
Abstract
Since the discovery of the significance of the cholesterol-carrying apolipoprotein E and cholesterolaemia as major risk factors for Alzheimer’s Disease (AD) there has been a mounting interest in the role of this lipid as a possible pathogenic agent. In this review we analyse the current evidence linking cholesterol metabolism and regulation in the CNS with the known mechanisms underlying the development of Alzheimer’s Disease. Cholesterol is known to affect amyloid-β generation and toxicity, although it must be considered that the results studies using the statin class of drugs to lower plasma cholesterol may be affected by other effects associated with these drugs. Finally, we report some of our results pointing at the interplay between neurons and astrocytes and NADPH oxidase activation as a new candidate mechanism linking cholesterol and AD pathology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s Disease (AD) is the most common form of dementia. It is characterized by deposition in the brain of neuritic plaques, mainly constituted of masses of fibrillary amyloid β (Aβ) peptide, and surrounded by dystrophic neurites (rich in phosphorylated tau), activated astrocytes and microglia. Apart from the small percentage of cases where there is a clear familial component due to a mutation in the genes either for the amyloid precursor protein (APP) or presenilins, for the vast majority of sporadic cases the primary causes of the disease remains elusive. Aβ is thought to be associated with neurodegeneration, and is neurotoxic in vitro and in vivo, but its exact role in the development of the disease is not fully understood. It has been linked with an increase in oxidative stress, dysregulation of calcium dynamics and inhibition of the activity of some enzymes, possibly through interaction with cellular membrane structures [1–3].
The cholesterol connection
Apart from mutations in the proteins involved in Aβ generation (APP, presenilins), the strongest known risk factor influencing the incidence of sporadic AD is the genotype for apolipoprotein E (ApoE), the major carrier of cholesterol in the CNS. Individuals carrying one or two copies of the ApoE ε4 allele have a higher risk of developing the disease [4], compared to those carrying the ε3 (the most common) or ε2 (which appears to be protective) forms. The “Rotterdam study” found an association between atherosclerosis and dementia, which was particularly strong in those with the apoE ε4 genotype [5]. In addition, it has been observed that patients with cardiovascular disease undergoing cholesterol lowering therapy with cholesterol synthesis inhibitors (statins) have a lower risk of developing AD [6, 7]. This has led to the hypothesis, now supported by further evidence, that cholesterol is somehow involved in AD pathogenesis, although the mechanism is at present not clear.
In this review we will focus on the growing evidence of an involvement of brain cholesterol in the pathogenesis of AD with particular reference to its interactions with Aβ.
Cholesterol metabolism
Cholesterol is a crucial component of mammalian membranes, as well as being an important precursor of steroid hormones and bile acids. The complex structure of cholesterol confers rigidity to lipid bilayers, affecting the fluidity, permeability and thickness of cell membranes. It is fundamental for the functionality of the membranes and many membrane-associated proteins (reviewed in [8]). Cholesterol is mainly concentrated in the detergent-resistant dynamic liquid-ordered domains in the plasma membrane called lipid rafts, which are also rich in sphingolipids and saturated phospholipids [9]. Cholesterol is also present in caveolae, membrane invaginations rich in the protein caveolin [10]. A number of proteins involved in signal transduction, cell adhesion and other functions are associated with the rafts, including the amyloid precursor protein and γ-secretase, one of the enzymes involved in APP cleavage to generate Aβ. Membranes of intracellular organelles such as the ER and mitochondria typically have low levels of cholesterol [11].
Cholesterol in the body originates partly from the diet, and partly from de novo synthesis, mainly in the liver and intestine. The biosynthetic pathway also generates intermediates used in the synthesis of ubiquinone and in the prenylation of proteins. The first steps of cholesterol synthesis (Fig. 1) are the conversion of three molecules of acetyl-CoA into one 3-hydroxy-3-methyglutaryl-CoA (HMG-CoA) in the cytoplasm. Then HMG-CoA is converted to mevalonate by the HMG-CoA reductase (HMGR), associated with the ER. This is the rate-limiting step, and is subject to complex regulation. High levels of cholesterol exert control by feedback inhibition and by stimulating HMGR ubiquitination and degradation by the proteasome. When levels are low, cleavage of the ER-bound sterol regulated element binding protein (SREBP) induces the generation of transcription factors which bind to the sterol regulatory element (SRE-1) which in turn controls the transcription of HMGR and other genes involved in the metabolism and transport of cholesterol and other lipids (Fig. 2).

Schematic diagram of the first steps of cholesterol biosynthesis

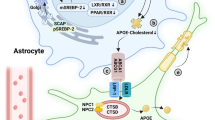
Schematic diagram of cholesterol metabolism in a model cell. Cholesterol bound to ApoE in LDLs is taken up into the cell via a LDLR-dependent mechanism, transported through the endosomal system and delivered to the ER via NPC-1. Endogenous synthesis of cholesterol also occurs in the ER. Excess cholesterol inhibits further synthesis by inhibiting HMGR while low levels stimulate cleavage of SRBP in the ER, to generate transcription factors which induce SRE-1-dependent transcription of HMGR, LRLR and other genes involved in cholesterol metabolism. Excess cholesterol can also be esterified by ACAT, oxidised to 24-OHC by CYP-46 (in neurons) or released together with ApoE (in astrocytes). ApoE transcription is enhanced by the nuclear LXR receptors, which are activated by 24-OHC
Cholesterol is transported in the serum mainly in esterified form, bound to apolipoproteins ApoB and ApoE, in low-density lipoproteins (LDLs). These deliver the lipid to cells by desorption or receptor-mediated internalization. Once in the cells it passes through the endosome system via a mechanism involving NPC1, the protein responsible for Niemann-Pick type C disease (NPC), a disorder which shares several features with AD (see below). Internalised cholesterol can then be cycled to the plasma membrane via vesicular transport [12].
Excess cholesterol is esterified in the ER by the Acyl-CoA acyltransferase (ACAT) and stored as lipid droplets, or released via a mechanism involving the ATP-binding cassette transporter ABCA-1. Cholesterol released in the serum is transported in high density lipoproteins (HDLs), and taken up by the liver which excretes it in the form of bile salts.
Cholesterol in the CNS
Cholesterol has a fundamental role in brain development and function. The CNS is particularly rich in cholesterol, containing a quarter of the total body content of this lipid, despite representing only 2% of body weight.
Brain cholesterol appears to be largely independent and unaffected by the serum levels [13], being impermeable to the blood brain barrier. Accordingly, cholesterol introduced with the diet has little or no impact on brain cholesterol, which is largely synthesised de novo within the organ.
Cholesterol turnover in the brain is slow, the half-life being estimated around 4–6 months in rodents [14] and 5 years in humans [15]. Release from the brain occurs only after oxidation to 24(S)-hydroxycholesterol (24-OHC) by the cholesterol 24-hydroxylase (also known as CYP46, a cytochrome P450 family member), a brain-specific, neuronal enzyme [16]. 24-OHC can cross the blood-brain barrier into the plasma, and is subsequently excreted by the liver. Oxysterols also have important regulatory functions on cholesterol metabolism within the brain. Circulating 24-OHC derives almost exclusively from the brain and is used as a marker of brain cholesterol metabolism [15]. Cholesterol metabolism in the brain has been recently reviewed by Dietschy [17].
Brain cholesterol is mainly unesterified; most of it is associated with myelin, but it is present also in the membranes of astrocytes and neurons.
Cholesterol synthesis occurs mainly in glial cells [18], together with synthesis of ApoE, the main lipoprotein in the CNS (which also has ApoA-I, ApoJ and ApoD [19]). The ApoE-cholesterol complex is secreted, in a process involving the ABCA-1 [20]. After the first stages of brain development, most mature neurons synthesise only small amounts of cholesterol, they become heavily dependent on uptake of cholesterol synthesised in astrocytes, which they internalize via a member of the LDL receptor (LDLR) family - mainly the LDLR itself and the LDL receptor-related protein, LRP, but also VLDL receptor, ApoE receptor 2, megalin and others [19] (Fig. 2). It has been shown that neurons require ApoE-cholesterol to develop numerous and efficient synapses in vitro [21].
Cholesterol homeostasis is regulated by the dynamic equilibrium of uptake, de novo synthesis, esterification, catabolism (oxidation) and release. The genes for both ApoE and ABCA-1 are among those controlled by the Liver X receptors (LXR) [22], nuclear receptors (originally identified in the liver) widely expressed throughout the body, which are activated by oxysterols [23]—these are therefore major regulators of cholesterol homeostasis, since they switch on the mechanism of efflux and excretion via ABCA-1 and ApoE [24], and also inhibit HMGR [25], therefore cholesterol synthesis (Fig. 2).
Cholesterol and AD
A number of studies point to a deleterious effect of cholesterol in the development of AD. As mentioned above, the risk of developing AD is increased in individuals expressing the ε4 form of the cholesterol-carrying ApoE, and is decreased in patients treated with cholesterol-lowering statins. Cholesterol accumulation has been observed in association with neuritic plaques in the brain in AD and in a transgenic AD mouse model [26]. In addition to the decreased AD incidence with statin treatment mentioned above, elevated total serum cholesterol has been associated with risk of AD in later life [27–29]. Increased cholesterol has been found in brain membranes in aging mice [30] and increased flux of cholesterol across the CNS in aging [16] and in early AD. 24-OHC levels increase in mild cases, while it decreases in more severe cases probably due to the loss of neurons expressing CYP46 [31]. Breaking down of neuronal membranes along with a reduction in the expression of CYP-46 would lead to accumulation of cholesterol in the brain [31]. A preliminary clinical study lowering plasma cholesterol with atorvastatin in mild-moderate AD indicated a positive effect on cognition and behaviour [32]. Staining of sections from AD cortex with a cholesterol-specific fluorescent probe revealed that tangle-bearing neurons contain more free cholesterol than adjacent tangle-free neurons [33].
However, the picture is more complex. On the other hand, cholesterol affects membrane fluidity and is essential for membrane function; adequate levels of cholesterol imported from astrocytes are necessary for synaptogenesis and neuronal functionality [21]. Other groups found that cholesterol synthesis decreases with age in human hippocampus, with no change in brain levels [34], or no change in hippocampal levels in AD [35]. In some cases increased levels of total plasma cholesterol in late life was associated with decreased risk of dementia [36], while in others no association was found [37, 38]. For a review, [39].
Recently it has been shown that polymorphisms in a cluster of genes related to cholesterol metabolism confer susceptibility to AD [40], including CYP46 [41] and ABCA1 [42], although other groups have found no link [43, 44]. Others have found that CYP46 expression shifts from neurons to astrocytes in AD [45].
Cholesterol and Aβ generation
The deleterious effects of cholesterol in AD have been attributed to its ability to increase Aβ generation. Aβ is produced by sequential cleavage of the precursor protein APP by the amyloidogenic β-secretase (as alternative to the non-amyloidogenic α-secretase) followed by cleavage by γ-secretases (presenilins). These processes take place mainly in the ER and plasma membranes.
High cholesterol diets caused higher brain accumulation of Aβ in rabbits [46, 47] and in a transgenic mouse model [48], while lowering cholesterol levels leads to an inhibition of Aβ generation both in cultured neurons and in vivo in guinea pigs and mice [49, 50]. In humans a reduction of Aβ in the CSF has been found with simvastatin in mild AD but not in more severe cases [51–53]. Lowered membrane cholesterol inhibits β-secretase activity in hippocampal neurons [54] and increases Aβ binding to membranes, and Aβ toxicity [55, 56].
At least a fraction of the cell APP, as well as the β-secretase and γ-secretases, are localised in cholesterol-rich lipid rafts [57–59], while the non-amyloidogenic α-secretase is associated with the membrane surface, outside raft domains [60]. A change in cholesterol levels or distribution within different membrane pools (i.e. raft vs. non-raft) alters the localization of APP molecules and their availability to the action of the various secretases [61, 62]. Lipophilic statins have been shown to decrease cholesterol levels in raft environment, and to decrease the expression of the raft marker protein flotillin [63]. However, drastic reductions (over 35%) in membrane cholesterol decreases Aβ generation [61], which may explain why some groups have found increased Aβ in the brain of transgenic mice treated with lovastatin [64]. APP processing can be altered by changes in the dietary cholesterol content in APP gene-targeted mice, by a mechanism dependent on ApoE [65]. This group found that high cholesterol diets in AD transgenic mouse models decreased brain Aβ [65].
Kalvodova and colleagues have found that the proteolitic activity of β-secretase is also directly increased by cholesterol, as well as by neutral glycosphingolipids and anionic glycerophospholipids [66].
Another mechanism linking cholesterol and Aβ secretion may be ABCA1, the transporter involved in cholesterol release, which is present in neurons. Oxysterols, as well as other agonists of the LXR receptors, which increase ABCA-1 expression, also increase Aβ secretion, especially the more hydrophobic Aβ1-42, suggesting that the peptide may be released bound to cholesterol [67]. Others however have found that increasing ABCA1 expression with oxysterols inhibits Aβ generation [68].
The levels of cholesteryl esters (CE) have also been linked to Aβ generation rather than free cholesterol (FC). In cells lacking ACAT, which cannot generate CE and accumulate FC, Aβ generation is inhibited [69]. Treatment with an ACAT inhibitor also leads to decreased brain Aβ load, as well as cognitive function, in a transgenic mouse model of AD [70].
Statin as a therapy for AD
As mentioned above, retrospective studies of cardiovascular patients taking statins have shown a decreased incidence of AD in this population [6, 7]; since then, a large part of the cholesterol inhibition research has been done using these drugs. Statins are inhibitors of HMG-CoA reductase, the regulatory step in the biosynthetic pathway for cholesterol; they have been used for a long time to treat hypercholesterolaemia, and are well tolerated. However, results in connection with AD have been inconsistent. Some recent prospective studies have found no association between statin treatment and AD risk [37, 71]. Statins also seem to have no effect on the Aβ burden in human AD brains [72].
Statins in mice decrease brain cholesterol by a mechanism that seems to involve ApoE, since no effect is observed in ApoE deficient animals [30]. Statins also decrease 24-OHC, indicating an effect on brain cholesterol [73, 74].
One problem in interpreting the statin treatment results is that where an effect is seen, all types of statins tested seem to have a similar impact on risk of AD and brain cholesterol [7, 74], irrespective of the fact that the more lipophilic drugs (lovastatin, simvastatin) cross the blood brain barrier much more easily than the more hydrophilic ones (pravastatin, atorvastatin) [75]—raising the question that the effect may be indirect.
It is possible that statin-induced reductions in peripheral cholesterol affect brain cholesterol via the degradation product 27-hydroxycholesterol (27-OHC), which can cross the blood brain barrier and is actively taken up by the brain [76].
Michikawa [77] ascribes the conflicting data to the existence of different cholesterol pools, pointing out that total serum cholesterol does not affect CSF or brain levels, and that statins affected brain cholesterol only at high doses. Cholesterol is transported in the CSF mainly in HDL, the form released from cells: this pool is low in AD patients and is increased by statins, both in serum and CSF. It is also the high HDL cholesterol levels that correlate with increased AD pathology, rather than the total or LDL pool [78]. The two pools are regulated in opposite ways also by the ApoE phenotype, i.e. higher total and LDL cholesterol and lower HDL cholesterol are found in the subjects with the ApoE4 phenotype compared to those with the ApoE3.
Another interesting set of data showed that cholesterol distribution in the plasma membrane was uneven and that statins caused cholesterol to translocate from the cytofacial leaflet to the exofacial leaflet. This change is accompanied by decreased Aβ levels in vivo, suggesting that cholesterol distribution and not total cholesterol levels may be important to Abeta production in the CNS [61, 79].
Finally, a side effect of statin treatment in a small proportion of cases is a myopathy, which seems to be related to the inhibition of the synthesis of ubiquinone, which shares the HMGR step with the biosynthetic pathway of cholesterol [80]. It is possible that an adverse effect on this important mitochondrial electron carrier may contribute to the variability of results obtained with statin therapies, especially in a situation such as AD where the antioxidant balance and mitochondrial function may already be compromised.
Statins have pleiotropic effects on top of lowering cholesterol. One of these is the prevention of the isoprenylation of small G-proteins such as RhoA and Rac1, which results in the inhibition of their translocation to the membrane [81]. This in turn regulates, among other things, the activities of NOS and NADPH oxidase, to generate NO and superoxide respectively [82, 83], resulting in alterations in the vascular tone and in the anti-inflammatory action. Statins have also direct immunological effects on microglia [84], and anti-apoptotic effects [85]. Some effects on APP processing have been attributed to inhibition of isoprenoid products [86]. Wolozin [87] has in fact proposed that the primary action of statins could be actually to reduce inflammation by inhibiting microglial activation, rather than to decrease the Aβ load. The implication would be that the treatment would prevent the progression of AD towards the more severe stages, rather than reducing the incidence, as the role of inflammation seems to be less important in the early stages.
However, at least part of the effects observed are attributable to statins’ cholesterol lowering action. In cultured neurons, lowering cholesterol with a completely different mechanism, i.e. binding the lipid with methyl-β-cyclodextrin, also lowered Aβ levels, indicating that at least in this model the effect is related to cholesterol levels [50]. In addition, a non-statin cholesterol lowering drug, BM15.766, also decreased brain Aβ in transgenic mice [88].
Micro-array experiments have revealed alterations in gene expression patterns in the brain of mice after statin treatment, mostly genes involved in cell growth and signaling and trafficking that were similarly changed by three drugs of this class [89].
Further clinical and laboratory trials are currently under way which will hopefully help clarify the mechanism of action of these drugs.
Niemann-Pick Disease type C
It is also worth pointing out the pathological similarities between AD and Niemann-Pick Disease type C (NPC), an autosomal recessive disorder caused by a failure in cholesterol trafficking, due to a mutation in the NPC-1 protein (Fig. 2). Both present neurofibrillary tangles and tauopathy, endosomal abnormalities, and increased Aβ generation [90]. In NPC cholesterol accumulates in late endosomes without being able to proceed towards the plasma membrane and ER, which as a result are cholesterol-poor. Mice expressing a pathogenic mutant NPC-1 display increased γ-secretase activity and accumulate Aβ1–40 and Aβ1–42 [91]. Cells treated with a drug which inhibits NPC-1, U18666A, also accumulate Aβ, and the effect disappears on removal of the drug [92].
Cholesterol and Aβ toxicity
Since Aβ toxicity is correlated to its state of aggregation, many studies have investigated the relationship between cholesterol and the toxic effects of Aβ. Aβ is known to interact with and disrupt the order and fluidity of lipid bilayers [93], according to their lipid composition, and this interaction also affects the rate of peptide aggregation. Cholesterol has been reported to favour the formation of amyloid polymerisation “seeds” which initiate peptide aggregation [94]. Arispe and Doh [95] found that lowering membrane cholesterol increased the membrane incorporation of Aβ and cytotoxicity. This is in agreement with the findings of many groups, who also showed that higher cholesterol reduced the effects of Aβ on calcium signalling and neurotoxicity [55, 56, 96–98].
On the other hand, cholesterol was necessary for Aβ binding and toxicity in other models [99, 100]. Oxidised cholesterol metabolites promote Aβ aggregation in vitro [101]. Recently, Schneider and colleagues [102] showed that low cholesterol reduces the ability of Aβ to form oligomeric aggregates, which are now considered the most toxic species of the peptide, without affecting the generation of Aβ monomers.
The interactions between membrane cholesterol and Aβ are undoubtedly very complex and subtle changes in the concentration and/or distribution of this lipid in the various cell compartments have often contrasting effects on Aβ binding and toxicity, depending also on the aggregation state of Aβ [103]. This has been brilliantly reviewed by Eckert and colleagues [104, 105].
Aβ on lipid metabolism
Interesting recent findings show that Aβ in turn also affects lipid metabolism. Aβ1–42 induces accumulation of cholesterol and ceramides in hippocampal neurons [106]. Ceramides are the products of sphingomyelin metabolism by sphingomyelinases (SM), and are involved in various signaling pathways including those leading to apoptosis. Aβ oligomers have been shown to induce neuronal apoptosis mediated by a SM-ceramide pathway [107]. Other groups have also shown that while Aβ1–42 stimulates sphingomyelinase, Aβ1-40 inhibits HMG reductase (the target of statins), and therefore inhibits cholesterol synthesis [108]. Changes in the 1–40/1–42 ratio in AD therefore may in turn alter membrane lipid composition, alter the activity of proteins associated with membranes, and affect cell viability, and initiate a vicious Aβ-cholesterol loop.
Grimm and collaborators have shown that detergent resistant membranes from presenilin KO mice differ from the wild type in the cholesterol content and also show altered membrane fluidity, demonstrating that Aβ and presenilin not only regulate lipid metabolism but also are involved in the regulation of membrane structure and function [109].
ApoE and neurodegeneration
The ApoE4 phenotype is associated with higher risk of AD [4], earlier age of onset of both AD [110] and Down’s syndrome (where there is an additional copy of the chromosome carrying the APP gene) [111], and also with a worse outcome after head trauma [112] and stroke, both in humans [113] and in transgenic mice expressing the human ApoE4 [114]. Individuals carrying ApoE4 have higher total and LDL cholesterol [115], greater amyloid and tangle pathology [116, 117] and show worse mitochondrial damage [118] compared to those carrying other forms.
In cultured neurons, cholesterol uptake is lower when the lipid is bound to ApoE4 compared to ApoE2 and ApoE3 [119]. ApoE4 is less efficient than the other forms in promoting cholesterol efflux from both neurons and astrocytes [120]. Lipidated ApoE binds aggregated Aβ in a isoform-specific manner, ApoE4 being much more effective than the other forms, and may cause enhanced deposition of the peptide [121].
ApoE knockout mice (ApoE−/−) have been used extensively for cardiovascular research. Brain cells from these animals are more sensitive to excitotoxic and age-related synaptic loss [122], while Aβ induced synaptosomal dysfunction was also exaggerated compared to material from control animals [123]. Human ApoE isoforms have been expressed in ApoE−/− mice. Expression of ApoE3, but not ApoE4, is protective against excitotoxicity and age related neurodegeneration [122] and Aβ toxicity [124]. Astrocytes from ApoE−/− mice expressing human ApoE3 release more cholesterol into the medium than those expressing ApoE4, and therefore may modulate the amount of the lipid available for neurons. In other cases ApoE3 was found to bind Aβ more avidly than ApoE4 when associated with lipids [125], and may therefore affect Aβ removal from the extracellular space.
The wide-ranging studies on the role of ApoE in AD have been reviewed extensively [19, 126, 127], including some suggestions that ApoE may be a direct pathogenic factor in AD independently from its effect on amyloid-related mechanisms [128], including neurotoxic effects of ApoE4 involving mitochondrial damage [129, 130].
Neuronal-glial interaction
Most of the research involving Aβ is focused on its interaction with neurons, since these are the cells that die in AD and after treatment with the peptide. However, the living brain is a complex structure where neurons depend heavily on surrounding glial cells for their survival. It is becoming increasingly clear that glial cells, notably astrocytes, far from having just a structural function, play pivotal roles in the metabolic support of neurons and in cell signaling. Walsh and collaborators [131] have found in retina that Aβ-induced dysfunction of glial cells caused secondary neuronal death in the ganglion cell layer.
We have found that in brain primary mixed cultures, Aβ selectively induced calcium oscillations surprisingly in astrocytes and not in neurons, following a delay of 10–15 min [132]. It has been shown previously that Aβ will insert into lipid bilayers and form calcium permeant channels [95, 133] and our data strongly suggested that the astrocytic calcium fluctuations were dependent on calcium influx from the extracellular space, probably through membrane channels formed by the peptide itself. In lipid bilayers, peptide aggregation and insertion depends strongly on the lipid composition of the membrane, in particular on the cholesterol content [95], raising the possibility that the lipid composition of neuronal and astrocytic membranes may differ and so determine the selective action of the peptide. At 24 h we observed cell death in neurons but not in astrocytes, although a decrease in glutathione content was observed in both cell types [132]. The functional importance of this response was underlined by the rescue of cells by glutathione precursors. Subsequent work showed that Aβ treatment also caused mitochondrial depolarisation but again only in astrocytes. This response was dependent on both the calcium response and on oxidative stress. We traced the source of oxidant species to the activation by Aβ of an NADPH oxidase expressed by astrocytes [134], which we described for the first time [135]. This would cause an oxidative stress in the astrocytes, followed by a secondary impact on neuronal viability, as neuronal antioxidant defences are maintained mainly by astrocytes [136]. Activation of this enzyme by Aβ has been observed in microglia [137], but has not been described before in astrocytes.
Aβ and NADPH oxidase
NADPH oxidase is primarily expressed in immune competent cells—neutrophils, monocytes and microglia—where it is responsible for the superoxide burst and bacterial killing. A range of isoforms have more recently been described in a wide range of cells and tissues. The microglial enzyme is activated by Aβ, as mentioned above [137], and expression is elevated in AD brains [138]. The enzyme consists of a membrane-bound cytochrome b558 (subunits p22phox and gp91phox), and cytosolic subunits (p40phox, p47phox, p67phox, Rac and Rap1A); the latter translocate to bind the membrane-bound subunits during activation.
We have characterized the properties of the astrocytic NADPH oxidase by Western blot analysis and by immunofluorescence which showed coexpression with the astrocyte-specific marker glial fibrillary acidic protein (GFAP), using antibodies against gp91 and p67, both in cultures and in vivo [135]. Recent studies [139, 140] have shown that the cytochrome b558 is localised in the cholesterol-rich lipid rafts, which are required for activation: cholesterol depletion reduces translocation of cytosolic subunits and superoxide generation. Statins also inhibit Rac1 independently from their effect on cholesterol [141]. Conversely, in mice lacking ApoE, which have higher levels of oxysterols, macrophages show increased p47 translocation and generation of reactive oxygen species [142].
It is also interesting to note that activation of sphingomyelinases and apoptosis by Aβ in neurons has been proposed to be mediated by activation of NADPH oxidase [143].
We believe that activation of NADPH oxidase may play a major role in the Aβ-induced neurodegeneration in Alzheimer’s Disease, raising the possibility that modulating the activity of this enzyme may prove beneficial.
Conclusion
There are multiple strands of research pointing at an important role for cholesterol in the pathogenesis of Alzheimer’s Disease. The picture is however far from clear, ad there is still disagreement in several areas, the difficulties rising among other things, from the fact that AD is a complex multifactorial disease, and that cholesterol metabolism is subject to a complex regulation at many different levels and in different compartments, so that increases or decreases in cholesterol levels bring about re-adjustments of the system in way that may not be immediately evident. Certainly more research is needed in this rapidly evolving and promising field.
References
Blass JP, Gibson GE (1991) The role of oxidative abnormalities in the pathophysiology of Alzheimer’s disease. Rev Neurol (Paris) 147:513–525
Canevari L, Abramov AY, Duchen MR (2004) Toxicity of amyloid beta peptide: tales of calcium, mitochondria, and oxidative stress. Neurochem Res 29:637–650
Mark RJ, Blanc EM, Mattson MP (1996) Amyloid beta-peptide and oxidative cellular injury in Alzheimer’s disease. Mol Neurobiol 12:211–224
Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261:921–923
Hofman A, Ott A, Breteler MM, Bots ML, Slooter AJ, van Hurskamp HF, van Duijn CN, Van Broeckhiven C, Grobbee DE (1997) Atherosclerosis, apolipoprotein E, and prevalence of dementia and Alzheimer’s disease in the Rotterdam Study. Lancet 349:151–154
Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G (2000) Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol 57:1439–1443
Jick H, Zornberg GL, Jick SS, Seshadri S, Drachman DA (2000) Statins and the risk of dementia. Lancet 356:1627–1631
Maxfield FR, Tabas I (2005) Role of cholesterol and lipid organization in disease. Nature 438:612–621
Simons K, Ikonen E (1997) Functional rafts in cell membranes. Nature 387:569–572
Rothberg KG, Heuser JE, Donzell WC, Ying YS, Glenney JR, Anderson RG (1992) Caveolin, a protein component of caveolae membrane coats. Cell 68:673–682
Schroeder F, Gallegos AM, Atshaves BP, Storey SM, McIntosh AL, Petrescu AD, Huang H, Starodub O, Chao H, Yang H, Frolov A, Kier AB (2001) Recent advances in membrane microdomains: rafts, caveolae, and intracellular cholesterol trafficking. Exp Biol Med (Maywood) 226:873–890
Brown MS, Goldstein JL (1986) A receptor-mediated pathway for cholesterol homeostasis. Science 232:34–47
Chobanian AV, Hollander W (1962) Body cholesterol metabolism in man. I. The equilibration of serum and tissue cholesterol. J Clin Invest 41:1732–1737
Serougne-Gautheron C, Chevallier F (1973) Time course of biosynthetic cholesterol in the adult rat brain. Biochim Biophys Acta 316:244–250
Björkhem I, Lütjohann D, Diczfalusy U, Stahle L, Ahlborg G, Wahren J (1998) Cholesterol homeostasis in human brain: turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J Lipid Res 39:1594–1600
Lütjohann D, Breuer O, Ahlborg G, Nennesmo I, Siden A, Diczfalusy U, Björkhem I (1996) Cholesterol homeostasis in human brain: evidence for an age-dependent flux of 24S-hydroxycholesterol from the brain into the circulation. Proc Natl Acad Sci U S A 93:9799–9804
Dietschy JM, Turley SD (2004) Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res 45:1375–1397
Saito M, Benson EP, Saito M, Rosenberg A (1987) Metabolism of cholesterol and triacylglycerol in cultured chick neuronal cells, glial cells, and fibroblasts: accumulation of esterified cholesterol in serum-free culture. J Neurosci Res 18:319–325
Beffert U, Danik M, Krzywkowski P, Ramassamy C, Berrada F, Poirier J (1998) The neurobiology of apolipoproteins and their receptors in the CNS and Alzheimer’s disease. Brain Res Rev 27:119–142
Wahrle SE, Jiang H, Parsadanian M, Legleiter J, Han X, Fryer JD, Kowalewski T, Holtzman DM (2004) ABCA1 is required for normal central nervous system apoE levels and for lipidation of astrocyte-secreted apoE. J Biol Chem 279:40987–40993
Mauch DH, Nagler K, Schumacher S, Goritz C, Muller EC, Otto A, Pfrieger FW (2001) CNS synaptogenesis promoted by glia-derived cholesterol. Science 294:1354–1357
Liang Y, Lin S, Beyer TP, Zhang Y, Wu X, Bales KR, DeMattos RB, May PC, Li SD, Jiang XC, Eacho PI, Cao G, Paul SM (2004) A liver X receptor and retinoid X receptor heterodimer mediates apolipoprotein E expression, secretion and cholesterol homeostasis in astrocytes. J Neurochem 88:623–634
Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ (1996) An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature 383:728–731
Abildayeva K, Jansen PJ, Hirsch-Reinshagen V, Bloks VW, Bakker AHF, Ramaekers FCS, de Vente J, Groen AK, Cheryl L, Kuipers F, Mulder M (2006) 24(S)-Hydroxycholesterol participates in a liver X receptor-controlled pathway in astrocytes that regulates apolipoprotein E-mediated cholesterol efflux. J Biol Chem 281:12799–12808
Sinensky M (1977) Isolation of a mammalian cell mutant resistant to 25-hydroxy cholesterol. Biochemical and Biophysical Research Communications 78:863–867
Mori T, Paris D, Town T, Rojiani AM, Sparks DL, Delledonne A, Crawford F, Abdullah LI, Humphrey JA, Dickson DW, Mullan MJ (2001) Cholesterol accumulates in senile plaques of Alzheimer disease patients and in transgenic APP(SW) mice. J Neuropathol Exp Neurol 60:778–785
Kivipelto M, Helkala EL, Hanninen T, Laakso MP, Hallikainen M, Alhainen K, Soininen H, Tuomilehto J, Nissinen A (2001) Midlife vascular risk factors and late-life mild cognitive impairment: A population-based study. Neurology 56:1683–1689
Pappolla MA, Bryant-Thomas TK, Herbert D, Pacheco J, Fabra GM, Manjon M, Girones X, Henry TL, Matsubara E, Zambon D, Wolozin B, Sano M, Cruz-Sanchez FF, Thal LJ, Petanceska SS, Refolo LM (2003) Mild hypercholesterolemia is an early risk factor for the development of Alzheimer amyloid pathology. Neurology 61:199–205
Whitmer RA, Sidney S, Selby J, Johnston SC, Yaffe K (2005) Midlife cardiovascular risk factors and risk of dementia in late life. Neurology 64:277–281
Eckert GP, Wood WG, Müller WE (2001) Effects of aging and beta-amyloid on the properties of brain synaptic and mitochondrial membranes. J Neural Transm 108:1051–1064
Papassotiropoulos A, Lütjohann D, Bagli M, Locatelli S, Jessen F, Rao ML, Maier W, Björkhem I, von Bergmann K, Heun R (2000) Plasma 24S-hydroxycholesterol: a peripheral indicator of neuronal degeneration and potential state marker for Alzheimer’s disease. Neuroreport 11:1959–1962
Sparks DL, Sabbagh MN, Connor DJ, Lopez J, Launer LJ, Browne P, Wasser D, Johnson-Traver S, Lochhead J, Ziolwolski C (2005) Atorvastatin for the treatment of mild to moderate Alzheimer disease: preliminary results. Arch Neurol 62:753–757
Distl R, Meske V, Ohm TG (2001) Tangle-bearing neurons contain more free cholesterol than adjacent tangle-free neurons. Acta Neuropathol (Berl) 101:547–554
Thelen KM, Falkai P, Bayer TA, Lütjohann D (2006) Cholesterol synthesis rate in human hippocampus declines with aging. Neurosci Lett 403:15–19
Eckert GP, Cairns NJ, Maras A, Gattaz WF, Müller WE (2000) Cholesterol modulates the membrane-disordering effects of beta-amyloid peptides in the hippocampus: Specific changes in Alzheimer’s disease. Dementia Geriatric Cogn Disord 11:181–186
Mielke MM, Zandi PP, Sjogren M, Gustafson D, Ostling S, Steen B, Skoog I (2005) High total cholesterol levels in late life associated with a reduced risk of dementia. Neurology 64:1689–1695
Li G, Shofer JB, Kukull WA, Peskind ER, Tsuang DW, Breitner JC, McCormick W, Bowen JD, Teri L, Schellenberg GD, Larson EB (2005) Serum cholesterol and risk of Alzheimer disease: a community-based cohort study. Neurology 65:1045–1050
Reitz C, Tang MX, Luchsinger J, Mayeux R (2004) Relation of plasma lipids to Alzheimer disease and vascular dementia. Arch Neurol 61:705–714
Wood WG, Igbavboa U, Eckert GP, Johnson-Anuna LN, Müller WE (2005) Is hypercholesterolemia a risk factor for Alzheimer’s disease? Mol Neurobiol 31:185–192
Papassotiropoulos A, Wollmer MA, Tsolaki M, Brunner F, Molyva D, Lütjohann D, Nitsch RM, Hock C (2005) A cluster of cholesterol-related genes confers susceptibility for Alzheimer’s disease. J Clin Psychiatry 66:940–947
Kolsch H, Lütjohann D, Ludwig M, Schulte A, Ptok U, Jessen F, von Bergmann K, Rao ML, Maier W, Heun R (2002) Polymorphism in the cholesterol 24S-hydroxylase gene is associated with Alzheimer’s disease. Mol Psychiatry 7:899–902
Katzov H, Chalmers K, Palmgren J, Andreasen N, Johansson B, Cairns NJ, Gatz M, Wilcock GK, Love S, Pedersen NL, Brookes AJ, Blennow K, Kehoe PG, Prince JA (2004) Genetic variants of ABCA1 modify Alzheimer disease risk and quantitative traits related to beta-amyloid metabolism. Hum Mutat 23:358–367
Chalmers KA, Culpan D, Kehoe PG, Wilcock GK, Hughes A, Love S (2004) APOE promoter, ACE1 and CYP46 polymorphisms and beta-amyloid in Alzheimer’s disease. Neuroreport 15:95–98
Ingelsson M, Jesneck J, Irizarry MC, Hyman BT, Rebeck GW (2004) Lack of association of the cholesterol 24-hydroxylase (CYP46) intron 2 polymorphism with Alzheimer’s disease. Neurosci Lett 367:228–231
Bogdanovic N, Bretillon L, Lund EG, Diczfalusy U, Lannfelt L, Winblad B, Russell DW, Björkhem I. (2001) On the turnover of brain cholesterol in patients with Alzheimer’s disease. Abnormal induction of the cholesterol-catabolic enzyme CYP46 in glial cells. Neuroscience Letters 314:45–48
Sparks DL, Scheff SW, Hunsaker JC III, Liu H, Landers T, Gross DR (1994) Induction of Alzheimer-like beta-amyloid immunoreactivity in the brains of rabbits with dietary cholesterol. Exp Neurol 126:88–94
Zatta P, Zambenedetti P, Stella MP, Licastro F (2002) Astrocytosis, microgliosis, metallothionein-I-II and amyloid expression in high cholesterol-fed rabbits. J Alzheimers Dis 4:1–9
Refolo LM, Malester B, LaFrancois J, Bryant-Thomas T, Wang R, Tint GS, Sambamurti K, Duff K, Pappolla MA (2000) Hypercholesterolemia accelerates the Alzheimer’s amyloid pathology in a transgenic mouse model. Neurobiol Dis 7:321–331
Chauhan NB, Siegel GJ, Feinstein DL (2004) Effects of lovastatin and pravastatin on amyloid processing and inflammatory response in TgCRND8 brain. Neurochem Res 29:1897–1911
Fassbender K, Simons M, Bergmann C, Stroick M, Lütjohann D, Keller P, Runz H, Kuhl S, Bertsch T, von Bergmann K, Hennerici M, Beyreuther K, Hartmann T (2001) Simvastatin strongly reduces levels of Alzheimer’s disease beta -amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc Natl Acad Sci U S A 98:5856–5861
Simons M, Schwarzler F, Lütjohann D, von Bergmann K, Beyreuther K, Dichgans J, Wormstall H, Hartmann T, Schulz JB (2002) Treatment with simvastatin in normocholesterolemic patients with Alzheimer’s disease: A 26-week randomized, placebo-controlled, double-blind trial. Ann Neurol 52:346–350
Hoglund K, Syversen S, Lewczuk P, Wallin A, Wiltfang J, Blennow K (2005) Statin treatment and a disease-specific pattern of beta-amyloid peptides in Alzheimer’s disease. Exp Brain Res 164:205–214
Sparks DL, Petanceska S, Sabbagh M, Connor D, Soares H, Adler C, Lopez J, Ziolkowski C, Lochhead J, Browne P (2005) Cholesterol, copper and Abeta in controls, MCI, AD and the AD cholesterol-lowering treatment trial (ADCLT). Curr Alzheimer Res 2:527–539
Simons M, Keller P, De Strooper B, Beyreuther K, Dotti CG, Simons K (1998) Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc Natl Acad Sci USA 95:6460–6464
Curtain CC, Ali FE, Smith DG, Bush AI, Masters CL, Barnham KJ (2003) Metal ions, pH, and cholesterol regulate the interactions of Alzheimer’s disease amyloid-beta peptide with membrane lipid. J Biol Chem 278:2977–2982
Yip CM, Elton EA, Darabie AA, Morrison MR, McLaurin J (2001) Cholesterol, a modulator of membrane-associated Abeta-fibrillogenesis and neurotoxicity. J Mol Biol 311:723–734
Riddell DR, Christie G, Hussain I, Dingwall C (2001) Compartmentalization of beta-secretase (Asp2) into low-buoyant density, noncaveolar lipid rafts. Curr Biol 11:1288–1293
Chen TY, Liu PH, Ruan CT, Chiu L, Kung FL (2006) The intracellular domain of amyloid precursor protein interacts with flotillin-1, a lipid raft protein. Biochem Biophys Res Commun 342:266–272
Wahrle S, Das P, Nyborg AC, McLendon C, Shoji M, Kawarabayashi T, Younkin LH, Younkin SG, Golde TE (2002) Cholesterol-dependent gamma-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiol Dis 9:11–23
Kojro E, Gimpl G, Lammich S, Marz W, Fahrenholz F (2001) Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha-secretase ADAM 10. Proc Natl Acad Sci U S A 98:5815–5820
Abad-Rodriguez J, Ledesma MD, Craessaerts K, Perga S, Medina M, Delacourte A, Dingwall C, De Strooper B, Dotti CG (2004) Neuronal membrane cholesterol loss enhances amyloid peptide generation. J Cell Biol 167:953–960
Ehehalt R, Keller P, Haass C, Thiele C, Simons K (2003) Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J Cell Biol 160:113–123
Kirsch C, Eckert GP, Mueller WE (2003) Statin effects on cholesterol micro-domains in brain plasma membranes. Biochem Pharmacol 65:843–856
Park IH, Hwang EM, Hong HS, Boo JH, Oh SS, Lee J, Jung MW, Bang OY, Kim SU, Mook-Jung I (2003) Lovastatin enhances Abeta production and senile plaque deposition in female Tg2576 mice. Neurobiol Aging 24:637–643
Howland DS, Trusko SP, Savage MJ, Reaume AG, Lang DM, Hirsch JD, Maeda N, Siman R, Greenberg BD, Scott RW, Flood DG (1998) Modulation of secreted beta-amyloid precursor protein and amyloid beta-peptide in brain by cholesterol. J Biol Chem 273:16576–16582
Kalvodova L, Kahya N, Schwille P, Ehehalt R, Verkade P, Drechsel D, Simons K (2005) Lipids as modulators of proteolytic activity of BACE—Involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. J Biol Chem 280:36815–36823
Fukumoto H, Deng A, Irizarry MC, Fitzgerald ML, Rebeck GW (2002) Induction of the cholesterol transporter ABCA1 in central nervous system cells by Liver X receptor agonists increases secreted Abeta levels. J Biol Chem 277:48508–48513
Brown J III, Theisler C, Silberman S, Magnuson D, Gottardi-Littell N, Lee JM, Yager D, Crowley J, Sambamurti K, Rahman MM, Reiss AB, Eckman CB, Wolozin B (2004) Differential expression of cholesterol hydroxylases in Alzheimer’s disease. J Biol Chem 279:34674–34681
Puglielli L, Konopka G, Pack-Chung E, Ingano LA, Berezovska O, Hyman BT, Chang TY, Tanzi RE, Kovacs DM (2001) Acyl-coenzyme A: cholesterol acyltransferase modulates the generation of the amyloid beta-peptide. Nat Cell Biol 3:905–912
Hutter-Paier B, Huttunen HJ, Puglielli L, Eckman CB, Kim DY, Hofmeister A, Moir RD, Domnitz SB, Frosch MP, Windisch M, Kovacs DM (2004) The ACAT inhibitor CP-113,818 markedly reduces amyloid pathology in a mouse model of Alzheimer’s disease. Neuron 44:227–238
Zandi PP, Sparks DL, Khachaturian AS, Tschanz J, Norton M, Steinberg M, Welsh-Bohmer KA, Breitner JC (2005) Do statins reduce risk of incident dementia and Alzheimer disease? The Cache County Study. Arch Gen Psychiatry 62:217–224
Wolozin B (2004) Cholesterol, statins and dementia. Curr Opin Lipidol 15:667–672
Locatelli S, Lütjohann D, Schmidt HH, Otto C, Beisiegel U, von Bergmann K (2002) Reduction of plasma 24S-hydroxycholesterol (cerebrosterol) levels using high-dosage simvastatin in patients with hypercholesterolemia: evidence that simvastatin affects cholesterol metabolism in the human brain. Arch Neurol 59:213–216
Vega GL, Weiner MF, Lipton AM, von Bergmann K, Lütjohann D, Moore C, Svetlik D (2003) Reduction in levels of 24S-hydroxycholesterol by statin treatment in patients with Alzheimer disease. Arch Neurol 60:510–515
Botti RE, Triscari J, Pan HY, Zayat J (1991) Concentrations of pravastatin and lovastatin in cerebrospinal fluid in healthy subjects. Clin Neuropharmacol 14:256–261
Heverin M, Meaney S, Lütjohann D, Diczfalusy U, Wahren J, Björkhem I (2005) Crossing the barrier: net flux of 27-hydroxycholesterol into the human brain. J Lipid Res 46:1047–1052
Michikawa M (2003) Cholesterol paradox: is high total or low HDL cholesterol level a risk for Alzheimer’s disease? J Neurosci Res 72:141–146
Launer LJ, White LR, Petrovitch H, Ross GW, Curb JD (2001) Cholesterol and neuropathologic markers of AD: a population-based autopsy study. Neurology 57:1447–1452
Burns MP, Igbavboa U, Wang LL, Wood WG, Duff K (2006) Cholesterol distribution, not total levels, correlate with altered amyloid precursor, protein processing in statin-treated mice. Neuromol Med 8:319–328
Paiva H, Thelen KM, Van Coster R, Smet J, De Paepe B, Mattila KM, Laakso J, Lehtimaki T, von Bergmann K, Lütjohann D, Laaksonen R (2005) High-dose statins and skeletal muscle metabolism in humans: a randomized, controlled trial. Clin Pharmacol Ther 78:60–68
Cordle A, Koenigsknecht-Talboo J, Wilkinson B, Limpert A, Landreth G (2005) Mechanisms of statin-mediated inhibition of small G-protein function. J Biol Chem 280:34202–34209
Endres M, Laufs U (2004) Effects of statins on endothelium and signaling mechanisms. Stroke 35:2708–2711
Nakagami H, Jensen KS, Liao JK (2003) A novel pleiotropic effect of statins: prevention of cardiac hypertrophy by cholesterol-independent mechanisms. Ann Med 35:398–403
Kuipers HF, Rappert AA, Mommaas AM, van Haastert ES, van der Vack P, Boddeke HW, Biber KP, van den Elsen PJ (2006) Simvastatin affects cell motility and actin cytoskeleton distribution of microglia. Glia 53:115–123
Tanaka K, Honda M, Takabatake T (2004) Anti-apoptotic effect of atorvastatin, a 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitor, on cardiac myocytes through protein kinase C activation. Clin Exp Pharmacol Physiol 31:360–364
Cole SL, Grudzien A, Manhart IO, Kelly BL, Oakley H, Vassar R (2005) Statins cause intracellular accumulation of amyloid precursor protein, beta-secretase-cleaved fragments, and amyloid beta-peptide via an isoprenoid-dependent mechanism. J Biol Chem 280:18755–18770
Wolozin B, Manger J, Bryant R, Cordy J, Green RC, McKee A (2006) Re-assessing the relationship between cholesterol, statins and Alzheimer’s disease. Acta Neurol Scand Suppl 185:63–70
Refolo LM, Pappolla MA, LaFrancois J, Malester B, Schmidt SD, Thomas-Bryant T, Tint GS, Wang R, Mercken M, Petanceska SS, Duff KE (2001) A cholesterol-lowering drug reduces beta-amyloid pathology in a transgenic mouse model of Alzheimer’s disease. Neurobiol Dis 8:890–899
Johnson-Anuna LN, Eckert GP, Keller JH, Igbavboa U, Franke C, Fechner T, Schubert-Zsilavecz M, Karas M, Müller WE, Wood WG (2005) Chronic administration of statins alters multiple gene expression patterns in mouse cerebral cortex. J Pharmacol Exp Therapeut 312:786–793
Nixon RA (2004) Niemann-Pick type C disease and Alzheimer’s disease - The APP-endosome connection fattens up. Am J Pathol 164:757–761
Burns M, Duff K (2003) Use of in vivo models to study the role of cholesterol in the etiology of Alzheimer’s disease. Neurochem Res 28:979–986
Yamazaki T, Chang TY, Haass C, Ihara Y (2001) Accumulation and aggregation of amyloid beta-protein in late endosomes of Niemann-pick type C cells. J Biol Chem 276:4454–4460
Kremer JJ, Sklansky DJ, Murphy RM (2001) Profile of changes in lipid bilayer structure caused by beta-amyloid peptide. Biochemistry 40:8563–8571
Mizuno T, Nakata M, Naiki H, Michikawa M, Wang R, Haass C, Yanagisawa K (1999) Cholesterol-dependent generation of a seeding amyloid beta-protein in cell culture. J Biol Chem 274:15110–15114
Arispe N, Doh M (2002) Plasma membrane cholesterol controls the cytotoxicity of Alzheimer’s disease A beta P (1–40) and (1–42) peptides. Faseb J 16:1526–1536
Hartmann H, Eckert A, Müller WE (1994) Apolipoprotein-E and cholesterol affect neuronal calcium signaling—the possible relationship to beta-amyloid neurotoxicity. Biochem Biophys Res Commun 200:1185–1192
Kawahara M, Kuroda Y (2001) Intracellular calcium changes in neuronal cells induced by Alzheimer’s beta-amyloid protein are blocked by estradiol and cholesterol. Cell Mol Neurobiol 21:1–13
Zhou Y, Richardson JS (1996) Cholesterol protects PC12 cells from beta-amyloid induced calcium disordering and cytotoxicity. Neuroreport 7:2487–2490
Subasinghe S, Unabia S, Barrow CJ, Mok SS, Aguilar MI, Small DH (2003) Cholesterol is necessary both for the toxic effect of Abeta peptides on vascular smooth muscle cells and for Abeta binding to vascular smooth muscle cell membranes. J Neurochem 84:471–479
Micelli S, Meleleo D, Picciarelli V, Gallucci E (2004) Effect of sterols on beta-amyloid peptide (AbetaP 1–40) channel formation and their properties in planar lipid membranes. Biophys J 86:2231–2237
Bieschke J, Zhang Q, Powers ET, Lerner RA, Kelly JW (2005) Oxidative metabolites accelerate Alzheimer’s amyloidogenesis by a two-step mechanism, eliminating the requirement for nucleation. Biochemistry 44:4977–4983
Schneider A, Schulz-Schaeffer W, Hartmann T, Schulz JB, Simons M (2006) Cholesterol depletion reduces aggregation of amyloid-beta peptide in hippocampal neurons. Neurobiol Dis 23:573–577
Sponne I, Fifre A, Koziel V, Oster T, Olivier JL, Pillot T (2004) Membrane cholesterol interferes with neuronal apoptosis induced by soluble oligomers but not fibrils of amyloid-beta peptide. FASEB J 18:836–838
Eckert GP, Kirsch C, Leutz S, Wood WG, Müller WE (2003) Cholesterol modulates amyloid beta-peptide’s membrane interactions. Pharmacopsychiatry 36(Suppl 2):S136–S143
Gibson-Wood W, Eckert GP, Igbavboa U, Müller WE (2003) Amyloid beta-protein interactions with membranes and cholesterol: causes or casualties of Alzheimer’s disease. Biochim Biophys Acta 1610:281–290
Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, Troncoso JC, Mattson MP (2004) Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc Natl Acad Sci U S A 101:2070–2075
Malaplate-Armand C, Florent-Bechard S, Youssef I, Koziel V, Sponne I, Kriem B, Leininger-Muller B, Olivier JL, Oster T, Pillot T (2006) Soluble oligomers of amyloid-beta peptide induce neuronal apoptosis by activating a cPLA(2)-dependent sphingomyelinase-ceramide pathway. Neurobiol Dis 23:178–189
Grimm MO, Grimm HS, Patzold AJ, Zinser EG, Halonen R, Duering M, Tschape JA, De Strooper B, Muller U, Shen J, Hartmann T (2005) Regulation of cholesterol and sphingomyelin metabolism by amyloid-beta and presenilin. Nat Cell Biol 7:1118–1123
Grimm MOW, Tschape JA, Grimm HS, Zinser EG, Hartmann T (2006) Altered membrane fluidity and lipid raft composition in presenilin-deficient cells. Acta Neurologica Scandinavica 114:27–32
Tang MX, Maestre G, Tsai WY, Liu XH, Feng L, Chung WY, Chun M, Schofield P, Stern Y, Tycko B, Mayeux R (1996) Relative risk of Alzheimer disease and age-at-onset distributions, based on APOE genotypes among elderly African Americans, Caucasians, and Hispanics in New York City. Am J Hum Genet 58:574–584
Schupf N, Sergievsky GH (2002) Genetic and host factors for dementia in Down’s syndrome. Br J Psychiatry 180:405–410
Nicoll JA, Roberts GW, Graham DI (1995) Apolipoprotein E epsilon 4 allele is associated with deposition of amyloid beta-protein following head injury. Nat Med 1:135–137
Liu Y, Laakso MP, Karonen JO, Vanninen RL, Nuutinen J, Soimakallio S, Aronen HJ (2002) Apolipoprotein E polymorphism and acute ischemic stroke: a diffusion- and perfusion-weighted magnetic resonance imaging study. J Cereb Blood Flow Metab 22:1336–1342
Horsburgh K, McCulloch J, Nilsen M, Roses AD, Nicoll JA (2000) Increased neuronal damage and apoE immunoreactivity in human apolipoprotein E, E4 isoform-specific, transgenic mice after global cerebral ischaemia. Eur J Neurosci 12:4309–4317
Sing CF, Davignon J (1985) Role of the apolipoprotein E polymorphism in determining normal plasma lipid and lipoprotein variation. Am J Hum Genet 37:268–285
Nagy Z, Esiri MM, Jobst KA, Johnston C, Litchfield S, Sim E, Smith AD (1995) Influence of the apolipoprotein E genotype on amyloid deposition and neurofibrillary tangle formation in Alzheimer’s disease. Neuroscience 69:757–761
McNamara MJ, Gomez-Isla T, Hyman BT (1998) Apolipoprotein E genotype and deposits of Abeta40 and Abeta42 in Alzheimer disease. Arch Neurol 55:1001–1004
Gibson GE, Haroutunian V, Zhang H, Park LC, Shi Q, Lesser M, Mohs RC, Sheu RK, Blass JP (2000) Mitochondrial damage in Alzheimer’s disease varies with apolipoprotein E genotype. Ann Neurol 48:297–303
Rapp A, Gmeiner B, Huttinger M (2006) Implication of apoE isoforms in cholesterol metabolism by primary rat hippocampal neurons and astrocytes. Biochimie 88:473–483
Michikawa M, Fan QW, Isobe I, Yanagisawa K (2000) Apolipoprotein E exhibits isoform-specific promotion of lipid efflux from astrocytes and neurons in culture. J Neurochem 74:1008–1016
Stratman NC, Castle CK, Taylor BM, Epps DE, Melchior GW, Carter DB (2005) Isoform-specific interactions of human apolipoprotein E to an intermediate conformation of human Alzheimer amyloid-beta peptide. Chem Phys Lipids 137:52–61
Buttini M, Orth M, Bellosta S, Akeefe H, Pitas RE, Wyss-Coray T, Mucke L, Mahley RW (1999) Expression of human apolipoprotein E3 or E4 in the brains of ApoE-/- mice: isoform-specific effects on neurodegeneration. J Neurosci 19:4867–4880
Keller JN, Lauderback CM, Butterfield DA, Kindy MS, Yu J, Markesbery WR (2000) Amyloid beta-peptide effects on synaptosomes from apolipoprotein E-deficient mice. J Neurochem 74:1579–1586
Jordan J, Galindo MF, Miller RJ, Reardon CA, Getz GS, LaDu MJ (1998) Isoform-specific effect of apolipoprotein E on cell survival and beta-amyloid-induced toxicity in rat hippocampal pyramidal neuronal cultures. J Neurosci 18:195–204
LaDu MJ, Pederson TM, Frail DE, Reardon CA, Getz GS, Falduto MT (1995) Purification of apolipoprotein E attenuates isoform-specific binding to beta-amyloid. J Biol Chem 270:9039–9042
Lane RM, Farlow MR (2005) Lipid homeostasis and apolipoprotein E in the development and progression of Alzheimer’s disease. J Lipid Res 46:949–968
Huang Y (2006) Apolipoprotein E and Alzheimer disease. Neurology 66:S79-S85
Crutcher KA (2004) Apolipoprotein E is a prime suspect, not just an accomplice, in Alzheimer’s disease. J Mol Neurosci 23:181–188
Chang S, ran MT, Miranda RD, Balestra ME, Mahley RW, Huang Y (2005) Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc Natl Acad Sci USA 102:18694–18699
Mahley RW, Weisgraber KH, Huang Y (2006) Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci USA 103:5644–5651
Walsh DT, Montero RM, Bresciani LG, Jen AY, Leclercq PD, Saunders D, EL-Amir AN, Gbadamoshi L, Gentleman SM, Jen LS (2002) Amyloid-beta peptide is toxic to neurons in vivo via indirect mechanisms. Neurobiol Dis 10:20–27
Abramov AY, Canevari L, Duchen MR (2003) Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J Neurosci 23:5088–5095
Quist A, Doudevski I, Lin H, Azimova R, Ng D, Frangione B, Kagan B, Ghiso J, Lal R (2005) Amyloid ion channels: a common structural link for protein-misfolding disease. Proc Natl Acad Sci U S A 102:10427–10432
Abramov AY, Canevari L, Duchen MR (2004) Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J Neurosci 24:565–575
Abramov AY, Jacobson J, Wientjes F, Hothersall J, Canevari L, Duchen MR (2005) Expression and modulation of an NADPH oxidase in mammalian astrocytes. J Neurosci 25:9176–9184
Keelan J, Allen NJ, Antcliffe D, Pal S, Duchen MR (2001) Quantitative imaging of glutathione in hippocampal neurons and glia in culture using monochlorobimane. J Neurosci Res 66:873–884
DellaBianca V, Dusi S, Bianchini E, Dal P I, Rossi F (1999) beta-amyloid activates the O- 2 forming NADPH oxidase in microglia, monocytes, and neutrophils. A possible inflammatory mechanism of neuronal damage in Alzheimer’s disease. J Biol Chem 274:15493–15499
Shimohama S, Tanino H, Kawakami N, Okamura N, Kodama H, Yamaguchi T, Hayakawa T, Nunomura A, Chiba S, Perry G, Smith MA, Fujimoto S (2000) Activation of NADPH oxidase in Alzheimer’s disease brains. Biochem Biophys Res Commun 273:5–9
Shao D, Segal AW, Dekker LV (2003) Lipid rafts determine efficiency of NADPH oxidase activation in neutrophils. FEBS Lett 550:101–106
Vilhardt F, van Deurs B (2004) The phagocyte NADPH oxidase depends on cholesterol-enriched membrane microdomains for assembly. EMBO J 23:739–748
Nakagami H, Jensen KS, Liao JK (2003) A novel pleiotropic effect of statins: prevention of cardiac hypertrophy by cholesterol-independent mechanisms. Ann Med 35:398–403
Rosenblat M, Aviram M (2002) Oxysterol-induced activation of macrophage NADPH-oxidase enhances cell-mediated oxidation of LDL in the atherosclerotic apolipoprotein E deficient mouse: inhibitory role for vitamin E. Atherosclerosis 160:69–80
Jana A, Pahan K (2004) Fibrillar amyloid-beta peptides kill human primary neurons via NADPH oxidase-mediated activation of neutral sphingomyelinase. Implications for Alzheimer’s disease. J Biol Chem 279:51451–51459
Acknowledgment
We wish to thank the Miriam Marks fund for supporting LC and the Division of Neurochemistry.
Author information
Authors and Affiliations
Corresponding author
Additional information
Special issue dedicated to John P. Blass.
Rights and permissions
About this article
Cite this article
Canevari, L., Clark, J.B. Alzheimer’s Disease and Cholesterol: The Fat Connection. Neurochem Res 32, 739–750 (2007). https://doi.org/10.1007/s11064-006-9200-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-006-9200-1