
Abstract
Chestnut blight, caused by Cryphonectria parasitica, is a severe disease that has devastated chestnut stands in North America and Europe. Genes encoding hydrolytic enzymes such as chitinases, which can degrade fungal cell wall components, are attractive candidates for improving disease resistance. This report describes a reliable and efficient protocol for the Agrobacterium-mediated transformation of somatic embryos of European chestnut with the endogenous CsCh3 gene that codes for chitinase. The transformation efficiency, determined on the basis of the fluorescence of surviving explants, was genotype-dependent. Although somatic embryos of all three lines evaluated were transformed, the best results were obtained with somatic embryos derived from line CI-9 (20 %). The addition of silver thiosulphate (20 or 40 μM) improved the transformation efficiency of somatic embryos derived from lines CI-3 and CI-9, although the differences were not significant. A total of 88 independent transformed lines were obtained. The presence of transgenes was confirmed by green fluorescent protein (GFP) expression, PCR and Southern blot analysis. Transgenic lines were maintained by secondary embryogenesis or cryopreservation following vitrification procedures. Maturation and germination of transformed somatic embryos yielded transgenic plants. Fluorescence indicating overexpression of the transgenes was observed in somatic embryos and also in shoots and leaves. No phenotypic differences were found relative to control plants, suggesting a lack of any cytotoxic effects of the GFP.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Chestnut blight, caused by the ascomycete fungus Cryphonectria parasitica (Murr.) Barr, affects both Castanea dentata (Marsh) Borkh (American chestnut) and C. sativa (Mill.) (European chestnut). The fungus invades wounds in the bark, destroys the cambial tissues and causes cankers of sunken appearance (Milgroom and Cortesi 2004). The fungus is transferred from tree to tree via spores carried out on the feet, fur and feathers of many animals and insects that walk across the cankers: sexual spores are also released into the air after autumnal rainstorms (Anagnostakis 2001). Chestnut blight is considered a major ecological disaster in the forests of eastern North America where the American chestnut has been transformed from a dominant tree to a minor and inconsequential component of the understorey (Anagnostakis 1995; Jacobs et al. 2013).
Unlike in the USA, the European chestnut can recover from the disease as a result of the natural occurrence of hypovirulence, a phenomenon in which the virulence of C. parasitica is reduced by fungal viruses such as the dsRNA hypovirus CH V1 (Van Alfen et al. 1975; Heiniger and Rigling 1994). However, the productivity and growth of surviving European chestnut trees are seriously affected by the fungal attack.
In order to mitigate the ecological catastrophe affecting American chestnut, a plan to breed a blight-resistant hybrid tree through backcross breeding with natural-resistant Chinese chestnut (Castanea mollissima Blume) was implemented in the US (Jacobs et al. 2013). As a result, a BC3F3 hybrid generation (BC refers to the backcross combination back to American chestnut) exhibiting 94.0 % American chestnut characteristics and 100 % blight resistance (Hebard 2006; Jacobs 2007) has been obtained, and operational releases of verified resistant material will be carried out in the next few years (Wheeler and Sederoff 2009). In contrast, no conventional breeding programmes have been carried out in Europe with the aim of producing blight resistant C. sativa trees.
The American breeding programme has been greatly aided by a genetic map, constructed by Kubisiak et al. (1997), showing molecular markers for the chromosomal regions associated with resistance to chestnut blight. At least two (unknown) genes are responsible for blight resistance (Kubisiak et al. 1997; Anagnostakis 2001); however, the genomic tools currently being developed (www.fagaceae.org) should help provide a better understanding of the disease (Wheeler and Sederoff 2009). The transcriptomes of American chestnut and Chinese chestnut produced in response to chestnut blight infection have been compared in a study that enabled identification of possible pathways involved in chestnut resistance to C. parasitica and of several candidate genes for resistance to necrotrophic fungal pathogens in trees (Barakat et al. 2009).
The genomic approach is currently under development, and genetic transformation may be useful as a complementary approach to obtaining disease-resistant chestnut trees. This particularly applies to European chestnut as conventional tree improvement programmes have not been carried out with this species. Although specific genes controlling blight are not yet available, the use of antifungal genes is a possible alternative approach, which has been successfully applied in plants. American chestnut somatic embryos have been transformed with a gene encoding the antifungal enzyme oxalate oxidase (OxO), isolated from wheat (Polin et al. 2006; Rothrock et al. 2007; Maynard et al. 2008). Resistance tests in these plants grown in the field showed enhanced blight resistance at a level intermediate between that of susceptible American chestnut and resistant Chinese chestnut (Zhan et al. 2013; Newhouse et al. 2014).
The use of different classes of pathogenesis-related (PR) proteins is also very promising. For example, the PR-5 family of proteins (thaumatin-like proteins) may produce transmembrane pores on fungal plasma membranes, thus promoting osmotic rupture (Roberts and Selitrennikoff 1990). Chitinases belonging to the PR-3 family of proteins hydrolyze the β-1,4 glycosidic bonds that link the N-acetylglucosamine residues of chitin and play a direct role in plant defence by hydrolyzing chitin (Veluthakkal and Dasgupta 2010). Four proteins with a chitin-binding domain have been purified from European chestnut plantlets grown in vitro, and three of these proteins inhibit hyphal growth of the C. parasitica fungus in culture (Vannini et al. 1999). In addition, a thaumatin-like protein (CsTL1) and the most abundant cotyledon endochitinase (CsCh3) have been isolated from seeds of European chestnut, and their antifungal activity has also been demonstrated under in vitro conditions (Collada et al. 1992; Allona et al. 1996; García-Casado et al. 2000). Genetic transformation of somatic embryogenic lines of European chestnut with the native CsTL1 gene has been reported (Corredoira et al. 2012). We have also performed experiments to determine whether or not overexpression of the gene increased the resistance of regenerated transgenic plants to the ink disease caused by Phytophthora cinnamomi Rand, although data are not yet available. Similarly, in American chestnut a Castanea mollissima thaumatin-like protein gene was also transformed into somatic embryos (Kong et al. 2014).
In order to obtain chestnut plants with potentially increased resistance/tolerance to blight disease, the aim of the present study was to investigate overexpression of the endogenous CsCh3 gene (encoding a chitinase-like protein) in embryogenic cultures of European chestnut (C. sativa) via Agrobacterium-mediated transformation.
Materials and methods
Plant material
Embryogenic cultures initiated from leaf explants obtained from shoot multiplication cultures (C12-H1) and zygotic embryos (CI-3 and CI-9) of European chestnut (Corredoira et al. 2003, 2006) were used in the transformation experiments. These embryogenic cultures were maintained by secondary embryogenesis with subculture at 6-week intervals onto standard proliferation medium (MP) consisting of MS (Murashige and Skoog 1962) vitamins and mineral salts (half-strength macronutrients), 3 mM glutamine, 0.1 mg/L benzyladenine (BA), 0.1 mg/L 1-naphthaleneacetic acid, 3 % sucrose (w/v), and 0.8 % Sigma-agar (w/v). The medium was sterilized by autoclaving at 121 °C for 20 min, after the pH was adjusted to 5.7. The embryogenic cultures were subjected to a 16-h photoperiod (provided by cool-white fluorescent lamps at a photon flux density of 50–60 μmol m−2 s−1) and 25 °C light/20 °C dark temperatures (standard conditions).
Construction of the binary vector and Agrobacterium transformation
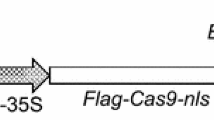
The chestnut gene encoding the endochitinase-like protein (designated the CsCh3 gene), and purified from mature chestnut cotyledons was used in the study (Collada et al. 1992; Allona et al. 1996). The CsCh3 gene was cloned into pK7WG2D under the CaMV35S promoter by using the Gateway cloning system (Invitrogen, USA). This gateway-compatible destination vector was obtained commercially (VIB, Brussels, Belgium). The pK7WG2D vector also contains the neomycin phosphotransferase (nptII) selectable marker and green fluorescence protein (egfp) reporter genes. For overexpression of the chestnut gene, the CsCh3 coding region was mobilized into the pENTR/D-TOPO intermediate vector (Invitrogen, Grand Island, NY) and pENTR/D-TOPO-CsCh3 was recombined with pK7WG2D using the LR recombination reaction in the Gateway LR Clonase Enzyme mix (Invitrogen, Grand Island, NY). The resulting plasmid, designated pK7WG2D-CsCh3 (Fig. 1), was transformed into Agrobacterium tumefaciens strain EHA105 (Hood et al. 1993) competent cells by the freeze–thaw method (Xu and Li 2008), and transformed colonies were selected by growing them for two–three days on semisolid LB medium (LB, 1 % tryptone, 0.5 % yeast extract, and 1 % NaCl, pH 7.0; Sambrook et al. 1989) containing 50 mg/L kanamicin (kan) and nalidixic acid. Independent colonies were picked and inoculated into liquid LB medium with 50 mg/L kan and nalidixic acid and grown at 28 °C until saturation. Cells were collected by centrifugation and plasmid DNA was obtained using the Qiagen DNeasy® Plasmid Mini Kit (Qiagen, Hamburg, Germany). The presence of the plasmid in the bacteria was verified by PCR and sequencing. Colonies of Agrobacterium confirmed as being transformed with the construct were selected, grown in liquid culture and stored at −80 °C in 50 % glycerol solution until required.

Schematic representation of the T-DNA region of the binary vector pK7WG2D-CsCh3. RB, right border; LB, left border; Nos-ter and Nos-pro, terminator and promoter of nopaline synthase gene, respectively; nptII, neomycin phosphotransferase marker gene; 35S-pro and T-35S, promoter and terminator of cauliflower mosaic virus gene, respectively; egfp, green fluorescence protein gene; CsCh3, gene encoding a endochitinase; prolD, rol root loci D promoter; EcoRI, restriction site; attR2 and attR1, reaction sites
Transformation procedures
For the transformation experiments, explants consisting of small clumps (4–7 mg) of two to three somatic embryos, at globular or early-torpedo stages, were dissected from embryogenic cultures 4 weeks after the previous subculture.
Cultures of Agrobacterium were initiated from a glycerol stock and grown for 2–3 days at 28 °C in LB semisolid medium containing kan and nalidixic acid (50 mg/L). A single colony was inoculated into 2 mL of LB medium containing 50 mg/L kan and nalidixic acid, and the culture was incubated overnight at 28 °C with shaking (at 150 rpm) in darkness. One mL of the bacterial suspension was used to inoculate 600 mL of LB liquid medium with the appropriate antibiotics, and the culture was incubated at 28 °C with shaking (at 100 rpm) until an OD600 = 0.6 was reached. The bacterial cultures were then centrifuged at 6500 rpm for 10 min at 10 °C and resuspended in 200 mL of MS liquid medium plus 5 % sucrose.
Somatic embryo clumps (20 clumps) were immersed in 20 mL of the bacterial suspension for 30 min, blot-dried on sterile filter paper and transferred to proliferation medium without growth regulators. After 5 days of co-cultivation in the dark at 25 °C, the embryos were washed for 30 min with sterilized water containing 500 mg/L cefotaxime. Somatic embryos were then blot-dried on sterile filter paper, transferred to Petri dishes containing proliferation medium supplemented with 300 mg/L carbenicillin, 200 mg/L cefotaxime and 150 mg/L kanamycin (selective medium) and incubated under standard conditions. To inhibit Agrobacterium overgrowth, cultures were transferred to fresh selection medium every 2 weeks. After 8 weeks on selective medium, kan-resistant explants were transferred to fresh selective medium. After a further four weeks (i.e. 12 weeks in total), putative transformants (identified by growth on selective medium) were evaluated by GFP-specific fluorescence.
In a first experiment, the effect of embryogenic genotype (CI-3, CI-9 and C12-H1) was investigated using 120 embryogenic clumps for each line (i.e. 360 explants in total). In a second experiment, we evaluated the effect of the antioxidant gluthatione and silver thiosulphate (STS), included in either the cocultivation or selection medium, on the transformation efficiency of lines CI-3 and CI-9. Gluthatione (0, 100, 200, 400 mg/L) and STS solution (0, 20, 40, 60 µM) were added, by filtration, to autoclaved cocultivation and selection media. In the second experiment, 60 explants were used per treatment and embryogenic line (i.e. 240 explants per embryogenic line).
In all transformation experiments, 20 non-inoculated (wild type, wt) embryo explants per embryogenic line were cultured on proliferation medium with or without antibiotics (negative and positive controls, respectively).
Selection and further development of transformants
After 12 weeks in culture, resistant somatic embryos were observed by GFP-specific fluorescence. GFP-specific fluorescence was evaluated in a Zeiss SV11 epi-fluorescence stereomicroscope equipped with a light source consisting of a 100-W mercury bulb and an FITC/GFP filter plus a 480 nm excitation filter and a 515 nm long-pass emission filter (Chroma Technology Corp., USA). The transformation efficiency was defined as the percentage of initial explants that developed GFP-positive (GFP+) embryogenic cultures. Regenerated cotyledonary embryos were isolated from GFP-positive lines and subcultured on selective medium to proliferate and establish different embryogenic transgenic lines. The transgenic embryogenic cultures obtained in different transformation experiments were routinely maintained by secondary embryogenesis with sequential subculture at 6-week intervals, under previously defined conditions (Corredoira et al. 2006).
Germination and plant regeneration of transgenic somatic embryos
Embryo germination was carried out as previously described (Corredoira et al. 2006, 2008). Cotyledonary somatic embryos (4-6 mm long) were isolated from transgenic cultures and cultured on maturation medium consisting of PGR-free MS (half strength macronutrients) medium supplemented with maltose (3 %). After 4 weeks of culture on maturation medium, somatic embryos were transferred to basal medium with 3 % sucrose and stored at 4 °C for 2 months, before being cultured for 8 weeks on germination medium (MS with half strength macronutrients, 0.1 mg/L BA, 0.1 mg/L indole-3-butyric acid and glutamine 200 mg/L). Shoots from germinating embryos exhibiting shoot development only were successfully multiplied by axillary shoot proliferation (Corredoira et al. 2015), which enables production of an unlimited number of transgenic shoots for rooting.
Analysis of transgenic somatic embryo lines and plant lines
PCR analysis
Chestnut genomic DNA was extracted from somatic embryos and leaf material from untransformed and potentially transgenic somatic embryos and plants by using the DNeasy®Plant Kit (Qiagen, Hamburg, Germany) according to the manufacturer’s instructions. The presence of transgenes (CsCh3, nptII and egfp genes) was confirmed by PCR analysis. Reactions were carried out in a 50 μL volume containing 1× supplied Taq buffer, 2.5 mM MgCl2, 200 μM dNTPs, 0.6 μM each primer, 1 U Taq DNA polymerase (Qiagen, Hamburg, Germany) and 100–200 ng of genomic DNA. PCR analysis was carried out with gene-specific primers: CsCh3 (forward: 5′-GATCTAACAGAACTCGCC-3′; reverse: 5′-CATGGCCAAGATTTTGAGGT-3′), nptII (forward: 5′-GTCATCTCACCTTGCTCCTGCC-3′; reverse: 5′-AAGAAGGCGATAGAAGCGA-3′) and egfp (forward: 5′-CACCGGGGTGGTGCCCAT-3′; reverse: 5′-CTAGTGGATCCCCCGGGC-3′). The expected sizes of PCR fragments were 1114 pb for CsCh3, 472 pb for nptII, and 740 pb for egfp. The fragments were amplified in a MJ Mini™ thermal cycler (Bio-Rad, Hercules, CA, USA) by use of the following programmes, after initial activation of polymerase at 95 °C for 3 min: 40 cycles at 94 °C for 40 s, 56 °C for 1 min and 72 °C for 40 s for CsCh3 gene, 35 cycles at 94 °C for 30 s, 60 °C for 30 s and 72 °C for 42 s for the nptII gene, and 40 cycles at 94 °C for 15 s, 56 °C for 30 s and 72 °C for 1 min for the egfp gene. The amplified products were resolved on 1.2 % (w/v) agarose gel and confirmed as only one single band of the expected size.
Southern blot
Genomic DNA of transgenic somatic embryos was isolated using the DNeasy Plant Maxi Kit (Qiagen, Hamburg, Germany). DNA (25–30 μg) was digested with EcoRI and then separated on a 0.8 % (w/v) agarose gel in 0.5× Tris–borate buffer. Following gel electrophoresis of digested genomic DNA and subsequent depurination, denaturation and neutralization, DNA was transferred onto a positively charged nylon membrane by capillary transfer. A DIG-labelled CsCh3 probe was produced by PCR labelling. Subsequent hybridization steps were carried out with the DIG High Prime DNA Labelling and Detection Starter Kit II (Roche Applied Science, USA). After hybridization to the probe, the chemiluminescence substrate CDP-Star was used to detect hybridization signals by X-ray film autography. Genomic DNA extracted from untransformed somatic embryos served as a negative control.
Statistical analysis
The statistical significance of differences between embryogenic lines and co-cultivation time was estimated using the test of independence (G-test) (Sokal and Rohlf 1981).
Results
The response of the three embryogenic lines to transformation after cocultivation for 5 days with strain EHA105pK7WG2D-CsCh3 was genotype-dependent. After the explants were cultured for two weeks in selection medium, new growth was only visible in line CI-9; the number of surviving explants (putative kanamycin-resistant) of this line was increased by increasing the length of time in selection medium. In lines CI-3 and C12-H1, the initial explants showed a higher degree of necrosis and new growth was not visible until eight weeks later. At this time, kanamycin-resistant explants were observed in all three embryogenic lines, although the highest rate of production was achieved with line CI-9. The resistant explants were isolated from the initial explants and subcultured for a further 4 week-period on selection medium to enable embryogenic growth to continue (Fig. 2a, b). All of the negative control cultures (untransformed) turned brown and died during the kanamycin selection period and neither yellow calluses nor embryo regeneration were observed.

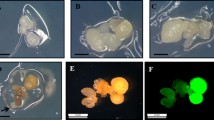
Transformation and regeneration of European chestnut somatic embryos using the vector pK7WG2D-CsCh3. a, b Transformed somatic embryos observed under white light after 8 weeks in selection medium. Bar 1 mm. c The same somatic embryos showed in a observed under blue light showing green fluorescence. Bar 1 mm. d Histodifferentiated transgenic cotyledonary embryo showing GFP expression. Bar 1 mm. e Transgenic plant derived from transgenic somatic embryo. GFP expression on leaf (f) and apex (g) of transgenic plant (right) and untransformed plant (left) visualized in an epi-fluorescence stereomicroscope. Note: untransformed tissues appear red under blue light due to autofluorescence of chlorophyll. Bar 1 mm
The percentage of kanamycin-resistant and GFP+ explants and the transformation efficiency were recorded 12 weeks after establishing the initial cultures (Table 1). The genotype significantly affected both parameters (p ≤ 0.0001; Table 1). The percentage of kanamycin-resistant explants was significantly higher in line CI-9 (26.7 %) than in lines CI-3 (5.8 %) and C21-H1(5.0 %). A similar trend was observed in the transformation efficiency parameter, which was defined as the percentage of initial explants that developed GFP+ embryogenic cultures (Table 1). The best results (20.0 %) were also achieved with line CI-9 and the efficiency was significantly higher than for GFP+ explants in line CI-3 (5.0 %) and in line C12-H1 (4.2 %). The selection efficiency (defined as the percentage of kan-resistant explants that also developed GFP-positive embryogenic cultures) ranged from 82.0 to 92.0 % (Table 1), indicating that kanamycin and GFP double selection appear to be appropriate for this transformation system, producing a relatively low number of escapes. These results demonstrated that GFP fluorescence is an effective and non-destructive marker for identifying transgenic somatic embryos in chestnut. In order to prevent the development of chimeras, only completely fluorescent embryos (Fig. 2c, d) were considered as GFP+ and those exhibiting partial fluorescence were eliminated. As a result of this experiment, 35 GFP positive transgenic lines were obtained. Twenty four of these lines were derived from CI-9, six lines were derived from CI-3 and five from C12-H1.
In order to prevent necrosis of target explants occurring in cocultivation and selection media and to optimize the transformation efficiency, the effect of the antioxidant compounds STS and gluthatione was evaluated during transformation of somatic embryos of lines CI-3 and CI-9. Neither of the two compounds affected growth of the bacteria during the co-culture period; however, the addition of gluthatione or STS to the cocultivation medium did not improve the transformation efficiency of either of the two embryogenic lines at any of the concentrations tested (data not shown). Although the presence of 100 mg/L gluthatione in the selection medium improved the transformation efficiency in line CI-3, the results obtained with lines CI-9 were inconclusive. However, the inclusion in the selection medium of STS 20 μM in line CI-3 or 40 μM in line CI-9 enhanced the transformation efficiency (Fig. 3), although with non-significant differences between these and other concentrations tested. As result of this experiment, 53 GFP positive transgenic lines were produced (14 transgenic lines for CI-3 and 39 for transgenic lines CI-9).

Effect of STS addition at the selection medium on the transformation efficiency of embryogenic lines CI-3 and CI-9 transformed with strain EHA105pK7WG2D-CsCh3
Maintenance and germination of transgenic somatic embryos
To ensure that each putative transformed line was derived from a unique transformation event, only one cotyledonary embryo was isolated from each GFP+ explant and was independently multiplied by secondary embryogenesis on selection medium for establishment and proliferation. Once the different embryogenic transgenic lines were established, they were maintained by secondary embryogenesis or were cryoconserved in liquid nitrogen following previously described protocols (Vieitez et al. 2011).
Following maturation and stratification of transgenic somatic embryos, as described in Materials and methods, embryos were cultured in germination medium for 8 weeks. The germination response (conversion into plantlets and/or shoot only development) ranged from 2.0 to 16.0 %, and no significant differences were detected between transgenic and wild type material. Germinated embryos exhibiting only shoot development were used to establish shoot cultures from each embryogenic line. Shoot cultures were multiplied by axillary branching, and the multiplication rates were very similar in both transgenic and wild type shoots (data not shown). A total of five shoot axillary lines were stabilized from germinated somatic embryos of three transgenic lines derived from CI-3 and two transgenic lines derived from CI-9. Rooting rates and performance were very similar in both transgenic (65.0–82.0 %) and untransformed shoots (90.0 %). Transgenic plants grew normally and were morphologically indistinguishable from non-transgenic chestnut controls. Fluorescence was observed in the leaves and shoots, confirming the non-toxicity of GFP in chestnut plantlets (Fig. 2f, g). The red autofluorescence from chlorophyll was not blocked by any of the interference filters. This allowed in vivo monitoring and easy discrimination of GFP positive and GFP negative developing shoots.
Molecular analysis
To check for the presence and integration of the transgenes, embryogenic transgenic lines were analyzed by PCR and Southern blot (Figs. 4, 5). Twenty-two transgenic lines (i.e. 25 % of all of the GFP+ isolated) were analyzed by PCR to confirm the presence of the genes nptII, egfp and CsCh3. Fragments of 472, 740 and 1114 pb, corresponding to the expected sizes of the nptII, egfp and CsCh3 genes were amplified in the transgenic embryos and in the plasmid (positive control), but not in the untransformed embryos (Fig. 4). Four PCR-positive lines were chosen at random for Southern blotting to verify stable integration of the CsCh3 transgene (Fig. 5). A DIG-labelled probe consisting of 658 bp fragment of the chitinase gene (including part of p35S promoter and the other (123 bp) an internal sequence of the CsCh3 gene) revealed that all of these lines contained the target gene in different integration patterns with between one and three copies (Fig. 5).

PCR analysis of European chestnut transgenic lines. a PCR amplification using specific primers for production of a 472-bp nptII fragment. b PCR amplification using specific primers for production of a 740-bp egfp fragment. c PCR amplification using primers specific for production of a 1114-bp CsCh3 fragment. M DNA ladder; 600 pb indicate DNA marker size in base pairs; Lanes 1–7 Transgenic lines derived from embryogenic line CI-9; Lanes 8, 9 Transgenic lines derived from embryogenic line CI-3; Lanes 10, 11 transgenic lines derived from embryogenic line C12-H1; U—untransformed somatic embryos (negative control); P—corresponding plasmid DNA (positive control)

Southern blot analysis of European chestnut transgenic lines. The number of bands reflects the number of transgene insertion sites. Lanes 1–3 transgenic lines derived from embryogenic line CI-9; L3 transgenic line derived from embryogenic line CI-3; P plasmid; U—untransformed somatic embryos
Discussion
Fungal pathogens are the cause of many serious diseases and have led to important agricultural and economic losses worldwide (Bezirganoglu et al. 2013). Introduction or overexpression of genes encoding PR-proteins is an important approach in the control of fungal diseases. Genes encoding endochitinases have already been used to enhance plant resistance to fungal diseases (Wally and Punja 2010; Ceasar and Ignacimuthu 2012). Chitinase catalyzes the hydrolysis of β-1,4 linkages of the N-acetyl-d-glucosamine (GlcNAc) polymer chitin (Veluthakkal et al. 2012). There is evidence supporting the hypothesis that plant chitinases have antibiotic functions and thus probably constitute a plant defence mechanism against pathogens (Pourhosseini et al. 2012). Recombinant DNA techniques enable isolation of specific genes and their inclusion in plants, which would not be possible with conventional breeding. Transgenic plants overexpressing these genes usually exhibit improved levels of resistance to fungal pathogens or delayed development of symptoms, relative to control plants such as peanut (Rohini and Rao 2001), cotton (Emani et al. 2003), tobacco (Corrado et al. 2008), tomato (Girhepuje and Shinde 2011), petunia (Khan et al. 2012) and banana (Kovács et al. 2013). There are very few examples of the introduction of chitinase genes in forest trees. Pasonen et al. (2004) reported the transformation of silver birch (Betula pendula) with a sugar beet class IV endochitinase and demonstrated increased resistance of few transgenic lines against rust disease caused by Melampsoridium betulinum under field conditions. Chitinase genes have also been used to enhance fungal resistance in hybrid poplar Populus simonii × P. nigra and in P. tomentosa (Zhiying et al. 2010; Jia et al. 2010).
The European chestnut is seriously threatened by the fungus Cryphonectria parasitica, which attacks the trunk and branches of the tree. The development of an improved transformation procedure for somatic embryos derived from European chestnut (Corredoira et al. 2004, 2007) provides the opportunity to transform this tree species with antifungal candidate genes, which may confer resistance to the fungal pathogen. Chestnut somatic embryos from three embryogenic lines were transformed by this procedure with the CsCh3 gene that codes for an endochitinase and was isolated from European chestnut cotyledons. This is the first report of genetic transformation in the genus Castanea with a gene that encodes an endochitinase.
As in previous reports, the transformation rates were genotype dependent, although transgenic somatic embryos were obtained from all three embryogenic lines used. Differences in transformation rates observed in the three chestnut lines confirm the importance of the genotype on genetic transformation, as reported in various woody species including the related species American chestnut (Polin et al. 2006; Kong et al. 2014), cork oak (Álvarez and Ordás 2007) and pedunculate oak (Mallón et al. 2014).
The transformation frequencies obtained in the present study were lower than that reported in a previous study in which chestnut somatic embryos were transformed with the thaumatin-like protein (Corredoira et al. 2012). In the present study, the transformation efficiency, also measured as the percentage of GFP+ explants, reached 20 % for line CI-9, 5.0 % for CI-3 and 4.2 % for C12-H1. The transformation efficiencies with the CsTL1 gene, which codes for a thaumatin-like protein, for the same embryogenic lines were respectively 32.5, 7.1 and 8.3 % (Corredoira et al. 2012). As the same strain was used and both genes were cloned in the same vector (pK7WG2D) in both the previous and present study, the differences in the transformation frequencies may be attributable to differences in the sizes of the CsCh3 and CsTL1 genes. Available evidence suggests that T-DNA processing may be affected by the size and/or organization of the T-DNA region, leading to the production of intact double-stranded or single-stranded forms of T-DNA, and this in turn has some effect on T-DNA mobility and delivery into plant tissue (Sharma et al. 2011).
The transformation frequencies achieved in the present study were relatively high in comparison with those reported for many woody species and with somatic embryos as the target explant. In American chestnut, somatic embryos engineered with a candidate anti-fungal gene oxalate oxidase (OxO) alone (Polin et al. 2006; Rothrock et al. 2007; Maynard et al. 2008) or a stacked combination of OxO and a synthetic antimicrobial peptide called ESF39 (Newhouse et al. 2014) were generated by A. tumefaciens-mediated transformation. Nevertheless, apparently only small numbers of plantlets were produced in these studies by germination of transformed embryos (Andrade et al. 2009). In species with very low embryo conversion capacity, such as chestnut, we understand that the use of shoot proliferation cultures as reported in this paper, is highly recommended for more efficient production of transgenic plants (see also Corredoira et al. 2012, 2015).
The GFP, encoded by the gfp reporter-gene from the jellyfish (Aequorea victoria), provides a useful, safe and easily detectable marker system for the development of chestnut transgenic somatic embryos and plants. The high selection efficiency reached (80–100 %) in the present study indicates that double selection with kanamycin and GFP imposes strong selection conditions, minimizing the number of escapes and preventing loss of samples, as when the GUS gene is used as the reporter gene. Selection of fluorescent embryos facilitates proliferation of transgenic embryos as it reduces subculture of escapes. In addition, the use of marker genes such as GFP will enable production of transgenic plants free of antibiotic resistant marker genes, as required by regulations controlling the production of genetic modified organisms in applied genetic engineering programmes (Leclercq et al. 2010).
During Agrobacterium-mediated transformation processes, browning and necrosis of transformed tissues, as a result of oxidative burst caused by Reactive Oxygen Species (ROS), severely reduces the transformation efficiency (De la Riva et al. 1998). The application of antioxidants to co-cultivation and/or selection media can help to prevent or reduce this problem (Dan 2008). Gluthatione is a redox-active molecule that is involved in various antioxidant reactions. It also functions as a cellular protectant and enhances cell division (Dutt et al. 2011 and references therein). The addition of glutathione increased plant regeneration and Agrobacterium-mediated transformation of Craterostigma plantagineum (Toldi et al. 2002). However, in the present study, addition of gluthatione to the selection medium was not very effective. Likewise, in previous studies, treatment of target explants with cysteine, another antioxidant, also did not improve the transformation efficiency in chestnut (Corredoira et al. 2012).
Ethylene is a plant growth regulator that may be produced in plant cell cultures due to wounding or the presence of auxins. Accumulation of this gas is associated with poor regeneration capacity and reduced gene transfer efficiency (Han et al. 2005; Nonaka et al. 2008; Nonaka and Ezura 2014). Silver thiosulphate, an ethylene inhibitor, is often used to block the action of this hormone. Application of this compound has been shown to increase the stable transformation efficiency of black cherry (Liu and Pijut 2010) and oilseed rape (Tang et al. 2011). Similarly, the presence of STS in selection media used for chestnut somatic embryos improved the transformation efficiency, although differences were not significant.
The process whereby an organism is genetically modified in order to overexpress genes isolated from the species itself or from species that are sexually compatible with the target species has been defined as cisgenesis or intragenesis (Schouten et al. 2006a, b; Jacobsen and Schouten 2008; Hou et al. 2014). Although cisgenic, intragenic or transgenic plants should undergo the similar type of controls before deployment, such controls would be less strict for cisgenic or intragenic plants. Various examples of the production of cisgenic plants have been reported in the last years (Dhekney et al. 2011; Vanblaere et al. 2011; Han et al. 2011; Joshi et al. 2011; Holme et al. 2012). Although the European chestnut plants obtained are not strictly cisgenic, as they contain foreign promoters and gene markers, their production can be considered as a first step in producing cisgenic plants of this species. We propose using co-transformation systems to eliminate the marker genes and to apply this procedure to transformed lines that show some type of resistance/tolerance to canker blight.
In conclusion, we have developed a reliable and efficient transformation system for three chestnut embryogenic lines by using an endogenous chitinase-encoding gene. Transgenic embryogenic lines grew normally and were morphologically indistinguishable from non-transgenic chestnut controls. We plan to evaluate chestnut plants transformed with the chitinase-encoding gene to determine the degree of resistance/tolerance to blight disease.
References
Allona I, Collada C, Casado R, Paz-Ares J, Aragoncillo C (1996) Bacterial expression of an active class Ib chitinase from Castanea sativa cotyledons. Plant Mol Biol 32:1171–1176
Álvarez R, Ordás RJ (2007) Improved genetic transformation protocol for cork oak (Quercus suber L.). Plant Cell, Tissue Organ Cult 91:45–52
Anagnostakis SL (1995) The pathogens and pests of chestnuts. In: Andrews JH, Tommerup I (eds) Advances in botanical research (Vol 21). Academic Press, New York, pp 125–145
Anagnostakis SL (2001) The effect of multiple importations of pests and pathogens on a native tree. Biol Invasions 3:245–254
Andrade GM, Nairn CJ, Le HT, Merkle SA (2009) Sexually mature transgenic American chestnut trees via embryogenic suspension-based transformation. Plant Cell Rep 28:1385–1397
Barakat A, DiLoreto DS, Zhang Y, Smith C, Baier K, Powell WA, Wheeler N, Sederoff R, Carlson JE (2009) Comparison of the transcriptomes of American chestnut (Castanea dentata) and Chinese chestnut (Castanea mollissima) in response to the chestnut blight infection. BMC Plant Biol 9:51
Bezirganoglu I, Hwang S-Y, Fang TJ, Shaw J-F (2013) Transgenic lines of melon (Cucumis melo L. var. makuwa cv. ‘Silver Light’) expressing antifungal protein and chitinase genes exhibit enhanced resistance to fungal pathogens. Plant Cell, Tissue Organ Cult 112:227–237
Ceasar SA, Ignacimuthu S (2012) Genetic engineering of crop plants for fungal resistance: role of antifugal genes. Biotechnol Lett 34:995–1002
Collada C, Casado R, Fraile A, Aragoncillo C (1992) Basic endochitinases are major proteins in Castanea sativa cotyledons. Plant Physiol 100:778–783
Corrado G, Arciello S, Fanti P, Fiandra L, Garonna A, Digilio MC, Lorito M, Giordana B, Pennacchio F, Rao R (2008) The Chitinase A from the baculovirus AcMNPV enhances resistance to both fungi and herbivorous pests in tobacco. Transgenic Res 17:557–571
Corredoira E, Ballester A, Vieitez AM (2003) Proliferation, maturation and germination of Castanea sativa Mill. somatic embryos originated from leaf explants. Ann Bot 92:129–136
Corredoira E, Montenegro D, San José MC, Vieitez AM, Ballester A (2004) Agrobacterium-mediated transformation of European chestnut embryogenic cultures. Plant Cell Rep 23:311–318
Corredoira E, Ballester A, Vieitez FJ, Vieitez AM (2006) Somatic embryogenesis in chestnut. In: Mujib A, Samaj J (eds) Plant cell monographs, Vol. 2, somatic embryogenesis. Springer, Berlin, pp 177–199
Corredoira E, San José MC, Vieitez AM, Ballester A (2007) Improving genetic transformation of European chestnut and cryopreservation of transgenic lines. Plant Cell, Tissue Organ Cult 91:281–288
Corredoira E, Valladares S, Vieitez AM, Ballester A (2008) Improved germination of somatic embryos and plant recovery of European chestnut. In Vitro Cell Dev Biol Plant 44:307–315
Corredoira E, Valladares S, Allona I, Aragoncillo C, Vieitez AM, Ballester A (2012) Genetic transformation of European chestnut somatic embryos with a native thaumatin-like protein (CsTL1) gene isolated from Castanea sativa seeds. Tree Physiol 32:1389–1402
Corredoira E, Valladares S, Vieitez AM, Ballester A (2015) Chestnut, European (Castanea sativa). In: Wang K (ed) Methods in molecular biology Agrobacterium protocols. Springer, New York, pp 163–176
Dan Y (2008) Biological functions of antioxidants in plant transformation. In Vitro Cell Dev Biol Plant 44:149–161
De la Riva G, Gónzalez-Cabrera J, Vázquez-Padron R, Ayra-Pardo C (1998) Agrobacterium tumefaciens: a natural tool for plant transformation. Electron J Biotechnol 1:1–15
Dhekney SA, Li ZT, Gray DJ (2011) Grapevines engineered to express cisgenic Vitis vinifera thaumatin-like protein exhibit fungal disease resistance. In Vitro Cell Dev Biol-Plant 47:458–466
Dutt M, Vasconcellos M, Grosser JW (2011) Effects of antioxidants on Agrobacterium-mediated transformation and accelerated production of transgenic plants of Mexican lime (Citrus aurantifolia Swingle). Plant Cell, Tissue Organ Cult 107:79–89
Emani C, García JM, Lopata-Finch E, Pozo MJ, Uribe P, Kim D-J, Sunilkumar G, Cook DR, Kenerly CM, Rathore KS (2003) Enhanced fungal resistance in transgenic cotton expressing an endochitinase gene from Trichoderma virens. Plant Biotechnol J 1:321–326
García-Casado G, Collada C, Allona I, Soto A, Casado R, Rodríguez-Cerezo E, Gómez L, Aragoncillo C (2000) Characterization of an apoplastic basic thaumatin-like protein from recalcitrant chestnut seeds. Physiol Plant 110:172–180
Girhepuje PV, Shinde GB (2011) Transgenic tomato plants expressing a wheat endochitinase gene demonstrate enhanced resistance to Fusarium oxysporum f. sp. lycopersici. Plant Cell, Tissue Organ Cult 105:243–251
Han JS, Kim CK, Park SH, Hirschi KD, Mok IG (2005) Agrobacterium-mediated transformation of bottle gourd (Lagenaria siceraria Standl). Plant Cell Rep 23:692–698
Han KM, Dharmawardhana P, Arias RS, Ma C, Busov V, Strauss SH (2011) Gibberellin-associated cisgenes modify growth, stature and wood properties in Populus. Plant Biotechnol J 9:162–178
Hebard FV (2006) The backcross breeding program of the American Chestnut Foundation. In: Steiner KC, Carlson JE (eds) Restoration of American chestnut to forest lands. Proceedings of a conference and workshop, May 4–6, 2004, The North Carolina Arboretum, Asheville. Natural Resources Report NPS/NCR/CUE/NRR – 2006/01. Washington, DC: National Park Service, pp 61–77
Heiniger U, Rigling D (1994) Biological control of chestnut blight in Europe. Ann Rev Phytopathol 32:581–599
Holme IB, Dionisio G, Brinch-Pedersen H, Wendt T, Madsen CK, Vincze E, Holm PB (2012) Cisgenic barley with improved phytase activity. Plant Biotechnol J 10:237–247
Hood EE, Gelvin SB, Melchers LS, Hoekema A (1993) New Agrobacterium helper plasmids for gene transfer to plants. Transgenic Res 2:208–218
Hou H, Atlihan N, Lu Z-X (2014) New biotechnology enhances the application of cisgenesis in plant breeding. Front Plant Sci 5:389
Jacobs DF (2007) Toward development of silvical strategies for forest restoration of American chestnut (Castanea dentata) using blight-resistant hybrids. Biol Conserv 137:497–506
Jacobs DF, Dalgleish HJ, Nelson CD (2013) A conceptual framework for restoration of threatened plants: the effective model of American chestnut (Castanea dentata) reintroduction. New Phytol 197:378–393
Jacobsen E, Schouten HJ (2008) Cisgenesis, a new tool for traditional plant breeding, should be exempted from the regulation of genetically modified organisms in a step by step approach. Potato Res 51:75–88
Jia Z, Sun Y, Yuan L, Tian Q, Luo K (2010) The chitinase gene (Bbchit1) from Beauveria bassiana enhances resistance to Cytospora chrysosperma in Populus tomentosa Carr. Biotechnol Lett 32:1325–1332
Joshi SG, Schaart JG, Groenwold R, Jacobsen E, Schouten HJ, Krens FA (2011) Functional analysis and expression profiling of HcrVf1 and HcrVf2 for development of scab resistant cisgenic and intragenic apples. Plant Mol Biol 75:579–591
Khan RS, Kameya N, Mii M, Nakamura I (2012) Transgenic Petunia hybrida expressing a synthetic fungal chitinase gene confers disease tolerance to Botrytis cinerea. Plant Biotechnol 29:285–291
Kong LK, Holtz CT, Nairn CJ, Houke H, Powell WA, Baier K, Merkle SA (2014) Application of airlift bioreactors to accelerate genetic transformation in American chestnut. Plan Cell Tissue Organ Cult 117:39–50
Kovács G, Sági L, Jacon G, Arinaitwe G, Busogoro J-P, Thiry E, Strosse H, Swennen R, Remy S (2013) Expression of a rice chitinase gene in transgenic banana (‘Gros Michel’, AAA genome group) confers resistance to black leaf streak disease. Transgenic Res 22:117–130
Kubisiak TL, Hebard FV, Nelson CD, Zhang J, Bernatzky R, Huang H, Anagnostakis SL, Doudrick RL (1997) Molecular mapping of resistance to blight in an interspecific cross in the genus Castanea. Phytopathology 87:751–759
Leclercq J, Lardet L, Martin F, Chapuset T, Oliver G, Montoro P (2010) The green fluorescent protein as an efficient selection marker for Agrobacterium tumefaciens-mediated transformation in Hevea brasiliensis (Müll. Arg). Plant Cell Rep 29:513–522
Liu X, Pijut PM (2010) Agrobacterium-mediated transformation of mature Prunus serotina (black cherry) and regeneration of transgenic shoots. Plant Cell, Tissue Organ Cult 101:49–57
Mallón R, Valladares S, Corredoira E, Vieitez AM, Vidal N (2014) Overexpression of the chestnut CsTL1 gene coding for a thaumatin-like protein in somatic embryos of Quercus robur. Plant Cell, Tissue Organ Cult 116:141–151
Maynard CA, Powell WA, Polin-McGuigan LD, Vieitez AM, Ballester A, Corredoira E, Merkle SA, Andrade A (2008) Chestnut. In: Kole C, Hall TC (eds) Compendium of transgenic crop plants: transgenic forest tree species. Blackwell, Chichester, pp 169–192
Milgroom MG, Cortesi P (2004) Biological control of chestnut blight with hypovirulence: a critical analysis. Ann Rev Phytopathol 42:311–338
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue culture. Physiol Plant 15:473–497
Newhouse AE, Polin-McGuigan LD, Baier KA, Valletta KER, Rottmann WH, Tschaplinski TJ, Maynard CA, Powell WA (2014) Transgenic American chestnut show enhanced blight resistance and transmit the trait to T1 progeny. Plant Sci 228:88–97
Nonaka S, Ezura H (2014) Plant-Agrobacterium interaction mediated by ethylene and super-Agrobacterium conferring efficient gene transfer. Front Plant Sci 5:681
Nonaka S, Yuhashi K, Takada K, Sugaware M, Minamisawa K, Ezura H (2008) Ethylene production in plants during transformation suppresses vir gene expression in Agrobacterium tumefaciens. New Phytol 178:647–656
Pasonen H-L, Seppänen S-K, Degefu Y, Rytkönen A, von Weissenberg K, Pappinen A (2004) Field performance of chitinase transgenic silver birches (Betula pendula): resistance to fungal diseases. Theor Appl Genet 109:562–570
Polin LD, Liang H, Rothrock RE, Nishii M, Diehl DL, Newhouse AE, Mairn CJ, Powell WA, Maynard CA (2006) Agrobacterium-mediated transformation of American chestnut (Castanea dentata (Marsh) Borkh) somatic embryos. Plant Cell, Tissue Organ Cult 84:69–79
Pourhosseini L, Habashi AA, Kermani MJ, Khalighi A, Tahmasbi Z (2012) Agrobacterium-mediated transformation of chitinase gene in Rosa damascena cv. Ghamsar. Ann Biol Res 3:2843–2850
Roberts WK, Selitrennikoff CP (1990) Zeamatin, an antifungal protein from maize with membrane-permeabilizing activity. J Gen Microbiol 136:1771–1778
Rohini VK, Rao KS (2001) Transformation of peanut (Arachis hypogea L.) with tobacco chitinase gene: variable response of transformants to leaf spot disease. Plant Sci 160:889–898
Rothrock RE, Polin-McGuigan LD, Newhouse AE, Powell WA, Maynard CA (2007) Plate flooding as an alternative Agrobacterium-mediated transformation method for American chestnut somatic embryos. Plant Cell, Tissue Organ Cult 88:93–99
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Schouten H, Krens FA, Jacobsen E (2006a) Cisgenic plants are similar to traditionally bred plants. EMBO Rep 7:750–753
Schouten HJ, Krens FA, Jacobsen E (2006b) Do cisgenic plants warrant less stringent oversight? Nat Biotechnol 24:753
Sharma M, Kothari-Chajer A, Jagga-Chugh S, Kothari SL (2011) Factors influencing Agrobacterium tumefaciens-mediated genetic transformation of Eleusine coracana (L.) Gaertn. Plant Cell, Tissue Organ Cult 105:93–104
Sokal RR, Rohlf FJ (1981) Biometry: the principles and practice of statistics and biological research. W. H. Freeman & Company, New York 776 pp
Tang G-X, Knecht K, Yang X-F, Qin YB, Zhou W-J, Cai D (2011) A two-step protocol for shoot regeneration from hypocotyl explants of oilseed rape and its application for Agrobacterium-mediated transformation. Biol Plant 55:21–26
Toldi O, Tóth S, Pónyi T, Scott P (2002) An effective and reproducible transformation protocol for the model resurrection plant Craterostigma plantagineum Hochst. Plant Cell Rep 211:63–69
Van Alfen NK, Jayners RA, Anagnostakis SL, Day PR (1975) Chestnut blight: biological control by transmissible hypovirulence in Endothia parasitica. Science 189:890–891
Vanblaere T, Szankowski I, Schaart J, Schouten H, Flachowsky H, Broggini GAL, Gessler C (2011) The development of a cisgenic apple plant. J Biotechnol 154:304–311
Vannini A, Caruso C, Leonardi L, Ruggini E, Chiarot E, Caporale C, Buonocore V (1999) Antifungal properties of chitinases from Castanea sativa against hypovirulent and virulent strains of the chestnut blight fungus Cryphonectria parasitica. Physiol Mol Plant Pathol 55:29–35
Veluthakkal R, Dasgupta MG (2010) Pathogenesis-related genes and proteins in forest tree species. Trees 24:993–1006
Veluthakkal R, Karpaga Raja Sundari B, Ghosh Dasgupta M (2012) Tree chitinases—stress- and developmental-driven gene regulation. For Path 42:271–278
Vieitez AM, San José MC, Corredoira E (2011) Cryopreservation of zygotic embryo axes and somatic embryos of European chestnut. In: Thorpe TA, Yeung EC (eds) Plant embryo culture: methods and protocols, methods in molecular biology, vol 710. Springer, New York, pp 201–213
Wally O, Punja ZK (2010) Genetic engineering for increasing fungal and bacterial disease resistance in crop plants. GM Crops 1:199–206
Wheeler N, Sederoff R (2009) Role of genomics in the potential restoration of the American chestnut. Tree Gen Genom 5:181–187
Xu R, Li QQ (2008) Protocol: Streamline cloning of genes into binary vectors in Agrobacterium via the Gateway®TOPO vector system. Plant Methods 4:4
Zhan B, Oakes AD, Newhouse AE, Baier KM, Maynard CA, Powell WA (2013) A threshold level of oxalate oxidase transgene expression reduces Cryphonectria parasitica-induced necrosis in a transgenic American chestnut (Castanea dentata) leaf bioassay. Transgenic Res 22:973–982
Zhiying W, Fuli Z, Zhanbin W (2010) Transformation of chitinase gene into Populus simonii × P. nigra and chitinase activity of transgenic plants. Sci Silvae Sin 46:147–151
Acknowledgments
The authors thank Dr Leandro Peña for valuable suggestions and help with the plasmid construction. The authors also thank M.J. Cernadas, R. Montenegro and J.C. Suárez for excellent technical assistance. This research was partially funded by Ministerio de Economía y Competitividad (Spain) through Project AGL2013-47400-C4-3-R.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Corredoira, E., San José, M.C., Vieitez, A.M. et al. Agrobacterium-mediated transformation of European chestnut somatic embryos with a Castanea sativa (Mill.) endochitinase gene. New Forests 47, 669–684 (2016). https://doi.org/10.1007/s11056-016-9537-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11056-016-9537-5