
Abstract
Patients with diffuse pontine gliomas have a median survival of less than one year and represent a challenge for pediatric oncologists, prompting them to attempt experimental therapies. From 1987 to 2005, 62 children with diffuse pontine glioma, not amenable to curative surgery, were treated according to four successive pilot protocols: (1) concomitant chemo–radiotherapy (etoposide, cytarabine, ifosfamide, cisplatin, and dactinomycin); (2) intensive high-dose courses chemotherapy (cisplatin/etoposide, cyclophosphamide/vincristine/methotrexate) and a subsequent course of myeloablative thiotepa followed by radiation and maintenance chemotherapy; (3) cisplatin/etoposide followed by isotretinoin before, during and after focal irradiation; and (4) iv vinorelbine before, during, and after irradiation. Considering all patients, 77% experienced a transient response to treatment, always detectable after radiotherapy. The progression-free survival (PFS) rate was 25 ± 6% at one year, median PFS was seven months; overall survival (OS) was 45 ± 6%, median OS was eleven months: no statistical differences in the four studies in terms of outcome were detected. Despite improved diagnostic, therapeutic, and supportive tools in pediatric neuro-oncology, little has been achieved for patients with diffuse pontine tumors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In more than 70% of cases, patients with diffuse pontine gliomas present with a brief clinical history characterized by cranial nerve involvement and pyramidal and cerebellar signs and symptoms. Typical MRI findings (an intrinsic, pontine-based infiltrative lesion hypodense in T1 and hypertense in the T2 sequences, with a significant mass effect on adjacent structures) are diagnostic, making histological confirmation unnecessary [1]. Surgical treatment with curative intent is feasible only for children with a long clinical history, focal neurological deficits, and focal, cystic, exophytic, or cervico-medullary tumors on MRI scan—these prognostically favorable features are, unluckily, seen in the minority of patients with diffuse pontine gliomas [2, 3]. Diffuse pontine gliomas pose a challenge to pediatric oncologists, making it worthwhile to attempt experimental treatments in this setting.
From 1987 to 2005, we accrued 62 children with diffuse pontine glioma not amenable to curative surgery. Given the dismal prognosis reported in the literature, we adopted four different pilot protocols in succession over the years, obtaining the results reported below.
Materials and methods
We retrospectively included in this analysis all newly-accrued patients whose clinical features and tumor radiology findings corresponded to the recently clarified criteria [4, 5], i.e. short-lived symptoms (median one month), at least two neurological signs and symptoms including cranial nerve deficits, cerebellar dysfunction and long-tract impairment, and MRI evidence of a large, expansile, pontine-based lesion with indistinct margins. We subsequently excluded six patients treated according to the protocols described here because of a focal pontine involvement enabling partial excision in four cases, and referral after brachytherapy in one (four of these six patients were alive after more than five years as at the time of this report).
All patients were studied at diagnosis with MRI, which included the entire CNS from 1991 onwards, biochemistry, and cerebro-spinal fluid cytology, when feasible without risk. All children were fitted with a central line before beginning any treatment. MRI was repeated during and after therapy, every three months or more frequently if the symptoms became worse. For children surviving more than one year, MRI was repeated every four to six months.
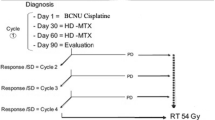
Figure 1 shows the four treatment protocols adopted over the years.

The four protocols. VP16, etoposide; ARA-C, cytarabine; IFO, ifosfamide; CDDP, cisplatin; DACT, actinomycin D; CPC, circulating progenitor cells; VCR, vincristine; EDX, cyclophosphamide; MTX, methotrexate; CCNU, lomustine; RT, radiotherapy; VNB, vinorelbine
The Ethical Committee’s approval was obtained and parents’ or tutors’ approval was required before beginning any diagnostic and therapeutic procedures.
Study 1. Concomitant chemo- and radiotherapy, 1987–1996, 21 patients. The schedule was the same for all malignant gliomas, using concomitant chemo and radiotherapy [6]. Treatment included etoposide (250 mg/m2, 30′ infusion for two doses, 8 h apart) on day 1, cytarabine (1.5 g/m2, 30′ infusion) on day 8, ifosfamide (5 g/m2, 24-h infusion) on day 20, cisplatin (100 mg/m2, 2-h infusion) on day 30, and dactinomycin (1 mg/m2 iv) on day 45. The scheduled duration of the whole study was 8 weeks. Radiotherapy was administered to the tumor bed plus 2 cm margins with a conventional fractionation of 180–200 cGy once a day for five days a week, up to a median dose of 54 Gy in 18 children; in three, it was hyperfractionated with two fractions of 1.1 Gy a day up to a total dose of 66 Gy.
Study 2. Pre-radiation sequential high-dose chemotherapy, 1996–2000. Ten consecutive children were treated with a pre-radiation sequential schedule that included high-dose chemotherapy and was adopted for all malignant gliomas [7]. The study consisted of induction with two courses of cisplatin (40 mg/m2) plus etoposide (150 mg/m2) for three days, and vincristine (1.4 mg/m2) plus cyclophosphamide (1.5 g/m2) and high-dose methotrexate (8 g/m2) for one day, followed by high-dose thiotepa (300 mg/m2 for three doses) then focal radiotherapy and maintenance with vincristine and lomustine for a total duration of one year. For most children, the planned total dose of radiation was 60 Gy, with a standard fractionation of 1.8 Gy a day, five days a week.
Study 3. Pre-radiation, concomitant and adjuvant cisplatin/etoposide plus isotretinoin, 2000–2002. Seventeen children received a combination of cisplatin (30 mg/m2) plus VP16 (150 mg/m2) on days 1–3 for ten monthly courses, each course being followed on days 10–25 by oral isotretinoin 100 mg/m2 divided into two daily doses. Focal radiotherapy was planned after the second course if the neurological signs were stable, using a schedule with a total dose of 54–60 Gy divided into daily fractions of 1.8–2 Gy.
Study 4. Pre-radiation, concomitant and adjuvant vinorelbine, 2002–2006. Fourteen children received vinorelbine before, during, and after radiotherapy, at a dose of 30 mg/m2 on days 1, 8, and 22 of each course (total duration ten months), reduced to 20 mg/m2 weekly when administered during radiotherapy. Radiation was delivered to the tumor bed 3–4 weeks after chemotherapy was started. The total dose delivered was 54–60 Gy in daily fractions of 200 cGy.
Statistical methods
The studies were observational, not randomized, retrospectively reported.
For each of the studies, progression-free survival (PFS) and overall survival (OS) were calculated in years according to the Kaplan and Meier method, considering failure and toxicity events from the date of first radiological evaluation and censoring data for progression and survival at the last follow-up visit [8]. Differences between patient groups were assessed using the log-rank test [9].
Results
All patients
The 62 patients included 39 females and were a median six years old (range 3–15). None suffered from neurofibromatosis type 1. Two patients presented at diagnosis with meningeal locations (both in the first study group). Classifying patients by the typical symptoms prompting the first radiological evaluation, i.e. cranial nerve deficits, cerebellar signs, and long tract impairment, 25 had all three symptoms, 15 had cranial nerve palsy and long tract impairment, 14 had cranial nerve palsy and cerebellar dysfunction, and eight had cranial nerve palsy alone. Twenty-two cases presented with ventricular enlargement at diagnosis, requiring a shunt in 13 instances.
Biopsy or resection of the minimal exophytic component was conducted for diagnostic purposes in 30 cases: 19 surgical procedures were performed during the first decade of the reported period (P < 0.01). Histological diagnoses were fibrillary astrocytoma in six children, anaplastic astrocytoma in 18 and glioblastoma multiforme in six.
Apart from different number of biopsies performed over the years, the four groups of patients were homogeneous for all other clinical features.
Study 1. Concomitant chemo and radiotherapy. More detailed results have already been published [6].
Progression of disease was evident clinically and on MRI in eight children, clinically evident but no MRI was performed in five, and only emerged on MRI in seven. In one child the tumor progressed locally and spread through the CNS.
PFS was 30 ± 10% at one year with a median PFS of seven months. Overall survival was 45 ± 6% at one year and 18 ± 5% at two, with a median survival of 12 months. There was one toxic death due to septic shock after etoposide neutropenia during irradiation.
Study 2. Pre-radiation sequential high-dose chemotherapy. The first four patients followed the chemotherapy schedule as outlined, but because pre-methotrexate hydration severely exacerbated the neurological symptoms in all cases, the next six children were given radiotherapy after the first chemotherapy course, and the remainder of the chemotherapy was administered after irradiation. One girl died of hemorrhagic interstitial pneumonia 13 months after diagnosis with no clinical signs of progression. All the other patients died of tumor progression.
Progression was clinically evident and confirmed radiologically in four, clinically evident but not confirmed by MRI in four, and first identified by MRI in one. Two children progressed with CNS dissemination alone.
PFS was 40 ± 15% at one year, with a median PFS of ten months. OS was 70 ± 14% at one year and 10 ± 9% at two, with a median survival of 13 months.
Study 3. Pre-radiation, concomitant and adjuvant cisplatin/etoposide plus isotretinoin. The treatment was easy to administer and well tolerated, without any major toxic events. Chemotherapy was always administered on an outpatient basis. Isotretinoin induced mild mucocutaneous dryness that did not develop into ulcerative mucositis because of the chemotherapeutic association.
Progression of the disease was clinically evident and confirmed radiologically in seven, clinically evident but not confirmed by MRI in six, and first became evident on MRI in four. Three children progressed with CNS dissemination alone.
PFS was 12 ± 8% at one year, with a median PFS of five months. OS was 29 ± 11% at one year and 12 ± 8% at two, with a median survival of nine months.
Study 4. Pre-radiation, concomitant and adjuvant vinorelbine. Vinorelbine was administered in a saline solution over 30 min through a central catheter; routinely-used antihemetics were given before its infusion. It was never necessary to hospitalize patients to administer the treatment. Two of the 14 patients subsequently developed multiple, transient episodes of monolateral peripheral facial nerve palsy during the treatment, which always regressed completely and spontaneously within about 30 min after completing the infusion. Macroscopic tumor progression was ruled out by MRI. The drug was subsequently administered as scheduled until the end of the study [10].
Progression of disease was clinically evident and also assessed by MRI in nine, clinically evident but not radiologically assessed in two, and emerged only on MRI in one. Two children progressed with CNS dissemination alone.
PFS was 21 ± 11% at one year, with a median PFS of seven months. OS was 43 ± 13% at one year and 21 ± 11% at two, with a median survival of nine months. Two children were alive without progression at 31 and 48 months, at the time of this report.
Considering all 62 patients treated between 1987 and 2005, 48/62 experienced a transient response to treatment in terms of an improvement in symptoms and/or radiologically evident tumor reduction that always became evident after completing the radiotherapy. There were no statistical differences between the four studies in terms of the responses obtained. We also compared the 30 children who had a histological diagnosis, to determine whether there was any difference according to grade; we found no difference, even when grouping patients according to sub-groups:
-
1
grade 2 patients versus grades 3 + 4; and
-
2
grades 2 + 3 against grade 4 (data not shown).
In the 41 patients studied with MRI during treatment or follow-up, eight patients had signs of dissemination and all the others had local progression.
In all, PFS was 25 ± 6% at one year and 3 ± 3% at two, with a median PFS of seven months (Fig. 2); OS was 45 ± 6% at one year and 14 ± 5% at two, with a median OS of 11 months (Fig. 3).

PFS for the four studies

OS for the four studies
There were two long-term survivors, at 31 and 48 months after their first MRI showing the tumor. None was submitted to a surgical approach, therefore, based only on the radiological and clinical features, we could not determine any peculiar feature to explain the different outcome.
Discussion
In the last 20 years, pediatric neuro-oncology has benefited from a number of advances. New neuroradiological tools have become available, such as MRI and PET, with their specific diagnostic potential. Sophisticated neurosurgical tools have been introduced, with neuronavigation, endoscopy, and the operating microscope. Radiotherapeutic techniques have improved, enabling the doses delivered to the tumor to be increased without further harming the healthy brain. New drugs have become available that can be administered alone or in polychemotherapy schedules, with the potential to overcome the blood–brain barrier. Myeloablative schedules have been implemented and, especially in the last five years, specific tumor subtypes have been distinguished, that demand specific therapy to suit their molecular profiles and their biological and prognostic features. Major progress has been made in survival rates and quality of survival, thanks to the efforts of highly specialized, dedicated pediatric neuro-oncology teams at work from the moment a first diagnostic image is recorded and throughout post-treatment rehabilitation.
These considerations apply to all pediatric CNS tumors except for intrinsic pontine glioma, which remains the grimmest and most frustrating disease pediatric oncologists have to face.
In our 20-year experience, we have more or less attempted all the reasonable strategies available at the time.
Radiotherapy is the only definitely-indicated treatment for diffusely infiltrating pontine gliomas and the standard treatment remains the best option with local field radiotherapy amounting to a total dose of 54–60 Gy in six weeks. Without radiation, the median survival rate is approximately 20 weeks [11], while radiotherapy achieves a worthwhile (albeit temporary) improvement in neurological functions in nearly 70% of patients [12]. Increased amounts of radiation and hyperfractionation have been unable to afford any additional survival benefit, however, while inducing more severe side-effects and longer steroid dependence [13].
As discussed in a previous paper [6], the rationale for using chemotherapy together with radiotherapy in our first protocol was based on the assumption of a better clinical outcome because of:
-
1
more tumor cells being killed; and
-
2
the blood–brain barrier against antiblastic drugs being overcome.
A more recent SFOP experience [14] exploiting the potentially synergic effect of carboplatin compounds with radiation therapy [15] in 38 patients produced results similar to those of our first study, with eleven-month survival of 47.4% and a median survival of eleven months. The situation was much the same when interferon, tamoxifen, or other chemotherapeutic schedules were tried. Freeman et al. also pointed out that combining cisplatin with radiation may have meant, if not a worse outcome for patients enrolled in POG study 9239, at least a deterioration in quality of life, as well as making it difficult to understand any imaging changes after treatment [16].
We consequently developed study 2 in an attempt to overcome the drug resistance of diffuse pontine tumors by means of sequential chemotherapy with non-cross-resistant antiblastics and with a myeloablative dose of thiotepa. Nobody has reported obtaining satisfactory results by applying this strategy to diffuse pontine glioma. Like increasing the radiotherapy dosage, so with increasing doses of chemotherapy also, the median OS has remained superimposable. In the study by Dunkel et al., six patients with newly diagnosed intrinsic pontine tumors received high-dose chemotherapy with carmustine, thiotepa, and etoposide followed by autologous bone marrow rescue and HFRT, achieving a median survival of 11.4 months after bone marrow reinfusion and, although there were no cases of fatal toxicity, all patients died of tumor progression [17]. Bouffet described the results obtained in a group of 24 patients given focal radiotherapy followed by myeloablative chemotherapy with busulfan and thiotepa, comparing them with the outcome in patients who, for various reasons, were not given the high-dose schedule. The median survival was 10 months and was identical for the two groups [18]. Using our strategy, we observed a comparable median PFS of ten months and OS of thirteen months.
We designed the subsequent protocol (study No. 3) with a view to combining radiation therapy, which was not to be delayed by hematological or subjective toxicity, with a schedule involving a pair of drugs—cisplatin and etoposide—that had proved active in a wide spectrum of low-grade gliomas [19], had induced an objective response in pontine tumors [20], and could be administered at length without causing cumulative bone marrow toxicity at the dosages considered. The addition of isotretinoin was prompted by studies on glioma cell lines showing the anti-proliferation and anti-migration effect of retinoids [21, 22]. We chose to administer retinoids after the nadir of the chemotherapy course to stop tumor cell proliferation and migration after antiblastic activity, and this was repeated after each course of chemotherapy [23]. The treatment was easy to administer and generally did not require hospitalization, but yet again none of the patients survived and the median time to progression was only five months, with more than half of the patients proving unable to complete the proposed treatment.
Finally, in protocol No. 4 we tried adding vinorelbine to increase the effect of radiotherapy. Vinorelbine is a semi-synthetic vinca alkaloid that has proved active against several tumor xenografts [24] and is less neurotoxic than vincristine [25]. Vinorelbine also has radio-enhancing activity when used against some adults tumors [26, 27]. By adopting vinorelbine during and after focal radiotherapy, we tried to exploit its known synergic effect for pontine tumor control. Despite two long-term survivors, the median PFS and OS remained invariably superimposable on the previous experience of our own and other authors.
The only advantage our patients have enjoyed over the years has been more limited toxicity of the adjuvant treatment administered. No sure effect of chemotherapy has been demonstrated.
The only experience published so far showing an advantage for patients given chemotherapy is a report from the HIT group [28], where one-year OS was 45.8 vs. 34.4% (P = 0.030) in the irradiated children who were or were not given chemotherapy, respectively. Since the set of patients not given chemotherapy was small it would have been interesting to know if they were characterized by any unfavorable features, e.g. hydrocephalus, age over four years, or cranial growth [29].
In the first period of our clinical experience, we included numerous patients who had undergone some sort of surgery without any clinical benefit. So far, treatment protocols for intrinsic pontine tumors have generally disregarded tumor histology [12] and the possibility of studying other biological features, but Gilbertson et al. performed molecular analyses on 28 brainstem glioma samples (partly deriving from autopsies) and found a large proportion of tumors expressing epidermal growth factor receptor (EGFR) with an intensity that correlated with tumor grade [30]. A recent trial by Bode et al. [31] consequently examined the effect of nimotuzumab (an anti-EGFR monoclonal antibody) in a subset of children with relapsing malignant gliomas and pontine tumors: among the 22 children with pontine tumors, ten showed stable disease and partial remission. Innovative approaches like these may benefit in future from small biopsies enabling the study of O-6-methylguanine-DNA methyltransferase status, EGFR status, PTEN loss, vascular endothelial growth factor expression, and a choice of therapy based on the molecular anomalies of individual tumors.
We may be able to associate these new approaches with standard radiation and non-toxic chemotherapy schedules to improve the prognosis for our patients, who deserve more from our present and future efforts.
References
Albright AL, Packer RJ, Zimmerman R et al (1999) Magnetic resonance scan should replace biopsies for the diagnosis of diffuse brain stem gliomas: a report from the Children’s Cancer Group. Neurosurgery 33:1026–1029
Shiminski-Maher T, Abbot R, Wisoff JH, Epstein FJ (1991) Current trends in the management of brainstem tumors in childhood. J Neurosci Nurs 23:356–362
Benesch M, Lackner H, Moser A et al (2001) Outcome and long-term side effects after synchronous radiochemotherapy for childhood brain stem gliomas. Pediatr Neurosurg 35:173–180
Korones DN (2007) Treatment of newly diagnosed diffuse brain stem gliomas in children: in search of the holy grail. Expert Rev Anticancer Ther 7:663–674
Hargrave D, Bartels U, Bouffet E (2006) Diffuse brainstem glioma in children: critical review of clinical trials. Lancet Oncol 7:241–248
Massimino M, Gandola L, Casanova M et al (2000) Concomitant chemoradiotherapy for childhood poor-prognosis gliomas. Med Pediatr Oncol 34:147–150
Massimino M, Gandola L, Luksch R et al (2005) Sequential chemotherapy, high-dose thiotepa, circulating progenitor cell rescue, and radiotherapy for childhood high-grade glioma. Neuro Oncol 7:41–48
Kaplan EL, Meier P (1958) Non-parametric estimation from incomplete observation. J Am Stat Assoc 53:457–481
Peto R, Peto J (1972) Asymptomatically efficient rank invariant test procedures. J R Stat Soc [A] 135:185–207
Massimino M, Simonetti F, Balestrini MR, Spreafico F et al (2006) Transitory, spontaneously recovering, peripheral facial nerve palsy after vinorelbine administration. Neurol Sci 27:110–113
Langmoen IA, Lundar T, Storm-Mathisen I et al (1991) Management of pediatric pontine gliomas. Childs Nerv Syst 7:13–15
Donaldson SS, Laningham F, Fisher PG (2006) Advances toward an understanding of brainstem gliomas. J Clin Oncol 24:1266–1272
Mandell LR, Kadota R, Freeman C et al (1999) There is no role for hyperfractionated radiotherapy in the management of children with newly diagnosed diffuse intrinsic brain stem tumors: results of a pediatric oncology group phase III trial comparing conventional vs hyperfractionated radiotherapy. Int J Radiat Oncol Biol Phys 43:959–964
Doz F, Neuenschwander S, Bouffet E et al (2002) Carboplatin before and during radiation therapy for the treatment of malignant brain stem tumours: a study by the Société Française d’Oncologie Pédiatrique. Eur J Cancer 38:815–819
Schwaofer JH, Crooijmans RP, Hoogenhout J, Kal HB, Theeuwes AG (1991) Effectiveness in inhibition of recovery of cell survival by cisplatin and carboplatin: influence of treatment sequence. Int J Radiat Oncol Biol Phys 20:1235–1241
Freeman CR, Kepner J, Kun LE et al (2000) A detrimental effect of a combined chemotherapy-radiotherapy approach in children with diffuse intrinsic brain stem gliomas? Int J Radiat Oncol Biol Phys 47:561–564
Dunkel IJ, O’Malley B, Finlay JL (1996) Is there a role for high-dose chemotherapy with stem cell rescue for brain stem tumors of childhood? Pediatr Neurosurg 24:263–266
Bouffet E, Raquin M, Doz F et al (2000) Radiotherapy followed by high dose busulfan and thiotepa. A prospective assessment of high dose chemotherapy in children with diffuse pontine gliomas. Cancer 88:685–692
Massimino M, Spreafico F, Cefalo G et al (2002) High response rate to cisplatin/etoposide regimen in childhood low-grade glioma. J Clin Oncol 20:4209–4216
Kretschmar CS, Tarbell N, Barnes P et al (1993) Pre-irradiation chemotherapy and hyperfractionated radiation therapy 66 Gy for children with brain stem tumors. Cancer 72:1404–1413
Bouterfa H, Picht T, Kess D et al (2000) Retinoids inhibit human glioma cell proliferation and migration in primary cell cultures but not in established cell lines. Neurosurgery 46:419–430
Yung WKA, Kyritsis AP, Gleason MJ et al (1996) Treatment of recurrent malignant gliomas with high dose 12-cis-retinoic acid. Clin Cancer Res 2:1031–1935
Puduvalli VK, Saito Y, Xu R et al (1999) Fenretinide activates caspases and induces apoptosis in gliomas. Clin Cancer Res 5:2230–2235
Hanley ML, Elion GB, Colvin OM et al (1998) Therapeutic efficacy of vinorelbine against pediatric and adult central nervous system tumors. Cancer Chemother Pharmacol 42:479–482
Binet S, Fellous A, Lataste H et al (1989) In situ analysis of the action of Navelbine on various types of microtubules using immunofluorescence. Semin Oncol 16(Suppl 4):5–8
Curran WJ (2002) New chemotherapeutic agents: update of major chemoradiation trials in solid tumors. Oncology 63(Suppl 2):29–38
Biassoni V, Casanova M, Spreafico F, Gandola L, Massimino M (2006) A case of relapsing glioblastoma multiforme responding to vinorelbine. J Neurooncol 80:195–201
Wagner S, Warmuth-Metz M, Emser A et al (2006) Treatment options in childhood pontine gliomas. J Neurooncol 79:281–287
Kornreich L, Schwarz M, Karmazyn B et al (2005) Role of MRI in the management of children with diffuse pontine tumors: a study of 15 patients and review of the literature Pediatr Radiol 35:872–879
Gilbertson RJ, Hill DA, Hernan R (2003) ERBB1 is amplified and overexpressed in high-grade diffusely infiltrative pediatric brain stem gliomas. Clin Cancer Res 9:3620–3624
Bode U, Buchen S, Warmuth-Metz M, Pietsch T, Bach F, Fleischhack G (2007) Final report of a phase II trial of nimotuzumab in the treatment of refractory and relapsed high-grade gliomas in children and adolescents. J Clin Oncol, 2007 ASCO Annual Meeting Proceedings; (abstract) 25, 18S (June 20 Supplement)
Acknowledgements
The authors are indebted to the Associazione “Bianca Garavaglia”, Busto Arsizio, Milano, for partial financial support (scholarships).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Massimino, M., Spreafico, F., Biassoni, V. et al. Diffuse pontine gliomas in children: changing strategies, changing results? A mono-institutional 20-year experience. J Neurooncol 87, 355–361 (2008). https://doi.org/10.1007/s11060-008-9525-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-008-9525-5