
Abstract
Introduction
Recently, the term “Diffuse glioma, BRAF V600E-mutant” has been recommended for IDH-wildtype gliomas with BRAF p.V600E mutation and without CDKN2A/B deletion. However, additional alterations in gliomas that coexist with BRAF-mutations are poorly defined.
Methods
We analyzed next-generation sequencing results in 315 cancer-associated genes for 372 gliomas from our institution (2010 to 2017). In addition, we reviewed IDH-WT gliomas with mutation and copy-number alterations available in cBioPortal, to further characterize BRAF-mutant gliomas.
Results
Seventeen (4.6%) showed BRAF mutations. Tumor types included 8 glioblastomas, 2 epithelioid glioblastomas (E-GBM), 2 pleomorphic xanthoastrocytomas (PXA), 1 anaplastic oligodendroglioma, 1 diffuse astrocytoma, and 3 pilocytic astrocytomas. Fifty-three percent (53%) of cases exhibited BRAF-alterations other than p.V600E. The majority of the tumors were localized in the temporal lobe (52.9%). In addition to BRAF mutations, glioblastomas showed concomitant mutations in TP53 (3/8), CDKN2A/B-loss (6/8), TERT-promoter (6/8), and/or PTEN (5/8). Both E-GBMs and PXAs showed CDKN2A/B-loss and BRAF p.V600E with absence of TERTp, TP53, and PTEN mutations. Similar findings were observed in BRAF-mutant infiltrating gliomas from cBioPortal.
Conclusions
Knowledge of additional alterations that co-occur with BRAF-mutations in gliomas may improve diagnosis and help identify patients that could benefit from targeted therapies. Furthermore, we provide examples of two patients whose tumors responded to BRAF pathway inhibitors, arguing in favor of these therapies in patients with BRAF-mutant gliomas.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Gliomas account for over 75% of all malignant primary central nervous system (CNS) tumors and have a variety of genomic alterations that may result in dysregulation of growth-factor signaling pathways and contribute to tumorigenesis. Glioblastoma (GBM) is the most aggressive and prevalent type of adult glioma [1]. The median overall survival (OS) is < 2 years despite maximal surgical resection and chemoradiation [2]. However, GBMs are a genetically heterogeneous group of tumors and understanding their molecular drivers is critical for accurate diagnosis, prognosis, and incorporation of targeted therapies.
The RAS–RAF–MEK extracellular signal-regulated kinase (ERK) signaling cascade is frequently mutated in human cancers. It transduces a growth signal from the cell membrane to the nucleus via a chain of protein kinases and is responsible for cellular proliferation and survival. A commonly altered gene in this pathway is v-raf murine sarcoma viral oncogene homolog B1 (BRAF), a RAF family serine/threonine kinase protein that is part of the RAS–RAF–MEK–ERK pathway [3]. BRAF alterations are common in several tumor types including melanoma, carcinomas from colorectal, thyroid and ovarian origins, and primary brain tumors [4]. BRAF alterations are most commonly found in circumscribed (or non-diffuse) gliomas like pilocytic astrocytoma (PA), ganglioglioma, pleomorphic xanthoastrocytoma (PXA), and anaplastic PXA, but have also been described in epithelioid glioblastoma (E-GBM) [5,6,7,8,9,10].
Interestingly, common clinical, histologic, immunohistochemical, and molecular features have been described for E-GBM and anaplastic PXAs, including the BRAF p.V600E mutation [11]. Moreover, methylation studies show that the majority of cases with a histologic diagnosis of E-GBM cluster with PXAs, obscuring the divide between these 2 apparently distinct entities [12]. While the genetic landscape of anaplastic PXAs shows recurring concomitant BRAF and CDKN2A/B alterations and frequent TERTp mutations [13], the additional genetic alterations that coexist with BRAF mutations in gliomas remain poorly understood. A better understanding of these additional alterations could improve the molecular classification of BRAF-mutant gliomas.
The majority of BRAF mutations in cancer affect the kinase domain at the 600th codon, where valine is substituted for glutamate (V600E). BRAF p.V600E confers approximately a tenfold greater kinase activity than wild-type (WT) BRAF [4]. While this mutation is often associated with poor prognosis in some gliomas [7, 14], it has also been shown to correlate with better prognosis in cases of isocitrate dehydrogenase wildtype glioblastomas (GBM IDH-WT) [15]. Immunohistochemistry with a BRAF p.V600E specific antibody is a useful tool for the detection of this mutation in gliomas. However, this approach is limited because it can only identify tumors with the V600E alteration. Targeted therapies, such as BRAF p.V600E (dabrafenib, vemurafenib) and MEK (trametinib, cobimetinib) inhibitors, have proven to delay progression in melanoma patients with BRAF p.V600 mutations [16]. Moreover, these drugs have been recently used in the treatment of BRAF-mutant high-grade gliomas and have shown responses, highlighting the importance of recognizing BRAF-altered gliomas in clinical practice [17,18,19,20].
The goal of this study is to evaluate additional genetic alterations in BRAF-mutant gliomas in a large glioma institutional registry (n = 372) and in cBioPortal. This study will allow a better understanding of additional genetic alterations that frequently coexist with BRAF mutations in gliomas and their diagnostic implications.
Methods
Patients and samples
372 consecutive glioma patients undergoing biopsy or surgical resection and whose tissue was evaluated by NGS (2010 to 2017) were included in this study, after approval by the institutional review board of the University of Texas Health Science Center at Houston (UTHealth) and Memorial Hermann Hospital, Houston, TX. Clinical data (age, gender, histologic diagnosis, imaging studies, recurrence, and survival) were collected using medical records and compiled using a REDCap database. Diagnosis were performed following the 2016 WHO Classification of Tumors of the CNS by board-certified neuropathologists.
Next-generation sequencing
Tumors were analyzed for genetic alterations by a targeted next-generation sequencing assay (NGS) interrogating 315 genes (FoundationOne®, Foundation Medicine Inc., Cambridge, MA, USA). The FoundationOne® assay was performed in a clinical laboratory improvement amendments (CLIA)-certified laboratory, as previously described [21, 22]. Tumor mutation burden (TMB) was calculated based on the number of somatic mutations in sequenced genes and extrapolating the value to the genome as a whole using a validated algorithm [23, 24]. TMB was reported as a number of mutations per megabase (mb) of genome.
cBioPortal for cancer genomics
We searched for BRAF-mutant gliomas with mutation and copy-number alterations available in cBioPortal (https://www.cbioportal.org/) (data accessed on August 2020) [25, 26]. We included cases from the following cBioPortal datasets: Brain Tumor PDXs (Mayo Clinic, 2019), Brain Lower Grade Glioma (TCGA, PanCancer Atlas), Glioblastoma Multiforme (TCGA, PanCancer Atlas), Memorial-Sloan Kettering Cancer Center (MSKCC, 2019), LGG University of California San Francisco (UCSF 2014), and GBM Columbia (Columbia, 2019) [27,28,29,30]. The cBioPortal Oncoprint tool was utilized to perform the oncoplots of both UTHealth and cBioPortal datasets [25, 26].
Results
Characteristics of the study cohort
The institutional database (UTHealth cohort) included 372 patients with a mean age of 57 years (range 2–87 years). There were 219 (58.9%) males, 263 (70.7%) Caucasians, 52 (14.0%) Hispanic, 36 (9.7%) African-American, 15 (4.0%) Asians, and 6 (1.6%) of other racial backgrounds. Histologic diagnoses in descending order of frequency include: GBM IDH-WT (249/372, 66.9%), GBM IDH-mutant (24/372, 6.5%), oligodendroglioma IDH-mutant and 1p/19q co-deleted (OD) (22/372, 5.9%), anaplastic astrocytoma (AA) IDH-mutant (16/372, 4.3%), diffuse astrocytoma (DA) IDH-mutant 15 (4.0%), AA, IDH-WT (13/372, 3.5%), anaplastic oligodendroglioma, IDH-mutant and 1p/19q codeleted (AO) (12/372, 3.2%), DA IDH-WT (9/372, 2.4%), pilocytic astrocytoma (PA) (6/372, 1.6%), PXA (3/372, 0.8%), E-GBM (2/372, 0.6%), and subependymal giant cell astrocytoma (1/372, 0.3%).
Characteristics of BRAF-mutant gliomas
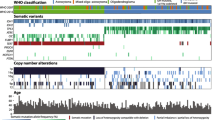
BRAF mutations were identified in 17/372 (4.6%) cases: 1/9 (11.1%) DA IDH-WT, 8/249 (3.2%) GBM IDH-WT, 2/2 (100%) E-GBMs, 1/12 (8.3%) AO, 3/6 (50%) PA, and 2/3 (66.7%) PXAs (Fig. 1). There were 8 females and 9 males with a median age of 50 years (range 2–83 years). The median age by diagnosis was: GBM IDH-WT (51.5; range 23–71 years), E-GBM (23.5; range 23–24 years), PXA (24; range 21–27 years), and PA (15; range from 2 to 26 years). Seizures were a presenting symptom in 5/17 (29.4%) patients. The anatomic distribution of BRAF-altered gliomas is shown in Fig. 2a. Lesions occurred in the temporal lobe in 9/17 (53%) cases; 4/8 GBM IDH-WT, 1/2 E-GBMs, 2/2 PXAs, 1/1 DA IDH-WT, 2/3 PAs. Two BRAF-altered GBM IDH-WT occurred in a midline location (#4 with brainstem involvement and #8 involving thalamus). The majority of patients with GBM IDH-WT (including both E-GBM cases) underwent resection followed by TMZ (10/10) and radiotherapy (RT) (9/10) according to the Stupp protocol [31], Online Resource 1. The median OS of patients with GBM IDH-WT with a BRAF alteration was 12.8-months and 7/8 (87.5%) patients are now deceased. All patients with PAs, PXAs, and E-GBM are alive at the moment of this report with an average OS of 52.4, 145.6, and 12.1 months, respectively. The DA IDH-WT and AO patients with a BRAF alteration are deceased with an OS of 16.1 and 22.7 months, respectively.

Mutations in cancer-related genes in 17 patients with BRAF-mutant gliomas. BRAF-mutant GBMs frequently showed TERTp, CDKN2A/B, PTEN, and/or TP53 mutations. E-GBMs and PXAs showed similar genomic alterations and similar age at diagnosis. PAs showed BRAF-KIAA1549 fusion even when located in the temporal lobe. WHO World Health Organization, PFS progression-free survival, OS overall survival, GBM IDH-WT glioblastoma IDH-wildtype, E-GBM epithelioid glioblastoma, PXA pleomorphic xanthoastrocytoma, OD oligodendroglioma, PA pilocytic astrocytoma. The GBM group includes one case with a histologic diagnosis of diffuse astrocytoma IDH-WT that showed molecular features of GBM WHO grade 4, according to cIMPACT-NOW Update 3 [46]

a Anatomic location for the 17 BRAF-mutant gliomas demonstrating that 52% (9/17) of the BRAF-mutated gliomas occurred in the temporal lobe. One BRAF-mutant glioma occurred in a midline location (thalamus) and one tumor showed frontal and brainstem involvement. b BRAF-mutant gliomas from the institutional database showing that 53% of patients have the V600E mutation. c BRAF-mutant gliomas from cBioPortal showing that 44% of cases have the V600E mutation. Other alterations included BRAF amplification, non V600E missense mutation, loss, and KLH7-BRAF fusion
Additional genomic alterations in BRAF-mutant gliomas
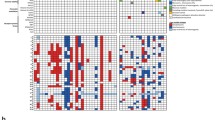
NGS identified 179 genomic alterations involving 99 genes. The median number of mutations per patient was 10 (range 7–17). Most alterations were reported as variants of unknown significance (VUS) (105/173 or 60.7%). As demonstrated in Fig. 2b, 9/17 (53%) patients had the BRAF p.V600E mutation and 9/17 (53%) had BRAF mutations distinct from p.V600E (one patient had two BRAF alterations, p.V600E and a non-V600E mutation). Other BRAF alterations included: BRAF-KIAA1549 fusion in PA, BRAF amplification, p.K483E, p.D594N, and p.N5811 in GBM IDH-WT, and BRAF p.L597Q in a patient with DA IDH-WT.
Genomic alterations for the 17 patients are reported in Fig. 1 and Online Resource 1. All cases except patient 14 (AO) were IDH-WT. In addition to BRAF alterations, BRAF-mutant gliomas showed mutations in TP53, TERTp, CDKN2A/B, and PTEN, among others. Online Resource 2 shows the mutations observed in these genes and their predicted effects on protein function (gain/loss of function).
Patients with BRAF-mutant GBM IDH-WT (n = 8) frequently showed additional alterations in PTEN (n = 5), CDKN2A/B (n = 6), and TERTp mutations (n = 6). In contrast, E-GBMs and PXAs exhibited BRAF p.V600E and CDKN2A/B mutations without TERTp, PTEN, or TP53 mutations. Additionally, MTAP was mutated in two patients, 1/2 E-GBM and 1/2 PXA. We compared the frequency of CDKN2A/B, TERTp, PTEN, and TP53 mutations between E-GBM/PXA (n = 4) and GBM/DA IDH-WT (n = 9). GBM/DA IDH-WT patients had increased frequency of concomitant CDKN2A/B and TERTp mutations compared to E-GBM/PXA (77.8% vs. 0%). Also, TP53 or PTEN mutations were more commonly present in GBM/DA compared to E-GBM/PXA (77.8% vs. 0%).
BRAF-mutant gliomas in cBioPortal datasets
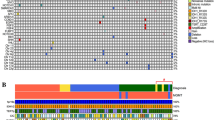
In cBioPortal, 82/2185 (3.8%) gliomas with a BRAF mutation were identified. Fifty-five out of the 82 patients were included in the study, as some patients were incompletely characterized, had unclear histologic diagnosis, or BRAF gene deletion, truncation, or variant of unknown significance (Online Resource 3). Fifty-six BRAF mutations were identified in the 55 patients (1 patient had two non-V600E point mutations). BRAF alterations included p.V600E (23/82, 41%), amplification (14/82, 25%), non p.V600E point mutations (11/82, 20%) and fusions (8/82, 14%) including KLHL7-BRAF, BRAF-TPR, BRAF-ATF7, FAM131B-BRAF, BRAF-UBE2H, BRAF-KIAA1549, and intragenic fusions (Fig. 2c). Most cases were classified as GBM, IDH-WT (33/55, 60%), while the remaining cases included 7 (12.8%) DA IDH-Mutant, 3 (5.6%) PA, 2 (3.6%) GBM IDH-Mutant, 2 (3.6%) AA IDH-Mutant, 2 (3.6%) DA IDH-WT, 2 (3.6%) ganglioma, 1 (1.8%) AA IDH-WT, 1 (1.8%) OD, 1 (1.8%) anaplastic PXA, and 1 (1.8%) PXA. The most common additional mutations in BRAF-mutant gliomas in cBioPortal were CDKN2A/B, TERTp, TP53, PTEN, TP53, IDH1/IDH2, and ATRX (Fig. 3). Considering the subgroup of patients with available TERTp status, all TERTp-mutant cases were diagnosed as GBM IDH-WT or OD. Interestingly, BRAF-amplification was the most common BRAF alteration in IDH-Mutant astrocytomas (7/11 63.7%) and PA showed BRAF fusions (BRAF-KIAA1549 or FAM131B-BRAF).

BRAF-mutant, IDH-WT, gliomas in cBioPortal. Fifty-five (55/2185, 2.5%) gliomas were identified to harbored BRAF activating mutations in six datasets available at cBioPortal. WHO World Health Organization, PFS progression-free survival, OS overall survival, WT wildtype, Mut mutant, GBM glioblastoma, AA anaplastic astrocytoma, DA diffuse astrocytoma, PXA pleomorphic xanthoastrocytoma, OD oligodendroglioma, PA pilocytic astrocytoma
Clinical use of BRAF inhibitors
Only two patients in our study (7 and 12) received targeted therapies with BRAF pathway inhibitors (cobimetinib, vemurafenib).
Case 7 is a 63-year-old female with an infiltrating BRAF-mutant (p.N581I—BRAF class 3 mutation), TP53-mutant (p.L257Q) glioma, diagnosed as GBM IDH-WT. After initial therapy and multiple therapeutic regimens for progressive disease, the patient’s MRI demonstrated worsening tumor burden (Fig. 4a, b). Therefore cobimetinib (MEK inhibitor) was initiated, as preclinical data suggest that class 3 BRAF mutations are resistant to RAF inhibitors like vemurafenib [32]. MRI 6-weeks after cobimetinib demonstrated a significant decrease in the enhancing tumor and vasogenic edema consisting with tumor response (Fig. 4c, d). The patient was deceased 5-months after initiating cobimetinib.

T1 post-contrast magnetic resonance imaging (T1c-MRI) and T2 Flair-MRI in a GBM patient with a BRAF p.N581I mutation (patient 7) before (a, b) and after (c, d) 6-weeks of treatment with cobimetinib. T1 post-contrast magnetic resonance imaging (T1c-MRI) and T2 Flair-MRI in a patient with PXA with BRAF p.V600E mutation (patient 12) before (e, f) and after (g, h) 10-weeks of cobimetinib and vemurafenib treatment. The post-treatment images exhibited a significant decrease in enhancement and vasogenic edema
Case 12 is a 21-year-old female who underwent resection of a relatively circumscribed, BRAF-mutant (p.V600E) and CDKN2A/B-mutant glioma, diagnosed as PXA, WHO grade II. After initial therapy and multiple therapeutic regimens for progressive disease, the patient’s MRI demonstrated tumor progression (Fig. 4e, f). Subsequently, cobimetinib and vemurafenib treatment (MEK and BRAF inhibitors) were added to the treatment regimen. MRI 10-weeks after cobimetinib/vemurafenib showed an excellent response (Fig. 4g, h). At the moment of this report, 10-months since the last recurrence, the patient is alive and continuing oral therapy with cobimetinib and vemurafenib with no disease progression.
Discussion
A growing body of genomic information has provided the opportunity to include genotype in the diagnosis and treatment of cancer patients [33]. Although previous studies have focused on BRAF p.V600E mutations in gliomas [3, 6, 9], the association of BRAF mutations with other genomic alterations in gliomas remains to be further refined.
The frequency of BRAF p.V600E in GBM IDH-WT, E-GBMs, DA IDH-WT, and PXAs in our study are similar to those previously described [6, 8, 34]. Consistent with previous studies [13, 34, 35], the mean age of diagnosis for BRAF-mutant PA and PXAs were 15 and 24 years, respectively. The median age of diagnosis for BRAF-mutant GBM IDH-WT was 51.5 years. This age is younger than the reported mean age for GBM IDH-WT (~ 65 years) [1, 8]. Unlike IDH mutations that are associated with infiltrating gliomas occurring at a younger age (< 55 years), BRAF mutations were associated with gliomas occurring at a wide range of ages (range 23–71 years) [36]. The median OS of the BRAF-mutant GBM IDH-WT gliomas was 12.8 months, lower than the median OS of 19.1 months for the remaining 241 GBM, IDH-WT (BRAF-wildtype) cases in our database. The average survival of patients with PA (52.4 months) and PXA (145.2 months) was consistent with the expected long survival for patients with these tumor types [13, 37, 38].
Epithelioid GBM vs. PXA
The age and the mutations in the two E-GBMs in our database strongly resembled those of the two PXAs. The four patients were diagnosed in early adulthood (21–27 years) and exhibited BRAF p.V600E and CDKN2A/B mutations, and lacked TERTp, PTEN, or TP53 mutations. In cBioPortal, there is 1 PXA with a BRAF-TPR fusion and 1 anaplastic PXA with BRAF p.V600E and a CIC mutation. Interestingly, a recent study of whole-genome methylation analysis of 64 E-GBMs concluded that the histopathologically defined E-GBM does not represent a single diagnostic entity. E-GBM cases could be classified into one of three molecularly and biologically distinct categories based on the genome methylation signature; (i) PXA with favorable prognosis, predominantly in children and young adults (38/64, 59.4%), (ii) GBM, IDH-WT with poor prognosis, mainly occurring in older adults, albeit with more frequent BRAF mutations (17/64, 26.6%), and (iii) pediatric GBM RTK1 of intermediate prognosis in children and young adults, associated with chromothripsis and frequent PDGFRA amplifications (9/64, 14%) [12]. Therefore, the two cases diagnosed as E-GBMs on histologic grounds alone (given the presence of necrosis) in our study might be better classified as PXAs, based on the current understanding of molecular alterations of these tumors [11, 12]. Although the follow up is limited, the average OS of the E-GBM patients was 12.1 months. At the time of this report, both E-GBM patients are alive and without recurrence, which would be uncommon for patients with GBM [31].
Other alterations in BRAF-mutant gliomas
In GBM IDH-WT, BRAF mutations are strongly associated with TERTp and PTEN mutations and mutations in either TP53 or CDKN2A/B. TP53 mutations and CDKN2A/B loss appear to be mutually exclusive in cases with BRAF missense mutations, as previously reported [39]. In agreement with this observation, in the cBioPortal dataset, TP53 mutation and CDKN2A/B loss are mutually exclusive events (p ≤ 0.001, n = 885) [25, 26, 40]. Our results show an association between coexisting BRAF p.V600E, CDKN2A/B loss, and TERTp mutations and a diagnosis of GBM, IDH-WT. In contrast, PXA/E-GBMs showed BRAF p.V600E and CDKN2A/B loss without TERTp mutations. These data suggest that TERTp mutations can help distinguish BRAF-mutant GBM from PXA/E-GBM.
We identified 2 oligodendrogliomas, IDH-mutant and 1p/19q codeleted with a BRAF mutation and 11 IDH-mutant astrocytomas with BRAF alterations. While BRAF alterations have been rarely detected in IDH-mutant tumors, the significance of this finding remains to be determined [41].
BRAF-mutant gliomas and the temporal lobe
The frequencies of PXAs and E-GBMs localizing to the temporal lobe in previous studies were similar to our cohort (3/4, 75%) [8, 35]. However, the previously reported frequency of GBMs occurring in the temporal lobe (28%) is lower than that of the BRAF-mutant GBM, IDH-WT cases in our study (4/8, 50%). This supports the observation that BRAF-mutant GBMs have a predilection for the temporal lobe [42, 43]. Our data did not show an association between PXA/E-GBM histology or CDKN2A/B loss and localization to the temporal lobe. However, the number of PXA/E-GBMs in our study is small.
BRAF p.V600E immunohistochemistry and targeting BRAF in gliomas
Two patients in our study (#7-GBM and #12-PXA) were treated with cobimetinib and vemurafenib and both responded favorably. Patient 7 had a non-V600E BRAF mutation, suggesting that MAPK pathway inhibitors could be incorporated in the treatment of gliomas with BRAF mutations other than the p.V600E. Importantly, in the present study, taking into account cases in our cohort and the cBioPortal dataset, 57% of BRAF-mutations were non-V600E. Recognizing BRAF-mutations opens the possibility of incorporating targeted therapy (BRAF and MEK inhibitors) in the treatment plan of glioma patients, which could be missed if BRAF status is only evaluated with the BRAF p.V600E specific antibody. Moreover, identifying the particular BRAF mutations might be crucial, as preclinical data has shown that some might not respond to BRAF inhibitors (class 3 mutations) and should be better treated with MEK inhibitors, as shown in case 7 [32].
BRAF mutant adults and pediatric gliomas
Our cohort includes mostly adult patients, which differs from most prior studies that include BRAF-mutant gliomas in the pediatric population [44]. This difference is driven by the clinical practice at our institution and it allows us to explore BRAF-mutant gliomas in an adult cohort. A recent study determined that nearly all pediatric low-grade gliomas harbored RAS/MAPK activation pathway alterations, with KIAA1549-BRAF fusion, BRAF p.V600E, or NF1 mutations accounting for 66% of cases [45]. Pediatric patients in our study included 1 PA with KIAA1549-BRAF and 2 gangliogliomas (p.V600E-mutant). In contrast to pediatric gliomas, BRAF alterations are infrequent in adults’ gliomas but can be detected in ~ 2–5% of patients with p.V600E being the most common.
Summary
In this study, we defined additional alterations in BRAF-mutant gliomas and their potential diagnostic implications. Our results demonstrate that gliomas with BRAF mutations frequently exhibit additional alterations in TP53, TERTp, CDKN2A/B, and PTEN and show a favorable response to BRAF and/or MEK inhibitors. Knowledge of alterations that co-occur with BRAF mutations in gliomas, in particular, alterations involving CDKN2A/B, TERTp, TP53, and PTEN may improve glioma diagnosis (Fig. 5). For example, BRAF-mutant gliomas with co-existing CDKN2A/B, TERTp, TP53, or PTEN mutations may be considered as GBM, while BRAF-mutant tumors with only CDKN2A/B alterations, but no mutations in TERTp, TP53, or PTEN appear to be better classified as PXAs. However, we recognize that our conclusions are limited by a small sample size (due to the rarity of BRAF-mutant infiltrating gliomas) and the absence of TERTp information in cBioPortal. Nonetheless, the additional genetic alterations that coexist with BRAF mutations can inform the diagnosis of gliomas including PXA, E-GBM, and “BRAF-mutant GBM IDH-WT”. Accurate diagnosis of these tumors is critical as their prognosis and treatment differs.

Possible molecularly informed diagnosis of BRAF-mutant gliomas. Incorporation of additional alterations that co-occur in BRAF-mutant gliomas may improve their molecularly informed diagnosis. Circumscribed and epithelioid BRAF-mutant gliomas with CDKN2A/B loss and intact TP53 and TERTp are most likely Pleomorphic xanthoastrocytomas. In contrast, BRAF mutant infiltrating gliomas with CDKN2A/B loss, TERTp mutations, and with or without PTEN or TP53 are most likely GBM. Similarly, BRAF-mutant infiltrating gliomas with TP53 mutation are most likely GBMs. Circumscribed gliomas with the KIAA1549-BRAF fusion are most likely pilocytic astrocytomas
Data availability
The datasets generated during and/or analyzed during the current study are available upon reasonable request to the corresponding authors.
References
Ostrom QT, Gittleman H, Truitt G et al (2018) CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015. Neuro Oncol 20:1–86. https://doi.org/10.1093/neuonc/noy131
Koshy M, Villano JL, Dolecek TA et al (2012) Improved survival time trends for glioblastoma using the SEER 17 population-based registries. J Neurooncol 107:207–212. https://doi.org/10.1007/s11060-011-0738-7
Maraka S, Janku F (2018) BRAF alterations in primary brain tumors. Discov Med 26:51–60
Davies H, Bignell GR, Cox C et al (2002) Mutations of the BRAF gene in human cancer. Nature 417:949–954. https://doi.org/10.1038/nature00766
Dias-Santagata D, Lam Q, Vernovsky K et al (2011) BRAF V600E mutations are common in pleomorphic xanthoastrocytoma: diagnostic and therapeutic implications. PLoS ONE 6:e17948. https://doi.org/10.1371/journal.pone.0017948
Schindler G, Capper D, Meyer J et al (2011) Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol 121:397–405. https://doi.org/10.1007/s00401-011-0802-6
Dahiya S, Haydon DH, Alvarado D et al (2013) BRAFV600E mutation is a negative prognosticator in pediatric ganglioglioma. Acta Neuropathol 125:901–910. https://doi.org/10.1007/s00401-013-1120-y
Kleinschmidt-DeMasters BK, Aisner DL, Birks DK, Foreman NK (2013) Epithelioid GBMs show a high percentage of BRAF V600E mutation. Am J Surg Pathol 37:685–698. https://doi.org/10.1097/PAS.0b013e31827f9c5e
Behling F, Barrantes-Freer A, Skardelly M et al (2016) Frequency of BRAF V600E mutations in 969 central nervous system neoplasms. Diagn Pathol 11:55. https://doi.org/10.1186/s13000-016-0506-2
Wesseling P, Capper D (2018) WHO 2016 classification of gliomas. Neuropathol Appl Neurobiol 44:139–150. https://doi.org/10.1111/nan.12432
Alexandrescu S, Korshunov A, Lai SH et al (2016) Epithelioid glioblastomas and anaplastic epithelioid pleomorphic xanthoastrocytomas—same entity or first cousins? Brain Pathol 26:215–223. https://doi.org/10.1111/bpa.12295
Korshunov A, Chavez L, Sharma T et al (2018) Epithelioid glioblastomas stratify into established diagnostic subsets upon integrated molecular analysis. Brain Pathol 26:656–662. https://doi.org/10.1111/neup.12459
Ida CM, Rodriguez FJ, Burger PC et al (2015) Pleomorphic xanthoastrocytoma: natural history and long-term follow-up. Brain Pathol 25:575–586. https://doi.org/10.1111/bpa.12217
Lassaletta A, Zapotocky M, Mistry M et al (2017) Therapeutic and prognostic implications of BRAF V600E in pediatric low-grade gliomas. J Clin Oncol 35:2934–2941. https://doi.org/10.1200/JCO.2016.71.8726
Chan A-Y, Zhang R-Q, Aibaidula A et al (2018) BRAF mutation marks out specific subgroups of glioma. Glioma 1:168. https://doi.org/10.4103/glioma.glioma_33_18
Ribas A, Gonzalez R, Pavlick A et al (2014) Combination of vemurafenib and cobimetinib in patients with advanced BRAFV600-mutated melanoma: a phase 1b study. Lancet Oncol 15:954–965. https://doi.org/10.1016/S1470-2045(14)70301-8
Matsumura N, Nakajima N, Yamazaki T et al (2017) Concurrent TERT promoter and BRAF V600E mutation in epithelioid glioblastoma and concomitant low-grade astrocytoma. Neuropathology 37:58–63. https://doi.org/10.1111/neup.12318
Kaley T, Touat M, Subbiah V et al (2018) BRAF inhibition in BRAF V600-mutant gliomas: results from the VE-BASKET study. J Clin Oncol 36:3477–3484. https://doi.org/10.1200/JCO.2018.78.9990
Johanns TM, Ferguson CJ, Grierson PM et al (2018) Rapid clinical and radiographic response with combined dabrafenib and trametinib in adults with BRAF-mutated high-grade glioma. J Natl Compr Cancer Netw 16:4–10. https://doi.org/10.6004/jnccn.2017.7032
Nicolaides TP, Li H, Solomon DA et al (2011) Targeted therapy for BRAFV600E malignant astrocytoma. Clin Cancer Res 17:7595–7604. https://doi.org/10.1158/1078-0432.CCR-11-1456
Frampton GM, Fichtenholtz A, Otto GA et al (2013) Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 31:1023–1031. https://doi.org/10.1038/nbt.2696
Dono A, Wang E, Lopez V et al (2020) Molecular characteristics and clinical features of multifocal glioblastoma. J Neurooncol. https://doi.org/10.1007/s11060-020-03539-z
Rosenberg JE, Hoffman-Censits J, Powles T et al (2016) Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 387:1909–1920. https://doi.org/10.1016/S0140-6736(16)00561-4
Goodman AM, Kato S, Bazhenova L et al (2017) Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther 16:2598–2608. https://doi.org/10.1158/1535-7163.MCT-17-0386
Gao J, Aksoy BA, Dogrusoz U et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6:1–20. https://doi.org/10.1126/scisignal.2004088
Cerami E, Gao J, Dogrusoz U et al (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2:401–404. https://doi.org/10.1158/2159-8290.CD-12-0095
Liu J, Lichtenberg T, Hoadley KA et al (2018) An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 173:400–416.e11. https://doi.org/10.1016/j.cell.2018.02.052
Jonsson P, Lin AL, Young RJ et al (2019) Genomic correlates of disease progression and treatment response in prospectively characterized gliomas. Clin Cancer Res 25:5537–5547. https://doi.org/10.1158/1078-0432.CCR-19-0032
Zhao J, Chen AX, Gartrell RD et al (2019) Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med 25:462–469. https://doi.org/10.1038/s41591-019-0349-y
Johnson BE, Mazor T, Hong C et al (2014) Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 343:189–193. https://doi.org/10.1126/science.1239947
Stupp R, Mason WP, van den Bent MJ et al (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996. https://doi.org/10.1056/NEJMoa043330
Yao Z, Yaeger R, Rodrik-Outmezguine VS et al (2017) Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 548:234–238. https://doi.org/10.1038/nature23291
Haber DA, Gray NS, Baselga J (2011) The evolving war on cancer. Cell 145:19–24. https://doi.org/10.1016/j.cell.2011.03.026
Jones DTW, Kocialkowski S, Liu L et al (2008) Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 68:8673–8677. https://doi.org/10.1158/0008-5472.CAN-08-2097
Phillips JJ, Gong H, Chen K et al (2019) The genetic landscape of anaplastic pleomorphic xanthoastrocytoma. Brain Pathol 29:85–96. https://doi.org/10.1111/bpa.12639
Parsons DW, Jones S, Zhang X et al (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812. https://doi.org/10.1126/science.1164382
Ohgaki H, Kleihues P (2005) Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol 64:479–489. https://doi.org/10.1093/jnen/64.6.479
Giannini C, Scheithauer BW, Burger PC et al (1999) Pleomorphic xanthoastrocytoma. Cancer 85:2033–2045. https://doi.org/10.1002/(SICI)1097-0142(19990501)85:9<2033:AID-CNCR22>3.0.CO;2-Z
Yan Y, Takayasu T, Hines G et al (2020) Landscape of genomic alterations in IDH wild-type glioblastoma identifies PI3K as a favorable prognostic factor. JCO Precis Oncol 3:575–584
McLendon R, Friedman A, Bigner D et al (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455:1061–1068. https://doi.org/10.1038/nature07385
Ahrendsen J, Torre M, Meredith D et al (2020) IDH-mutant gliomas with other class-defining events—an argument for broad genetic testing. J Neuropathol Exp Neurol 79:656. https://doi.org/10.1093/jnen/nlaa036
Larjavaara S, Mäntylä R, Salminen T et al (2007) Incidence of gliomas by anatomic location. Neuro Oncol 9:319–325. https://doi.org/10.1215/15228517-2007-016
Tandon N, Esquenazi Y (2013) Resection strategies in tumoral epilepsy: is a lesionectomy enough’. Epilepsia 54:72–78. https://doi.org/10.1111/epi.12448
Vuong HG, Altibi AMA, Duong UNP et al (2018) BRAF mutation is associated with an improved survival in glioma—a systematic review and meta-analysis. Mol Neurobiol 55:3718–3724. https://doi.org/10.1007/s12035-017-0599-y
Ryall S, Zapotocky M, Fukuoka K et al (2020) Integrated molecular and clinical analysis of 1,000 pediatric low-grade gliomas. Cancer Cell 37:569–583.e5. https://doi.org/10.1016/j.ccell.2020.03.011
Brat DJ, Aldape K, Colman H et al (2018) cIMPACT-NOW update 3: recommended diagnostic criteria for “Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV”. Acta Neuropathol 136:805–810. https://doi.org/10.1007/s00401-018-1913-0
Acknowledgements
We would like to thank Melissa Stephens, Ayna Matyakuba, and Kimberly Fontenot for their assistance with this project.
Funding
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number K08CA241651 (LYB). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
Experimental design: YE, LYB. Collection and assembly of data: AD, JV, MA, GH, TT, JJZ. Analysis and interpretation of the data: AD, JV, MA, GH, YY, YE, LYB. Manuscript writing: AD, JV, MA, GH, NT, YE, LYB. Final approval of manuscript: all authors.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declared no conflict of interest.
Ethical approval
This retrospective study was approved by the institutional review board of The University of Texas Health Science Center at Houston and Memorial Hermann Hospital, Houston, TX, following the 1964 Helsinki Declaration and its later amendments.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Dono, A., Vu, J., Anapolsky, M. et al. Additional genetic alterations in BRAF-mutant gliomas correlate with histologic diagnoses. J Neurooncol 149, 463–472 (2020). https://doi.org/10.1007/s11060-020-03634-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-020-03634-1