity plays a vital role in hyperalgesia resulted from opioid withdrawal [93]. Cannabis has two receptors like cannabinoid receptor subtype 1 and 2, both of which are Gαi/o-coupled receptors and are expressed in many types of neurons including TRPV1 positive ones. Cannabis has been prescribed to treat many refractory pain disorders including headache, arthritis, and post-operation pain, indicating the importance of cannabinoid receptors activation in pain relief [94]. Endogenous cannabinoids are bioactive lipids known as endocannabinoids and are derived from dietary omega-3 and omega-6 polyunsaturated fatty acids [95]. The endocannabinoid-TRPV1 axis represents an intrinsic prologetic pathway. Recently, a study identified epoNADA (epoxidation of N-arachidonic acid-dopamine) and epoNA5HT (epoxidation of N-arachidonic acid-serotonin), which are formed by CYP peroxygenases under inflammatory conditions, are bifunctional rheostat modulators of the endocannabinoid-TRPV1 axis. They demonstrated that epoNADA and epoNA5HT are more potent than their precursors (NADA and NA5HT respectively) in modulating TRPV1 activity. Their capability as strong antagonists is shown by suppressing intracellular calcium response and membrane currents provoked by capsaicin in sensory neurons. In addition, epoNA5HT is also a complete cannabinoid receptor subtype 1 agonist. These molecules can further effectively reduce pro-inflammatory biomarkers such as IL-6, IL-1β, and TNF-α. Collectively, these results indicate that cannabinoids are potential candidates in the development of anti-pain and anti-inflammation therapeutics [96].
3.4.3 CaMKII-Dependent Phosphorylation
CaMKII, refers to Ca2+-calmodulin-dependent kinase II, is essential for synaptic plasticity in peripheral TRPV1+ fibers. CaMKII phosphorylates TRPV1, thereby regulates vanilloid agonist binding to the receptor. Studies have shown that the dephosphorylation of TRPV1 by the protein phosphatase 2B, also known as calcineurin, leads to a desensitization of the receptor [97]. Moreover, point mutations in TRPV1 at two putative consensus sites (Ser502 and Thr704) that are required for CaMKII binding lead to the failure of capsaicin-stimulate currents and cause a concomitant reduction in TRPV1 phosphorylation. In a word, CaMKII and calcineurin could control the balance between activation and desensitization in nociceptors by regulating TRPV1 binding [98]. A further study also revealed an interesting phenomenon named potentiation related to CaMKII. It is shown that activation of CaMKII and extracellular signal-regulated kinase (ERK)1/2 contribute to a time-dependent potentiation of Ca2+ responses elicited by repeated application of capsaicin during a 40-min interval. However, pretreatment with deltamethrin to block calcineurin and tachyphylaxis can enhance the potentiation effect. Therefore, potentiation may be an important peripheral auto sensitization mechanism that needs further investigation [99].
3.5 Other Modulators
3.5.1 Protons
Acidosis is the feature of many pathological microenvironments, hence acid-sensing ion channels represent the ion channel families that detect noxious chemical stimuli. It has been validated that the proton-sensing channels named ASIC1a and ASIC3, are preferentially expressed in TRPV1+ sensory neurons and function as nociceptive sensors [100]. In addition to binding its specific acid channels, protons could act as TRPV1 agonists by acting on the S6 domain or modulators by combining the S5 domain. Thermal hypersensitivity mainly attributes to TRPV1 sensitization by acidosis in pathological conditions [31]. By lowering pH value, TRPV1 capability responsive to heat and capsaicin is potentiated in a dose-dependent manner. When the extracellular pH decreases to 6.4, spontaneous openings of the TRPV1 channel are observed even at body temperature. The TRPV1 binding site of E600, but not E648, is responsible for this type of regulation and serves as a regulator in the acid environment rather than heat- or capsaicin-evoked responses [24]. The potent effect of protons to lower the nociceptive threshold for temperature directly drives cation entry via TRPV1 opening at body temperature and contributes to the mechanisms of thermal pain (or hypersensitivity) in the context of tissue injury. This phenomenon can explain the clinical observation in which thermal hypersensitivity is usually exhibited in the context of infection, inflammation, and ischemia situations.
3.5.2 Pirt
Pirt is a membrane protein that is expressed in all peripheral neural tissues. It can bind both PIP2 and TRPV1 and is a positive regulator for TRPV1 [101]. Studies have shown that Pirt-deficient mice showed impaired responsiveness to noxious heat and capsaicin and responsible currents were significantly attenuated. Gain-of-function approaches indicated that TRPV1-mediated currents are enhanced by Pirt co-expression in a heterologous expression system. To achieve the modulation mechanism, the C terminus of Pirt directly binds to TRPV1 and PIP2 to strengthen TRPV1 activity [101].
Pirt regulation for TRPV1-dependent neuropathic pain has been observed in a chronic constriction injury model, which is a neuropathic pain model widely utilized in murine. In this context, pain behavior and TRPV1 over-expression were attenuated by knocking out Pirt gene [102]. In a visceral pain model that mimics uterine contraction-induced pain, Pirt expression and capsaicin-mediated activity were enhanced, leading to distinct pain behavior featured as writhing responses. However, this pain behavior was significantly attenuated in Pirt deficient-mice, indicating that Pirt might be involved in visceral pain that is mediated by TRPV1 [103].
In addition to pain, Pirt is also involved in itch modulation. Pirt deficit attenuates cellular and behavioral responses to various pruritogens such as histamine and CQ in animals. It has been shown that both TRPV1-dependent and TRPV1-independent itch likely need Pirt, suggesting its broad function in itch transmission [104].
3.5.3 GABA-Autocrine Feedback
Interestingly, TRPV1 activation can also trigger GABA release from peripheral nerve endings. GABA release serves as an autocrine feedback mechanism exclusively limiting TRPV1 sensitization in the setting of pathological inflammation. This feedback effect did not affect the TRPV1 response to capsaicin under physiological status. Also, this effect is independent of canonical G protein signaling but relies on the close juxtaposition of the GABAB1 receptor subunit and TRPV1 [105]. These modulation effects are essential for signaling transmission, helping expand neuroimmune interactions, and demonstrating self-protection by autocrine feedback mechanisms.
3.6 Pain-to-Itch Switch
In order to distinguish pain and itch phenotype in experimental mice, researchers have identified that upon intradermal injection of aversive chemicals into the cheek, pruritogens induce mice scratching behavior using hind paws, while pain signaling induces wiping behavior with forepaws [106]. Via behavior studies, substances like histamine, 5-HT, and agonists of protease-activated receptors PAR-2 and PAR-4, have been identified as itch-inducers. In contrast, capsaicin, AITC, and bradykinin that usually elicit dose-related forelimb wiping contribute to pain behavior [107]. However, under pathological conditions like allergic contact dermatitis in which bovine adrenal medulla 8–22 (a peptide that elicits histamine-independent itch via MrgprC11)-related scratching is enhanced, bradykinin that is supposed to cause pain behavior evokes both scratching behavior and wiping behavior, indicating a phenotype named as pain-to-itch switch [108]. This phenomenon has been confirmed in a dry skin mouse model, in which the increased innervation of TRPV1+ fibers in dry skin was demonstrated [81]. Although the precise mechanisms remain undetermined, this phenomenon implies TRPV1 may contribute to neural function plasticity in pathological conditions.
A research group has carried out studies to test human sensation evoked by capsaicin and histamine spicules in volunteer subjects [109, 110]. They observed that while single spicule of capsaicin, histamine, or cowhage induced dose-dependent pruritic sensations that were usually accompanied by pricking/stinging and burning, a wider and deeper application by capsaicin intradermal injection only induced pain with hyperalgesia but not itch or alloknesis [109, 110]. Although why capsaicin could induce different sensations by different application methods (punctate v.s injection) remains unknown, it provides clues to further study the phenomenon of pain-to-itch switch.
4 TRPV1+ Neurons as the Center of Neuroimmune Interactions
While pain and itch allow the host to sense and expel environmental stimuli, the immune system provides a following protective mechanism by fighting pathogens. Emerging evidences have revealed that immunity and nociception have crosstalk particularly in the skin and mucous barriers. On one hand, cytokines derived from immune cells or even environmental materials alone could act on sensory neurons. On the other hand, sensory neurons particularly nociceptors in turn release neuropeptides and cause immunity remodeling. In the sections above, we talk about mechanisms of pain and itch that are mediated by inflammatory mediators. In this section, we will focus on how the peripheral sensory nervous system regulates immune responses via neuropeptide releasing (Fig. 12.3).

Neuroimmune interactions mediated by TRPV1+ neurons in the context of pain and itch. TRPV1+ fibers in the peripheral tissues directly sense pain and itch mediators, evoking action potential to transmit the nociceptive signaling to feed into the central circuitry. On the other hand, TRPV1+ peripheral axons release abundant neuropeptides to provoke neuroimmune interactions
Neuropeptides are signaling peptides that are mainly made by and released from neurons. Neuropeptides can affect both neurons and non-neuronal cells and are considered to be key mediators in the communication between neurons (in particular sensory neurons) and effector cells. A number of candidate neurotransmitters/neuropeptides released from peripheral sensory neurons towards the spinal cord include GRP, glutamate, SP, BNP, and neuromedin B (NMB), etc. [44, 55, 111,112,113,114,115]. Meanwhile, activated nociceptors are capable of releasing neuropeptides to act on non-neural cells. These processes are particularly significant in pathologic conditions such as infectious immunity and allergic reactions.
4.1 Mast Cells: A Classic Neuroimmune Paradigm
The axon reflex represents a classic neuroimmune paradigm. The activation of nociceptors not only leads to the transmission of action potentials toward the central nervous system but also causes neuropeptide releasing along its fibers back to skin endings. These released peptides mainly include CGRP and SP. Both CGRP and SP have been shown to act on the vasculature and mast cells to induce vasodilatation, edema, degranulation, and consequent immune cell recruitment [116]. Vasodilatation caused by CGRP is mediated by receptors at precapillary arterioles, whereas SP induces plasma extravasation from venoles. Unlike neurokinin-1 receptor (NK1R) which is the receptor for SP in the spinal cord, recent studies have shown that MrgprB2 in mice (MRGPRX2 in humans) is the key receptor that induces SP-driven activation of mast cells [117, 118]. Therefore, mast cells appear to be the key cellular mechanism that bridges immunity and somatosensation. On the one hand, they release inflammatory mediators to promote pain and itch upon stimuli. On the other hand, they accept neuropeptide signaling from sensory nerves to further exemplify the noxious responses. Thus, the reaction of axon reflex is also called “neurogenic inflammation.”
More interestingly, a recent study uncovered that common environmental allergens that have cysteine protease activity, e.g., house dust mites, can directly activate TRPV1+ neurons to release SP. Further, SP acts on MrgprB2 on mast cells to promote type 2 inflammation in the skin. These findings elucidate how environmental stimuli directly activate nociceptors and regulate inflammation via the sensory system [119].
4.2 Beyond Mast Cells: Other Immune Cells Regulated by Nociceptors
In addition to mast cells, more innate immune cells have been found capable to respond to neuropeptides. Neutrophils are an innate immune cell population that is well known for rapidly responding to infections, helping resolve infections, and healing the damaged tissue. A number of bacteria infections provoke host pain sensation, but the precise mechanisms are not fully understood. In 2013, Chiu et al. firstly found that bacterial infections cause pain by directly activating nociceptors [120]. Later in 2018, the Chiu group identified that TRPV1+ nociceptors suppressed protective immunity against lethal Staphylococcus aureus pneumonia through suppression of the recruitment and surveillance of neutrophils and alteration of lung γδ T cell numbers [121]. Further, in the same year, the same research team reported that on the basis of nociceptor activation stimulated by Streptococcus pyogenes-released streptolysin S to produce pain, nociceptors, in turn, secrete neuropeptide CGRP to inhibit the recruitment of neutrophils and the killing of bacteria [122]. In the above studies, they employed the approach of RTX administration and TRPV1-DTR strain to demonstrate the role of TRPV1+ nociceptors in the context of bacterial infections. Taken together, it appears that nociceptors and their related neuropeptide CGRP are likely negative factors to defense bacterial pathogens across barriers from the skin to the lung.
Group 2 innate lymphoid cells (ILC2) belong to the family of innate lymphocytes and are a prominent source of type 2 cytokines [123]. ILC2s are found constitutively at the skin and mucosal barriers and in the spleen, fat-associated lymphoid clusters, and lymph nodes. The significant role of ILC2 plays in type 2 immunity like AD, metabolic homeostasis, and chronic pathologies like fibrosis has been demonstrated in both patients and animal models [124]. So far, the identified neuropeptides that can regulate ILC2s include vasoactive intestinal peptide (VIP), neuromedin U (NMU), and NMB [125,126,127,128,129]. However, the most relevant neuropeptide released from nociceptors is VIP. VIP was first shown to stimulate gut ILC2s to release IL-5 through the VPAC2 receptor and cause eosinophil accumulation [130]. Another study conducted by Talbot et al. identified that IL-5 directly activates nociceptors to induce the secretion of VIP. Then VIP further activates resident ILC2s and effector CD4+ T cells via VPAC2. This positive feedback loop formed by cytokine, nociceptors, and immune cells significantly amplifies the pathological allergic conditions, which suggests targeting the nervous system as a treatment option in allergies. NMU and NMB have been, respectively, found to promote lung ILC2 and helminth-induced ILC2 responses [127,128,129]. However, while the main cellular source of NMU is cholinergic neurons, the major source of NMB in the setting of helminth infection remains unclear. Notwithstanding this, these studies highlight how significant the nervous system is in innate immunity regulation and imply that nervous systems might also play a role in the link between innate immunity and adaptive immune responses.
Dendritic cells (DCs) are important antigen-presenting cells and are resident in multiple barriers. They are responsible to process and present antigens or allergens to adaptive immune cells like T cells and promote their differentiation. Candida albicans is the most common pathogen that causes cutaneous candidiasis. In a murine model of cutaneous candidiasis, Kashem et al. discovered that IL-23-derived from CD301+ dermal DCs could augment γδ T cells to release IL-17A and inhibit cutaneous Candida albicans infection. Interestingly, CGRP released from TRPV1+ neurons that are directly activated by Candida albicans is sufficient to promote IL-23 releasing from dermal DCs [131]. The role of the other neuropeptide SP released from TRPV1+ neurons play in DCs and antigen-presenting processes were reported recently. In the study published by Perner et al. [132], authors identified that environmental allergens like papain and Alternaria extract were sufficient to provoke scratching behavior in mice and this phenotype was dependent on TRPV1+ sensory neurons. Intriguingly, using TRPV1-DTR mouse strain, they surprisingly found that allergen-induced CD301b+ DC migration required the TRPV1+ neuron subset as well as SP. Collectively, these studies highlight the significance of TRPV1+ sensory neurons in antigen-presenting process for both infectious and allergic immune responses.
Studies to explore somatosensory neurons in adaptive immune cells are not as many as in innate immune responses. In 2019, Cohen et al. employed a novel optogenetic approach to activate TRPV1+ neurons. They generated mice (TRPV1-Ai32) in which TRPV1+ neurons in the skin could express the light-gated cation channel channelrhodopsin-2 and be activated by cutaneous light stimulation [133]. Strikingly, this optogenetic stimulation is capable of initiating skin inflammation which is predominated by Th17 (type 3) immune responses. The biological effect of such skin immunity was further shown to enhance host local defense to Candida albicans and Staphylococcus aureus. More importantly, this defense effect can be propagated to adjacent, unstimulated skin areas through a nerve reflex arc. Thus, although the concept of “neurogenic inflammation” was established a century ago, this study demonstrated that activation of sensory neurons alone is sufficient to results in inflammation and might be a future target to augment local immune responses in fighting pathogens.
5 Therapeutic Strategy and Perspectives
5.1 Agonist
The basic concept for analgesic effects of capsaicin refers to the selective excitation and subsequent desensitization of TRPV1+ fibers (pain and itch sensing fibers). This is widely used in relieving pain and itch related to many sensory disorders. This acute desensitization occurs rapidly within 20 s upon activation and is dependent on the calcium influx, which represents a feedback mechanism protecting nociceptors from toxic calcium overload [87]. This effect of “activation triggers sequential desensitization” has been repeatedly verified in capsaicin topical application to alleviate pain and itch. In pain clinic, 8% capsaicin topical patch has been used in the painful disorders, such as diabetic neuropathy, chemotherapy-induced peripheral neuropathy, and peripheral neuropathy associated with hypereosinophilic syndrome [134,135,136,137,138]. Furthermore, direct capsaicin injection into the joint has been used to relieve osteoarthritis-related pain [139]. Unlike “activation triggers sequential desensitization,” tachyphylaxis is another type of desensitization and refers to the reduced responses due to repeated activation. Further studies are needed to verify its anti-itch or anti-pain effects in clinical practice.
Although topical capsaicin has been approved by Food and Drug Administration (FDA) for neuropathic pain treatment, its advantages are significant as it not only desensitizes the TRPV1 channel-related pain sensation, but also disrupts TRPV1+ peripheral terminals to abolish the potential nociception transmission via other co-expressed channels or receptors. However, one major concern is the disruption of physiological functions mediated by neuropeptides released TRPV1+ peripheral nerves. Disrupting or desensitizing the TRPV1+ fibers in a period of time will inevitably lead to the absence of neuropeptide releasing in various noxious conditions such as axon reflex. Furthermore, long-term use of the capsaicin patch might null the somatosensation of the treated skin, which might attenuate the response upon harmful stimulation, such as noxious heat. Finally, activation of TRPV1 in skin lesions causes painful sensation before desensitization occurs, which is not acceptable to some fatigue patients or children.
5.2 Antagonist
TRPV1 antagonists are expected to treat pain and itch sensation by inhibiting receptor potential and preventing convey of nociceptive signaling from peripheral sites to the central nervous system. A series of novel drugs have been tested in clinical trials. GlaxoSmithKline SB-705498 is a small molecular and a potent, selective, orally bioavailable antagonist for TRPV1 [140]. The anti-pain effect of SB-705498 has been tested in a cohort of 19 healthy volunteers who had pain symptom that was evoked by noxious heat, capsaicin or ultraviolet radiation B (UVB) irradiation [141]. The results appear positive as SB-705498 was revealed safe and well-tolerated and exhibited pharmacodynamic activities in alleviating pain and hyperalgesia.
However, some tested antagonists were found to potentiate hyperthermia or hypothermia. Given hyperthermia/hypothermia as the most common side effects, the pharmaceutical value of TRPV1 agonists was questioned. Thermoregulatory effects of TRPV1 antagonists in humans have been intensively studied. Controversial views are emerging as the tested compounds in vitro may have different binding potency in vivo and thus, the safety and efficacy of new TRPV1 antagonists should be reexamined. Accordingly, clinical trials revealed that polymodal TRPV1 antagonists (ABT-102, AZD1386, and V116517) cause an increase of body temperature, which was not shown in patients treated with mode-selective blocker NEO6860 [142]. In a Phase I clinical trial in which AMG517 is examined, subjects exhibited marked dose-dependent hyperthermia that might be related to the vasoconstriction and increasing thermogenesis [143]. Another potent and selective TRPV1 channel antagonist, JNJ-38893777, was tested in a single-center, double-blind, placebo-controlled, sequential group, single-ascending-dose phase 1 study. Results turned out promising as increases in body temperature or changes in Fridericia-corrected QT interval (QTcF) were not observed [144].
While systemic application of TRPV1 antagonist may face unexpected thermogenesis problems, topical administration could be a safer strategy. To treat itch-related skin problem, TRPV1-inhibitor 4-t-butylcyclohexanol based skin care cream has been tested to treat perioral dermatitis [145]. Perioral dermatitis is a skin problem and characterized by impaired skin barrier function that results in redness, dryness, burning, and pruritus. After TRPV1-inhibitor treatment, skin barrier function was surprisingly restored, featured as the decreased transepidermal water loss values and increased stratum corneum hydration [145]. These results support that topical application of TRPV1 antagonists is better tolerated.
More profound TRPV1 antagonists in pain- and itch-associated phenotypes have been tested in laboratory animals. For example, pharmacological blockade of TRPV1 using AMG9810 (a potent and selective TRPV1 antagonist [146, 147]) significantly decreased calcium responses in sensitized neurons, suggesting pharmacological effects in hyperalgesia conditions. Indeed, in an allergic conjunctivitis mouse model, antagonizing TRPV1 by AMG9810 could effectively disrupt histamine-dependent itch behavior [148], indicating the topical application of AMG9810 might be effective in dealing with ocular discomforts.
The most common concern about the overall antagonization of the TRPV1 channel is that the normal physiological function is likely disrupted. Thus, more studies are still needed to explore how to reserve TRPV1 physiological function when targeting pathologic pain and itch [149].
5.3 TRPV1 Activity-Dependent Silencing by QX-314
In addition to molecular antagonists, cell-specific design and synthesis may open a new avenue to save the protective role of pain and itch. The newly proof-of-concept to silence a specific activated nociceptive population has become available when QX-314 was introduced into the field. QX-314 is a charged, membrane-impermeant lidocaine derivative and can only penetrate into the neurons via a hydrophilic pore in the cell membrane. After entering cells, QX-314 sequentially blocks sodium channels to inhibit the overall neural activity and thus functions as local anesthetics. This specific characteristic enables the selective effect in which nociceptors are silenced via an activity-dependent manner. In 2007, Binshtok, Woolf, and colleagues generated a novel strategy to inhibit pain sensation [150]. They simultaneously applied QX-314 and capsaicin into rat hind paws or sciatic nerve and found that pain sensitivity was suppressed and the effect lasted for greater than 2 h without motor or tactile function impact [150]. This strategy opens a door to inhibit pain-responsible firing that is mediated by a broad range of factors. Since the opening of TRPV1 or/and TRPA1 channels is widely exhibited upon stimulation of pain mediators, which provides the critical condition of QX-314 entry. Besides for inflammatory mediators, pain induced by bacterial infections can also be blocked by QX-314. Three classes of PFTs-alpha-hemolysin (Hla), phenol-soluble modulins (PSMs), and the leukocidin HlgAB associated with methicillin-resistant Staphylococcus aureus (MRSA) have been identified to induce pain during the infection. Strikingly, QX-314 application revealed immediate and long-lasting blockade of pain sensation in a murine model infected by MRSA. Moreover, this approach is even more effective than analgesic reagents like lidocaine or ibuprofen [151].
Similarly, the effect of QX-314 was also confirmed in itch-related study. By applying QX-314 and histamine or chloroquine together, itch behavior was significantly inhibited [152]. Importantly, blocking itch-transmitting fibers did not reduce pain-associated behavior. Overall, this strategy not only helps identify the distinct populations for pain and itch mediators, but also provides a clinical anti-pruritic therapeutic approach for both histaminergic and non-histaminergic pruritus [152]. A specific case is the distinguishment of ocular pain and itch sensation. Although ocular discomforts can present as pain or itch, whether these signals are transmitted by the same or different sensory neurons remains not clear. Via tracing approaches, we found that pain and itch sensing sensory fibers have different anatomic territories. To be specific, while ocular itch is mediated by a subset of conjunctival-selective sensory fibers marked by MrgprA3, pain-sensing fibers were only observed in corneal innervations. By selectively silencing MrgprA3-expressing conjunctiva sensory fibers by using QX-314 and CQ (the agonist of MrgprA3), we observed that the itching behavior was markedly reduced in both pruritogen-stimulated and allergic-related conditions [153]. Taken together, these results indicate that TRPV1 activity-dependent silencing by QX-314 may have great significance in research and development for anti-pruritic medications.
6 Summary
The broad integration of physiological and pathophysiological of TRPV1 has been well studied and documented in pain- and itch-related fields. Over time, advanced research progresses emerge and indicate TRPV1 as a potential pharmaceutical target for treating pathologic pain and itch. The resolution of TRPV1 structure in the steady-state and the observation and mechanisms of dynamic change in TRPV1 upon stimulation makes it possible to modulate TRPV1 in a variety of conditions [3, 82]. Of note, although adjustment of expression level and the channel activity have revealed prosperity in pain and itch relieving, TRPV1 antagonists in clinical trials have encountered difficulties due to their adverse effects such as hyperthermia or hypothermia [142, 143]. Notwithstanding this, how to modulate TRPV1 activity in specific tissues and avoid affecting its basic physiological function remains an active research field to achieve the anti-pain and anti-itch effects. Excitingly, the novel concept of TRPV1 activity-dependent silencing by QX-314 has provided a promising approach to block the selective pain sensation. Over time, the consistent studies of TRPV1 in pain and itch will lead a new era to solve debilitating sensory disorders.
References
Venkatachalam K, Montell C (2007) TRP channels. Annu Rev Biochem 76:387–417
Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D (1997) The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389:816–824
Liao M, Cao E, Julius D, Cheng Y (2013) Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 504:107–112
Dhaka A, Uzzell V, Dubin AE, Mathur J, Petrus M, Bandell M, Patapoutian A (2009) TRPV1 is activated by both acidic and basic pH. J Neurosci 29:153–158
Kobayashi K, Fukuoka T, Obata K, Yamanaka H, Dai Y, Tokunaga A, Noguchi K (2005) Distinct expression of TRPM8, TRPA1, and TRPV1 mRNAs in rat primary afferent neurons with adelta/c-fibers and colocalization with trk receptors. J Comp Neurol 493:596–606
Kuruvilla M, Kalangara J, Lee FEE (2019) Neuropathic pain and itch mechanisms underlying allergic conjunctivitis. J Investig Allergol Clin Immunol 29:349–356
Jara-Oseguera A, Simon SA, Rosenbaum T (2008) TRPV1: on the road to pain relief. Curr Mol Pharmacol 1:255–269
McKemy DD (2011) A spicy family tree: TRPV1 and its thermoceptive and nociceptive lineage. EMBO J 30:453–455
Mishra SK, Tisel SM, Orestes P, Bhangoo SK, Hoon MA (2011) TRPV1-lineage neurons are required for thermal sensation. EMBO J 30:582–593
Kim YS, Chu Y, Han L, Li M, Li Z, LaVinka PC, Sun S, Tang Z, Park K, Caterina MJ, Ren K, Dubner R, Wei F, Dong X (2014) Central terminal sensitization of TRPV1 by descending serotonergic facilitation modulates chronic pain. Neuron 81:873–887
Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D (2000) Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 288:306–313
Raja SN, Carr DB, Cohen M, Finnerup NB, Flor H, Gibson S, Keefe FJ, Mogil JS, Ringkamp M, Sluka KA, Song XJ, Stevens B, Sullivan MD, Tutelman PR, Ushida T, Vader K (2020) The revised International Association for the Study of Pain definition of pain: concepts, challenges, and compromises. Pain 161:1976–1982
Ikoma A, Steinhoff M, Stander S, Yosipovitch G, Schmelz M (2006) The neurobiology of itch. Nat Rev Neurosci 7:535–547
Saloman JL, Chung MK, Ro JY (2013) P2X(3) and TRPV1 functionally interact and mediate sensitization of trigeminal sensory neurons. Neuroscience 232:226–238
Barabas ME, Stucky CL (2013) TRPV1, but not TRPA1, in primary sensory neurons contributes to cutaneous incision-mediated hypersensitivity. Mol Pain 9:9
Li CL, Li KC, Wu D, Chen Y, Luo H, Zhao JR, Wang SS, Sun MM, Lu YJ, Zhong YQ, Hu XY, Hou R, Zhou BB, Bao L, Xiao HS, Zhang X (2016) Somatosensory neuron types identified by high-coverage single-cell RNA-sequencing and functional heterogeneity. Cell Res 26:967
Li C, Wang S, Chen Y, Zhang X (2018) Somatosensory neuron typing with high-coverage single-cell RNA sequencing and functional analysis. Neurosci Bull 34:200–207
Usoskin D, Furlan A, Islam S, Abdo H, Lonnerberg P, Lou D, Hjerling-Leffler J, Haeggstrom J, Kharchenko O, Kharchenko PV, Linnarsson S, Ernfors P (2015) Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat Neurosci 18:145–153
Zeisel A, Hochgerner H, Lonnerberg P, Johnsson A, Memic F, van der Zwan J, Haring M, Braun E, Borm LE, La Manno G, Codeluppi S, Furlan A, Lee K, Skene N, Harris KD, Hjerling-Leffler J, Arenas E, Ernfors P, Marklund U, Linnarsson S (2018) Molecular architecture of the mouse nervous system. Cell 174:999–1014.e22
Treede RD, Rief W, Barke A, Aziz Q, Bennett MI, Benoliel R, Cohen M, Evers S, Finnerup NB, First MB, Giamberardino MA, Kaasa S, Kosek E, Lavand’homme P, Nicholas M, Perrot S, Scholz J, Schug S, Smith BH, Svensson P, Vlaeyen JWS, Wang SJ (2015) A classification of chronic pain for ICD-11. Pain 156:1003–1007
Treede RD, Rief W, Barke A, Aziz Q, Bennett MI, Benoliel R, Cohen M, Evers S, Finnerup NB, First MB, Giamberardino MA, Kaasa S, Korwisi B, Kosek E, Lavand’homme P, Nicholas M, Perrot S, Scholz J, Schug S, Smith BH, Svensson P, Vlaeyen JWS, Wang SJ (2019) Chronic pain as a symptom or a disease: the IASP Classification of Chronic Pain for the International Classification of Diseases (ICD-11). Pain 160:19–27
Nugraha B, Gutenbrunner C, Barke A, Karst M, Schiller J, Schafer P, Falter S, Korwisi B, Rief W, Treede RD (2019) Pain ITftCoC: the IASP classification of chronic pain for ICD-11: functioning properties of chronic pain. Pain 160:88–94
Basbaum AI, Bautista DM, Scherrer G, Julius D (2009) Cellular and molecular mechanisms of pain. Cell 139:267–284
Jordt SE, Tominaga M, Julius D (2000) Acid potentiation of the capsaicin receptor determined by a key extracellular site. Proc Natl Acad Sci U S A 97:8134–8139
Siemens J, Zhou S, Piskorowski R, Nikai T, Lumpkin EA, Basbaum AI, King D, Julius D (2006) Spider toxins activate the capsaicin receptor to produce inflammatory pain. Nature 444:208–212
Bohlen CJ, Priel A, Zhou S, King D, Siemens J, Julius D (2010) A bivalent tarantula toxin activates the capsaicin receptor, TRPV1, by targeting the outer pore domain. Cell 141:834–845
Caterina MJ, Julius D (2001) The vanilloid receptor: a molecular gateway to the pain pathway. Annu Rev Neurosci 24:487–517
Pogatzki-Zahn EM, Shimizu I, Caterina M, Raja SN (2005) Heat hyperalgesia after incision requires TRPV1 and is distinct from pure inflammatory pain. Pain 115:296–307
Chen SR, Pan HL (2006) Loss of TRPV1-expressing sensory neurons reduces spinal mu opioid receptors but paradoxically potentiates opioid analgesia. J Neurophysiol 95:3086–3096
Tang HB, Inoue A, Oshita K, Nakata Y (2004) Sensitization of vanilloid receptor 1 induced by bradykinin via the activation of second messenger signaling cascades in rat primary afferent neurons. Eur J Pharmacol 498:37–43
Abdelhamid RE, Sluka KA (2015) ASICs mediate pain and inflammation in musculoskeletal diseases. Physiology (Bethesda) 30:449–459
Bautista DM, Jordt SE, Nikai T, Tsuruda PR, Read AJ, Poblete J, Yamoah EN, Basbaum AI, Julius D (2006) TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell 124:1269–1282
Bautista DM, Movahed P, Hinman A, Axelsson HE, Sterner O, Hogestatt ED, Julius D, Jordt SE, Zygmunt PM (2005) Pungent products from garlic activate the sensory ion channel TRPA1. Proc Natl Acad Sci U S A 102:12248–12252
Macpherson LJ, Dubin AE, Evans MJ, Marr F, Schultz PG, Cravatt BF, Patapoutian A (2007) Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature 445:541–545
Trevisani M, Siemens J, Materazzi S, Bautista DM, Nassini R, Campi B, Imamachi N, Andre E, Patacchini R, Cottrell GS, Gatti R, Basbaum AI, Bunnett NW, Julius D, Geppetti P (2007) 4-Hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1. Proc Natl Acad Sci U S A 104:13519–13524
Taylor-Clark TE, McAlexander MA, Nassenstein C, Sheardown SA, Wilson S, Thornton J, Carr MJ, Undem BJ (2008) Relative contributions of TRPA1 and TRPV1 channels in the activation of vagal bronchopulmonary C-fibres by the endogenous autacoid 4-oxononenal. J Physiol 586:3447–3459
Bautista DM, Pellegrino M, Tsunozaki M (2013) TRPA1: a gatekeeper for inflammation. Annu Rev Physiol 75:181–200
Taylor-Clark TE, Undem BJ, Macglashan DW Jr, Ghatta S, Carr MJ, McAlexander MA (2008) Prostaglandin-induced activation of nociceptive neurons via direct interaction with transient receptor potential A1 (TRPA1). Mol Pharmacol 73:274–281
McMahon SB, Wood JN (2006) Increasingly irritable and close to tears: TRPA1 in inflammatory pain. Cell 124:1123–1125
Akopian AN, Ruparel NB, Jeske NA, Hargreaves KM (2007) Transient receptor potential TRPA1 channel desensitization in sensory neurons is agonist dependent and regulated by TRPV1-directed internalization. J Physiol 583:175–193
Weng HJ, Patel KN, Jeske NA, Bierbower SM, Zou W, Tiwari V, Zheng Q, Tang Z, Mo GC, Wang Y, Geng Y, Zhang J, Guan Y, Akopian AN, Dong X (2015) Tmem100 is a regulator of TRPA1-TRPV1 complex and contributes to persistent pain. Neuron 85:833–846
Moon EH, Kim MJ, Ko KS, Kim YS, Seo J, Oh SP, Lee YJ (2010) Generation of mice with a conditional and reporter allele for Tmem100. Genesis 48:673–678
Weyer AD, Stucky CL (2015) Loosening pain’s grip by tightening TRPV1-TRPA1 interactions. Neuron 85:661–663
Sun YG, Chen ZF (2007) A gastrin-releasing peptide receptor mediates the itch sensation in the spinal cord. Nature 448:700–703
Liu Q, Tang Z, Surdenikova L, Kim S, Patel KN, Kim A, Ru F, Guan Y, Weng HJ, Geng Y, Undem BJ, Kollarik M, Chen ZF, Anderson DJ, Dong X (2009) Sensory neuron-specific GPCR Mrgprs are itch receptors mediating chloroquine-induced pruritus. Cell 139:1353–1365
Dong X, Han S, Zylka MJ, Simon MI, Anderson DJ (2001) A diverse family of GPCRs expressed in specific subsets of nociceptive sensory neurons. Cell 106:619–632
Han L, Ma C, Liu Q, Weng HJ, Cui Y, Tang Z, Kim Y, Nie H, Qu L, Patel KN, Li Z, McNeil B, He S, Guan Y, Xiao B, Lamotte RH, Dong X (2013) A subpopulation of nociceptors specifically linked to itch. Nat Neurosci 16:174–182
Liu Q, Sikand P, Ma C, Tang Z, Han L, Li Z, Sun S, LaMotte RH, Dong X (2012) Mechanisms of itch evoked by β-alanine. J Neurosci 32:14532–14537
Mishra SK, Hoon MA (2010) Ablation of TrpV1 neurons reveals their selective role in thermal pain sensation. Mol Cell Neurosci 43:157–163
Cevikbas F, Wang X, Akiyama T, Kempkes C, Savinko T, Antal A, Kukova G, Buhl T, Ikoma A, Buddenkotte J, Soumelis V, Feld M, Alenius H, Dillon SR, Carstens E, Homey B, Basbaum A, Steinhoff M (2014) A sensory neuron-expressed IL-31 receptor mediates T helper cell-dependent itch: involvement of TRPV1 and TRPA1. J Allergy Clin Immunol 133:448–460
Meng J, Moriyama M, Feld M, Buddenkotte J, Buhl T, Szollosi A, Zhang J, Miller P, Ghetti A, Fischer M, Reeh PW, Shan C, Wang J, Steinhoff M (2018) New mechanism underlying IL-31-induced atopic dermatitis. J Allergy Clin Immunol 141:1677–1689.e8
Wang F, Trier AM, Li F, Kim S, Chen Z, Chai JN, Mack MR, Morrison SA, Hamilton JD, Baek J, Yang TB, Ver Heul AM, Xu AZ, Xie Z, Dong X, Kubo M, Hu H, Hsieh CS, Dong X, Liu Q, Margolis DJ, Ardeleanu M, Miller MJ, Kim BS (2021) A basophil-neuronal axis promotes itch. Cell 184:422–440.e17
Dimitriadou V, Rouleau A, Dam Trung Tuong M, Newlands GJ, Miller HR, Luffau G, Schwartz JC, Garbarg M (1994) Functional relationship between mast cells and C-sensitive nerve fibres evidenced by histamine H3-receptor modulation in rat lung and spleen. Clin Sci (Lond) 87:151–163
Shim WS, Tak MH, Lee MH, Kim M, Kim M, Koo JY, Lee CH, Kim M, Oh U (2007) TRPV1 mediates histamine-induced itching via the activation of phospholipase A2 and 12-lipoxygenase. J Neurosci 27:2331–2337
Mishra SK, Hoon MA (2013) The cells and circuitry for itch responses in mice. Science 340:968–971
Campion M, Smith L, Gatault S, Metais C, Buddenkotte J, Steinhoff M (2019) Interleukin-4 and interleukin-13 evoke scratching behaviour in mice. Exp Dermatol 28:1501–1504
Oetjen LK, Mack MR, Feng J, Whelan TM, Niu H, Guo CJ, Chen S, Trier AM, Xu AZ, Tripathi SV, Luo J, Gao X, Yang L, Hamilton SL, Wang PL, Brestoff JR, Council ML, Brasington R, Schaffer A, Brombacher F, Hsieh CS, Gereau RW, Miller MJ, Chen ZF, Hu H, Davidson S, Liu Q, Kim BS (2017) Sensory neurons co-opt classical immune signaling pathways to mediate chronic itch. Cell 171:217–228.e13
Stanger R, Rivera-Oyola R, Lebwohl M (2020) Dupilumab as a treatment for generalized idiopathic pruritus: a report of two cases. Br J Dermatol 182:1494–1495
Zhai LL, Savage KT, Qiu CC, Jin A, Valdes-Rodriguez R, Mollanazar NK (2019) Chronic pruritus responding to dupilumab-a case series. Medicines (Basel) 6(3):72
Calugareanu A, Jachiet M, Tauber M, Nosbaum A, Aubin F, Misery L, Droitcourt C, Barbarot S, Debarbieux S, Saussine A, Bagot M, de Masson A, Seneschal J, Staumont-Salle D, Bouaziz JD, French Group of Research and Study in Atopic Dermatitis from the French Society of Dermatology (2020) Effectiveness and safety of dupilumab for the treatment of prurigo nodularis in a French multicenter adult cohort of 16 patients. J Eur Acad Dermatol Venereol 34:e74–e76
Giura MT, Viola R, Fierro MT, Ribero S, Ortoncelli M (2020) Efficacy of dupilumab in prurigo nodularis in elderly patient. Dermatol Ther 33:e13201
Napolitano M, Fabbrocini G, Scalvenzi M, Nistico SP, Dastoli S, Patruno C (2020) Effectiveness of dupilumab for the treatment of generalized prurigo nodularis phenotype of adult atopic dermatitis. Dermatitis 31:81–84
Tanis R, Ferenczi K, Payette M (2019) Dupilumab treatment for prurigo nodularis and pruritis. J Drugs Dermatol 18:940–942
Jeon J, Wang F, Badic A, Kim BS (2021) Treatment of patients with chronic pruritus of unknown origin with dupilumab. J Dermatolog Treat:1–4
Imamachi N, Park GH, Lee H, Anderson DJ, Simon MI, Basbaum AI, Han SK (2009) TRPV1-expressing primary afferents generate behavioral responses to pruritogens via multiple mechanisms. Proc Natl Acad Sci U S A 106:11330–11335
Kim BM, Lee SH, Shim WS, Oh U (2004) Histamine-induced Ca(2+) influx via the PLA(2)/lipoxygenase/TRPV1 pathway in rat sensory neurons. Neurosci Lett 361:159–162
Han SK, Mancino V, Simon MI (2006) Phospholipase Cbeta 3 mediates the scratching response activated by the histamine H1 receptor on C-fiber nociceptive neurons. Neuron 52:691–703
Jian T, Yang N, Yang Y, Zhu C, Yuan X, Yu G, Wang C, Wang Z, Shi H, Tang M, He Q, Lan L, Wu G, Tang Z (2016) TRPV1 and PLC participate in histamine H4 receptor-induced itch. Neural Plast 2016:1682972
Villarino AV, Kanno Y, O’Shea JJ (2017) Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat Immunol 18:374–384
Wang F, Morris C, Bodet ND, Kim BS (2019) Treatment of refractory chronic pruritus of unknown origin with tofacitinib in patients with rheumatoid arthritis. JAMA Dermatol 155(12):1426–1428
Salmon JA, Higgs GA (1987) Prostaglandins and leukotrienes as inflammatory mediators. Br Med Bull 43:285–296
O’Byrne PM, Israel E, Drazen JM (1997) Antileukotrienes in the treatment of asthma. Ann Intern Med 127:472–480
Luster AD, Tager AM (2004) T-cell trafficking in asthma: lipid mediators grease the way. Nat Rev Immunol 4:711–724
Andoh T, Kuraishi Y (1998) Intradermal leukotriene B4, but not prostaglandin E2, induces itch-associated responses in mice. Eur J Pharmacol 353:93–96
Fernandes ES, Vong CT, Quek S, Cheong J, Awal S, Gentry C, Aubdool AA, Liang L, Bodkin JV, Bevan S, Heads R, Brain SD (2013) Superoxide generation and leukocyte accumulation: key elements in the mediation of leukotriene B(4)-induced itch by transient receptor potential ankyrin 1 and transient receptor potential vanilloid 1. FASEB J 27:1664–1673
Solinski HJ, Kriegbaum MC, Tseng PY, Earnest TW, Gu X, Barik A, Chesler AT, Hoon MA (2019) Nppb neurons are sensors of mast cell-induced itch. Cell Rep 26:3561–3573.e4
Voisin T, Perner C, Messou MA, Shiers S, Ualiyeva S, Kanaoka Y, Price TJ, Sokol CL, Bankova LG, Austen KF, Chiu IM (2021) The CysLT2R receptor mediates leukotriene C4-driven acute and chronic itch. Proc Natl Acad Sci U S A 118(13):e2022087118
Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ (2002) p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron 36:57–68
Cheng JK, Ji RR (2008) Intracellular signaling in primary sensory neurons and persistent pain. Neurochem Res 33:1970–1978
Li F, Yang W, Jiang H, Guo C, Huang AJW, Hu H, Liu Q (2019) TRPV1 activity and substance P release are required for corneal cold nociception. Nat Commun 10:5678
Yu G, Yang N, Li F, Chen M, Guo CJ, Wang C, Hu D, Yang Y, Zhu C, Wang Z, Shi H, Gegen T, Tang M, He Q, Liu Q, Tang Z (2016) Enhanced itch elicited by capsaicin in a chronic itch model. Mol Pain 12:1744806916645349
Cao E, Liao M, Cheng Y, Julius D (2013) TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 504:113–118
Lishko PV, Procko E, Jin X, Phelps CB, Gaudet R (2007) The ankyrin repeats of TRPV1 bind multiple ligands and modulate channel sensitivity. Neuron 54:905–918
Salzer I, Ray S, Schicker K, Boehm S (2019) Nociceptor signalling through ion channel regulation via GPCRs. Int J Mol Sci 20(10):2488
Zhang N, Inan S, Cowan A, Sun R, Wang JM, Rogers TJ, Caterina M, Oppenheim JJ (2005) A proinflammatory chemokine, CCL3, sensitizes the heat- and capsaicin-gated ion channel TRPV1. Proc Natl Acad Sci U S A 102:4536–4541
Chung MK, Guler AD, Caterina MJ (2008) TRPV1 shows dynamic ionic selectivity during agonist stimulation. Nat Neurosci 11:555–564
Touska F, Marsakova L, Teisinger J, Vlachova V (2011) A “cute” desensitization of TRPV1. Curr Pharm Biotechnol 12:122–129
Cao E, Cordero-Morales JF, Liu B, Qin F, Julius D (2013) TRPV1 channels are intrinsically heat sensitive and negatively regulated by phosphoinositide lipids. Neuron 77:667–679
Sun X, Zakharian E (2015) Regulation of the temperature-dependent activation of transient receptor potential vanilloid 1 (TRPV1) by phospholipids in planar lipid bilayers. J Biol Chem 290:4741–4747
Xiao R, Xu XZS (2021) Temperature sensation: from molecular thermosensors to neural circuits and coding principles. Annu Rev Physiol 83:205–230
Mohapatra DP, Nau C (2003) Desensitization of capsaicin-activated currents in the vanilloid receptor TRPV1 is decreased by the cyclic AMP-dependent protein kinase pathway. J Biol Chem 278:50080–50090
Vetter I, Wyse BD, Monteith GR, Roberts-Thomson SJ, Cabot PJ (2006) The mu opioid agonist morphine modulates potentiation of capsaicin-evoked TRPV1 responses through a cyclic AMP-dependent protein kinase A pathway. Mol Pain 2:22
Spahn V, Fischer O, Endres-Becker J, Schafer M, Stein C, Zollner C (2013) Opioid withdrawal increases transient receptor potential vanilloid 1 activity in a protein kinase A-dependent manner. Pain 154:598–608
Baron EP, Lucas P, Eades J, Hogue O (2018) Patterns of medicinal cannabis use, strain analysis, and substitution effect among patients with migraine, headache, arthritis, and chronic pain in a medicinal cannabis cohort. J Headache Pain 19:37
Freitas HR, Isaac AR, Malcher-Lopes R, Diaz BL, Trevenzoli IH, De Melo Reis RA (2018) Polyunsaturated fatty acids and endocannabinoids in health and disease. Nutr Neurosci 21:695–714
Arnold WR, Carnevale LN, Xie Z, Baylon JL, Tajkhorshid E, Hu H, Das A (2021) Anti-inflammatory dopamine- and serotonin-based endocannabinoid epoxides reciprocally regulate cannabinoid receptors and the TRPV1 channel. Nat Commun 12:926
Por ED, Samelson BK, Belugin S, Akopian AN, Scott JD, Jeske NA (2010) PP2B/calcineurin-mediated desensitization of TRPV1 does not require AKAP150. Biochem J 432:549–556
Jung J, Shin JS, Lee SY, Hwang SW, Koo J, Cho H, Oh U (2004) Phosphorylation of vanilloid receptor 1 by Ca2+/calmodulin-dependent kinase II regulates its vanilloid binding. J Biol Chem 279:7048–7054
Zhang X, Daugherty SL, de Groat WC (2011) Activation of CaMKII and ERK1/2 contributes to the time-dependent potentiation of Ca2+ response elicited by repeated application of capsaicin in rat DRG neurons. Am J Physiol Regul Integr Comp Physiol 300:R644–R654
Ugawa S, Ueda T, Yamamura H, Shimada S (2005) In situ hybridization evidence for the coexistence of ASIC and TRPV1 within rat single sensory neurons. Brain Res Mol Brain Res 136:125–133
Kim AY, Tang Z, Liu Q, Patel KN, Maag D, Geng Y, Dong X (2008) Pirt, a phosphoinositide-binding protein, functions as a regulatory subunit of TRPV1. Cell 133:475–485
Wang C, Gu L, Ruan Y, Gegen T, Yu L, Zhu C, Yang Y, Zhou Y, Yu G, Tang Z (2018) Pirt together with TRPV1 is involved in the regulation of neuropathic pain. Neural Plast 2018:4861491
Wang C, Wang Z, Yang Y, Zhu C, Wu G, Yu G, Jian T, Yang N, Shi H, Tang M, He Q, Lan L, Liu Q, Guan Y, Dong X, Duan J, Tang Z (2015) Pirt contributes to uterine contraction-induced pain in mice. Mol Pain 11:57
Patel KN, Liu Q, Meeker S, Undem BJ, Dong X (2011) Pirt, a TRPV1 modulator, is required for histamine-dependent and -independent itch. PLoS One 6:e20559
Hanack C, Moroni M, Lima WC, Wende H, Kirchner M, Adelfinger L, Schrenk-Siemens K, Tappe-Theodor A, Wetzel C, Kuich PH, Gassmann M, Roggenkamp D, Bettler B, Lewin GR, Selbach M, Siemens J (2015) GABA blocks pathological but not acute TRPV1 pain signals. Cell 160:759–770
Shimada SG, LaMotte RH (2008) Behavioral differentiation between itch and pain in mouse. Pain 139:681–687
Akiyama T, Carstens MI, Carstens E (2010) Differential itch- and pain-related behavioral responses and micro-opoid modulation in mice. Acta Derm Venereol 90:575–581
Fu K, Qu L, Shimada SG, Nie H, LaMotte RH (2014) Enhanced scratching elicited by a pruritogen and an algogen in a mouse model of contact hypersensitivity. Neurosci Lett 579:190–194
Sikand P, Shimada SG, Green BG, LaMotte RH (2009) Similar itch and nociceptive sensations evoked by punctate cutaneous application of capsaicin, histamine and cowhage. Pain 144:66–75
Sikand P, Shimada SG, Green BG, LaMotte RH (2011) Sensory responses to injection and punctate application of capsaicin and histamine to the skin. Pain 152:2485–2494
Akiyama T, Tominaga M, Takamori K, Carstens MI, Carstens E (2014) Roles of glutamate, substance P, and gastrin-releasing peptide as spinal neurotransmitters of histaminergic and nonhistaminergic itch. Pain 155:80–92
Brumovsky P, Watanabe M, Hokfelt T (2007) Expression of the vesicular glutamate transporters-1 and -2 in adult mouse dorsal root ganglia and spinal cord and their regulation by nerve injury. Neuroscience 147:469–490
Sun YG, Zhao ZQ, Meng XL, Yin J, Liu XY, Chen ZF (2009) Cellular basis of itch sensation. Science 325:1531–1534
Carstens EE, Carstens MI, Simons CT, Jinks SL (2010) Dorsal horn neurons expressing NK-1 receptors mediate scratching in rats. Neuroreport 21:303–308
Liu Y, Abdel Samad O, Zhang L, Duan B, Tong Q, Lopes C, Ji RR, Lowell BB, Ma Q (2010) VGLUT2-dependent glutamate release from nociceptors is required to sense pain and suppress itch. Neuron 68:543–556
Achanta S, Chintagari NR, Brackmann M, Balakrishna S, Jordt SE (2018) TRPA1 and CGRP antagonists counteract vesicant-induced skin injury and inflammation. Toxicol Lett 293:140–148
McNeil BD, Pundir P, Meeker S, Han L, Undem BJ, Kulka M, Dong X (2015) Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature 519:237–241
Meixiong J, Anderson M, Limjunyawong N, Sabbagh MF, Hu E, Mack MR, Oetjen LK, Wang F, Kim BS, Dong X (2019) Activation of mast-cell-expressed Mas-related G-protein-coupled receptors drives non-histaminergic itch. Immunity 50:1163–1171.e5
Serhan N, Basso L, Sibilano R, Petitfils C, Meixiong J, Bonnart C, Reber LL, Marichal T, Starkl P, Cenac N, Dong X, Tsai M, Galli SJ, Gaudenzio N (2019) House dust mites activate nociceptor-mast cell clusters to drive type 2 skin inflammation. Nat Immunol 20:1435–1443
Chiu IM, Heesters BA, Ghasemlou N, Von Hehn CA, Zhao F, Tran J, Wainger B, Strominger A, Muralidharan S, Horswill AR, Bubeck Wardenburg J, Hwang SW, Carroll MC, Woolf CJ (2013) Bacteria activate sensory neurons that modulate pain and inflammation. Nature 501:52–57
Baral P, Umans BD, Li L, Wallrapp A, Bist M, Kirschbaum T, Wei Y, Zhou Y, Kuchroo VK, Burkett PR, Yipp BG, Liberles SD, Chiu IM (2018) Nociceptor sensory neurons suppress neutrophil and gammadelta T cell responses in bacterial lung infections and lethal pneumonia. Nat Med 24:417–426
Pinho-Ribeiro FA, Baddal B, Haarsma R, O’Seaghdha M, Yang NJ, Blake KJ, Portley M, Verri WA, Dale JB, Wessels MR, Chiu IM (2018) Blocking neuronal signaling to immune cells treats streptococcal invasive infection. Cell 173:1083–1097.e22
Spits H, Di Santo JP (2011) The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol 12:21–27
McKenzie AN (2014) Type-2 innate lymphoid cells in asthma and allergy. Ann Am Thorac Soc 11(Suppl 5):S263–S270
Talbot S, Abdulnour RE, Burkett PR, Lee S, Cronin SJ, Pascal MA, Laedermann C, Foster SL, Tran JV, Lai N, Chiu IM, Ghasemlou N, DiBiase M, Roberson D, Von Hehn C, Agac B, Haworth O, Seki H, Penninger JM, Kuchroo VK, Bean BP, Levy BD, Woolf CJ (2015) Silencing nociceptor neurons reduces allergic airway inflammation. Neuron 87:341–354
Cardoso V, Chesne J, Ribeiro H, Garcia-Cassani B, Carvalho T, Bouchery T, Shah K, Barbosa-Morais NL, Harris N, Veiga-Fernandes H (2017) Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature 549:277–281
Wallrapp A, Riesenfeld SJ, Burkett PR, Abdulnour RE, Nyman J, Dionne D, Hofree M, Cuoco MS, Rodman C, Farouq D, Haas BJ, Tickle TL, Trombetta JJ, Baral P, Klose CSN, Mahlakoiv T, Artis D, Rozenblatt-Rosen O, Chiu IM, Levy BD, Kowalczyk MS, Regev A, Kuchroo VK (2017) The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature 549:351–356
Klose CSN, Mahlakoiv T, Moeller JB, Rankin LC, Flamar AL, Kabata H, Monticelli LA, Moriyama S, Putzel GG, Rakhilin N, Shen X, Kostenis E, Konig GM, Senda T, Carpenter D, Farber DL, Artis D (2017) The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature 549:282–286
Inclan-Rico JM, Ponessa JJ, Valero-Pacheco N, Hernandez CM, Sy CB, Lemenze AD, Beaulieu AM, Siracusa MC (2020) Basophils prime group 2 innate lymphoid cells for neuropeptide-mediated inhibition. Nat Immunol 21:1181–1193
Nussbaum JC, Van Dyken SJ, von Moltke J, Cheng LE, Mohapatra A, Molofsky AB, Thornton EE, Krummel MF, Chawla A, Liang HE, Locksley RM (2013) Type 2 innate lymphoid cells control eosinophil homeostasis. Nature 502:245–248
Kashem SW, Riedl MS, Yao C, Honda CN, Vulchanova L, Kaplan DH (2015) Nociceptive sensory fibers drive interleukin-23 production from CD301b+ dermal dendritic cells and drive protective cutaneous immunity. Immunity 43:515–526
Perner C, Flayer CH, Zhu X, Aderhold PA, Dewan ZNA, Voisin T, Camire RB, Chow OA, Chiu IM, Sokol CL (2020) Substance P release by sensory neurons triggers dendritic cell migration and initiates the type-2 immune response to allergens. Immunity 53:1063–1077.e7
Cohen JA, Edwards TN, Liu AW, Hirai T, Jones MR, Wu J, Li Y, Zhang S, Ho J, Davis BM, Albers KM, Kaplan DH (2019) Cutaneous TRPV1(+) neurons trigger protective innate type 17 anticipatory immunity. Cell 178:919–932.e14
Anand P, Elsafa E, Privitera R, Naidoo K, Yiangou Y, Donatien P, Gabra H, Wasan H, Kenny L, Rahemtulla A, Misra P (2019) Rational treatment of chemotherapy-induced peripheral neuropathy with capsaicin 8% patch: from pain relief towards disease modification. J Pain Res 12:2039–2052
Cabezon-Gutierrez L, Custodio-Cabello S, Palka-Kotlowska M, Khosravi-Shahi P (2020) High-dose 8% capsaicin patch in treatment of chemotherapy-induced peripheral neuropathy. A systematic review. J Pain Symptom Manage 60:1047–1054.e1
Abrams RMC, Pedowitz EJ, Simpson DM (2021) A critical review of the capsaicin 8% patch for the treatment of neuropathic pain associated with diabetic peripheral neuropathy of the feet in adults. Expert Rev Neurother 21:259–266
Galvez R, Navez ML, Moyle G, Maihofner C, Stoker M, Ernault E, Nurmikko TJ, Attal N (2017) Capsaicin 8% patch repeat treatment in nondiabetic peripheral neuropathic pain: a 52-week, open-label, single-arm, safety study. Clin J Pain 33:921–931
Ruivo EFM, Gestosa SVS, Mulenas NME, Lares AMG (2020) Peripheral neuropathy associated with hypereosinophilic syndrome: a clinical therapeutic success with capsaicin 8% patch. J Pain Palliat Care Pharmacother 34:155–158
Campbell JN, Stevens R, Hanson P, Connolly J, Meske DS, Chung MK, Lascelles BDX (2021) Injectable capsaicin for the management of pain due to osteoarthritis. Molecules 26(4):778
Rami HK, Thompson M, Stemp G, Fell S, Jerman JC, Stevens AJ, Smart D, Sargent B, Sanderson D, Randall AD, Gunthorpe MJ, Davis JB (2006) Discovery of SB-705498: a potent, selective and orally bioavailable TRPV1 antagonist suitable for clinical development. Bioorg Med Chem Lett 16:3287–3291
Chizh BA, O’Donnell MB, Napolitano A, Wang J, Brooke AC, Aylott MC, Bullman JN, Gray EJ, Lai RY, Williams PM, Appleby JM (2007) The effects of the TRPV1 antagonist SB-705498 on TRPV1 receptor-mediated activity and inflammatory hyperalgesia in humans. Pain 132:132–141
Garami A, Shimansky YP, Rumbus Z, Vizin RCL, Farkas N, Hegyi J, Szakacs Z, Solymar M, Csenkey A, Chiche DA, Kapil R, Kyle DJ, Van Horn WD, Hegyi P, Romanovsky AA (2020) Hyperthermia induced by transient receptor potential vanilloid-1 (TRPV1) antagonists in human clinical trials: insights from mathematical modeling and meta-analysis. Pharmacol Ther 208:107474
Gavva NR, Treanor JJ, Garami A, Fang L, Surapaneni S, Akrami A, Alvarez F, Bak A, Darling M, Gore A, Jang GR, Kesslak JP, Ni L, Norman MH, Palluconi G, Rose MJ, Salfi M, Tan E, Romanovsky AA, Banfield C, Davar G (2008) Pharmacological blockade of the vanilloid receptor TRPV1 elicits marked hyperthermia in humans. Pain 136:202–210
Manitpisitkul P, Mayorga A, Shalayda K, De Meulder M, Romano G, Jun C, Moyer JA (2015) Safety, tolerability and pharmacokinetic and pharmacodynamic learnings from a double-blind, randomized, placebo-controlled, sequential group first-in-human study of the TRPV1 antagonist, JNJ-38893777, in healthy men. Clin Drug Investig 35:353–363
Srour J, Bengel J, Linden T, Jovanovic Z, Roggenkamp D, Reinholz M, Rothenberger C, Neufang G, Wollenberg A (2020) Efficacy of a skin care cream with TRPV1 inhibitor 4-t-butylcyclohexanol in the topical therapy of perioral dermatitis. J Cosmet Dermatol 19:1409–1414
Gavva NR, Tamir R, Qu Y, Klionsky L, Zhang TJ, Immke D, Wang J, Zhu D, Vanderah TW, Porreca F, Doherty EM, Norman MH, Wild KD, Bannon AW, Louis JC, Treanor JJ (2005) AMG 9810 [(E)-3-(4-t-butylphenyl)-N-(2,3-dihydrobenzo[b][1,4] dioxin-6-yl)acrylamide], a novel vanilloid receptor 1 (TRPV1) antagonist with antihyperalgesic properties. J Pharmacol Exp Ther 313:474–484
Norman MH, Zhu J, Fotsch C, Bo Y, Chen N, Chakrabarti P, Doherty EM, Gavva NR, Nishimura N, Nixey T, Ognyanov VI, Rzasa RM, Stec M, Surapaneni S, Tamir R, Viswanadhan VN, Treanor JJ (2007) Novel vanilloid receptor-1 antagonists: 1. Conformationally restricted analogues of trans-cinnamides. J Med Chem 50:3497–3514
Huang CC, Kim YS, Olson WP, Li F, Guo C, Luo W, Huang AJW, Liu Q (2016) A histamine-independent itch pathway is required for allergic ocular itch. J Allergy Clin Immunol 137:1267–1270.e6
Moore C, Gupta R, Jordt SE, Chen Y, Liedtke WB (2018) Regulation of pain and itch by TRP channels. Neurosci Bull 34:120–142
Binshtok AM, Bean BP, Woolf CJ (2007) Inhibition of nociceptors by TRPV1-mediated entry of impermeant sodium channel blockers. Nature 449:607–610
Blake KJ, Baral P, Voisin T, Lubkin A, Pinho-Ribeiro FA, Adams KL, Roberson DP, Ma YC, Otto M, Woolf CJ, Torres VJ, Chiu IM (2018) Staphylococcus aureus produces pain through pore-forming toxins and neuronal TRPV1 that is silenced by QX-314. Nat Commun 9:37
Roberson DP, Gudes S, Sprague JM, Patoski HA, Robson VK, Blasl F, Duan B, Oh SB, Bean BP, Ma Q, Binshtok AM, Woolf CJ (2013) Activity-dependent silencing reveals functionally distinct itch-generating sensory neurons. Nat Neurosci 16:910–918
Huang CC, Yang W, Guo C, Jiang H, Li F, Xiao M, Davidson S, Yu G, Duan B, Huang T, Huang AJW, Liu Q (2018) Anatomical and functional dichotomy of ocular itch and pain. Nat Med 24:1268–1276
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Li, F., Wang, F. (2021). TRPV1 in Pain and Itch. In: Zhou, L. (eds) Ion Channels in Biophysics and Physiology. Advances in Experimental Medicine and Biology, vol 1349. Springer, Singapore. https://doi.org/10.1007/978-981-16-4254-8_12
Download citation
DOI: https://doi.org/10.1007/978-981-16-4254-8_12
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-4253-1
Online ISBN: 978-981-16-4254-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)