
Abstract
The objective of this study was to investigate the loading capacity and release profiles of curcumin (CUR), as a hydrophobic drug molecule, and doxorubicin hydrochloride (DOX), as a hydrophilic drug molecule, into hollow mesoporous silica nanoparticles (HMSNs) through a co-delivery system. The drug loading and release studies were conducted and analyzed by HPLC–UV. The hydrodynamic size and zeta potential of HMSNs were 206.8 nm and − 28.8 mV respectively. Drug loadings of DOX into HMSN-DOX NPs and HMSN-DOX-CUR NPs were 37.51 ± 2.07% and 18.57 ± 6.22% respectively, and the loading capacities of CUR in HMSN-CUR NPs and HMSN-DOX-CUR NPs were 61.57 ± 2.82% and 43.73 ± 8.3% respectively. The release study of DOX in phosphate buffer saline (PBS, pH = 7.4) showed that 10.36 ± 0.7% and 13.06 ± 0.26% of loaded DOX in HMSN-DOX NPs and HMSN-DOX-CUR NPs were released after 24 h, respectively, whereas 92.04 ± 3.89% and 95.53 ± 0.29% of the loaded CUR in HMSN-CUR NPs and HMSN-DOX-CUR NPs were released after 6 h in albumin 3%, pH = 7.4, respectively. The result of this study suggests that HMSNs can be used as a suitable candidate system for the delivery of hydrophilic drugs in cancer therapies, thanks to their ability to prevent premature drug release from NPs. On the other hand, the release study of CUR as a hydrophobic drug model suggests that the considered NPs may not be a suitable candidate for the delivery of such drugs in cancer therapies due to the rapid release of compounds in a simulated plasma model.
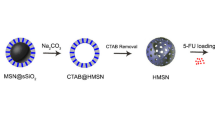
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nowadays, various types of nanoparticles are being studied to provide a suitable carrier for delivering chemotherapy medicine to overcome the side effects on healthy cells as a premier goal (Akhter et al. 2018). In the research process, nanoparticles are evaluated both in vitro and in vivo in terms of efficacy and mechanism of action. In vitro studies provide an efficient and rapid tool for researchers to assess nano-systems. Many of these studies eliminate the need for in vivo investigations (Huang et al. 2010). Moreover, release evaluations among in vitro studies might play a fundamental role before cytotoxicity investigations (D’Souza 2014).
For example, release studies may reveal that a high percentage of the drug is released from nanoparticles in a simulated plasma environment during the initial hours. Therefore, the hypothesis of using these nano-systems for cancer therapy would be rejected because, at the time of reaching the site of action, the nanoparticles practically lack the medicine due to drug release during the first hours after administration. Consequently, investigations of cellular lines somehow would be almost useless without considering the release profile of the designed nanoparticle. Hence, the appropriate drug selection for the designed nanoparticles would be of value by performing release studies before other in vitro studies.
The present study has aimed to clarify the importance of appropriate drug selection for each nano-system through loading and release studies. In this regard, among various properties of pharmaceutical agents, lipophilicity has been chosen as the main physicochemical characteristic to assess whether two drugs with completely different lipophilicity characteristics demonstrate a similar behavior in the same system or not. Thus, the loading capacity and release profiles of curcumin (CUR) and doxorubicin hydrochloride (DOX) have been studied in hollow mesoporous silica nanoparticles (HMSNs) as a nano delivery system. Although the benefits of DOX and CUR co-delivery in cancer therapy have been discussed in several studies (Zhang et al. 2016; Zhao et al. 2014; Karavasili et al. 2019), these compounds have been just selected to observe their behavior in a specific nanoparticle (HMSNs) to predict whether the nano-drug system can be used as a potential candidate in cancer therapy or not.
Mesoporous silica nanoparticles (MSNs) are among the developing nano-systems which are widely anticipated in many applications including drug delivery, gene delivery, chemotherapy, ultrasound therapy, photothermal therapy, and imaging (Liu et al. 2018; Maghsoudnia et al. 2020). MSNs demonstrate excellent structure with adjustable pore sizes, making this system an appropriate candidate for drug delivery due to the good control on drug loading and release (Vallet-Regí et al. 2018). These nanoparticles can also preserve the loaded drug for a long time with low burst releases before reaching the targeted site, which increases the chance of their use in the treatment of cancer (Vallet-Regí et al. 2018). Researchers have also used MSNs as a carrier to deliver a variety of chemotherapy drugs with a wide range of physicochemical properties, including 5-fluorouracil, curcumin, docetaxel, doxorubicin, quercetin, topotecan, and sunitinib (Narayan et al. 2018).
The HMSNs are introduced as a new generation of MSNs with loading capacity for both types of hydrophilic and hydrophobic drugs due to their hollow interiors and porous shells (Narayan et al. 2018). The high loading capacity of these NPs is directly associated with the hollow nature and the size of the interior cavity that is directly corresponding to the particle diameter which could be of great importance to load different active agents simultaneously in a carrier (Abdelaal and Shaikjee 2020; Zhou et al. 2018; Bharti et al. 2015). Moreover, the mesoporous nature of their shells can control the behavior of drug release as a sustained manner (Abdelaal and Shaikjee 2020; Deepika and PonnanEttiyappan 2018).
CUR is a natural polyphenol derived from traditional medicine known as turmeric (Curcuma longa). This compound has been documented to have poor aqueous solubility resulting in its poor bioavailability (Log P range from 3.62 to 4.12) (Yen et al. 2010; Nelson et al. 2017). On the contrary, DOX is a chemotherapeutic agent of the anthracycline group which is believed to dissolve in water with Log P ranging from 0.49 to 1.3 (Alrushaid et al. 2017).
Materials and methods
Materials
Tetraethyl orthosilicate (TEOS), bis [3-(triethoxysilyl) propyl] disulfide (BTESPD), cetyltrimethylammonium chloride (CTAC, 25 wt%), sodium chloride, sodium carbonate, triethylamine (TEA), and sodium lauryl sulfate (SLS) were purchased from Merck, Inc. (Germany). Doxorubicin hydrochloride salt and curcumin were from Pfizer® Inc. (USA) and Sabinsa Corporation Inc. (India), respectively. Human albumin 20% was acquired from Biotest, Inc. All chemicals were used as received without further purification.
Synthesis of HMSNs
Three steps were involved for fabricating the hollow nanostructures by the modified Stӧber method (Fig. 1a) (Chen et al. 2014). Briefly, uniform dense silica dSiO2 NPs were synthesized using a TEOS precursor as the first step. Then, dSiO2 NPs were coated with a shell of MSN, forming dSiO2@MSN using a surfactant-based mesoporous shell and TEOS and bis [3-(triethoxysilyl) propyl] disulfide BTESPD precursors. Finally, a high concentration of Na2CO3 was utilized to selectively etch dSiO2 NPs, resulting in uniform HMSNs.

a Different steps of the preparation procedure for HMSNs adapted from Chen et al. (Mistiran et al. 2010). b Size and morphology of HMSNs that were investigated by transmission electron microscopy (TEM)
Physicochemical characterization of HMSNs
The hydrodynamic size and zeta potential of dSiO2 NPs and HMSNs were acquired by a dynamic light scattering instrument (DLS and Zetasizer Malvern Instruments Ltd., Worcestershire, UK). The sizes and morphologies of the nanoparticles were investigated by transmission electron microscopy (TEM, JEM-1400, Japan). FT-IR spectra were evaluated by a Fourier transform infrared spectrometer (FT-IR, PerkinElmer Frontier, UK). Thermogravimetric analysis (TGA) was conducted using a Mettler Toledo Gas Controller GC 100 TGA instrument under N2 atmosphere from 25 to 600 °C at a heating rate of 10 °C/min. Nitrogen adsorption–desorption isotherm analysis was carried out on a Micromeritics NOVA 2000e QuantaChrome instrument (Anton-Paar, USA) for measuring surface area and pore size. All samples were dried at 120 °C for 2 h prior to analysis. The total pore area (ap,BJH), total pore volume (vp,BJH), and pore diameter (rp,BJH) were obtained from an adsorption branch by using the Barrett-Joyner-Halenda (BJH) method. The Brunauer–Emmett–Teller (BET) specific surface area (as,BET) was investigated via adsorption data. Images and spectra of the nanoparticles were acquired by field emission scanning electron microscopy (FESEM, Tescan, Czech) with a dual-energy dispersive X-ray spectrometer (EDXS) detector at an electron beam energy of 200 kV.
Apparatus and chromatographic conditions
At first, a reported UV spectroscopic method by Zhang et al. (2016) was used for simultaneous determination of both compounds for the loading capacity and release studies. Therefore, the standard curves of CUR and DOX in PBS (pH = 7.4) were plotted at a UV wavelength of 421 nm. As shown in Fig. S1-a, both compounds have absorbance at the same concentrations (DOX Ab ≈ 1/3 CUR Ab). To confirm this finding, the UV absorption spectra of DOX and CUR were plotted in the range of 200–500 nm. As can be seen in Figs. S1-b and S1-c, both drugs have absorbance at the same wavelengths. Thus, a UV-HPLC method was replaced to ensure the accuracy of the analysis method.
The HPLC method was performed in accordance with a previous method described by Mistiran et al. (2010) but with a slight modification. Briefly, the analyses of DOX and CUR (entrapped in or released from NPs) were conducted on a K-1001 HPLC pump coupled with a UV–visible detector (Knauer, Germany), equipped with a C18 column (12.5 cm × 4.6 mm × 5 µm) and a guard column with a maximum UV absorbance of 242 nm and 421 nm for DOX and CUR respectively, under an isocratic condition.
The mobile phases consisted of 30:70 (v/v) and 60:40 (v/v) mixtures of acetonitrile and acetic acid (2%) for DOX and CUR respectively, eluting at 1.0 mL/min. The column temperature was kept at 25 °C. Chromatographic data were acquired and processed using the EZChrom Elite software (version 3.2.1; Knauer, Germany).
Drug loading study
DOX, as a hydrophilic drug, was loaded into HMSNs at various nanoparticles-to-drug ratios such as the ratio 2:1, 1:1, and 1:2 in triplicate according to the following procedures, respectively: Firstly, 1 mL of 2 mg/mL DOX solution and 4, 2, and 1 mg of HMSNs were mixed separately, the reactions of which were conducted under stirring (500 RPM) for 72 h at room temperature in light-sealed vials. After 72 h, DOX-loaded HMSNs were settled by centrifugation at 7000 RPM for 10 min and washed with DI water to remove free DOX completely. The above procedures were conducted for CUR, as a hydrophobic drug, in the same nanoparticle-to-drug ratios, except that 2 mg of CUR was dissolved in 600 μL of ethanol 96% and reached 1 mL by DI water for preparation of the CUR solution and mixing with NPs. At the final stage, washing was done by DI water:ethanol (60:40, v/v) to remove free CUR.
For DOX and CUR co-loading, 1 mL of each solution was mixed, after which the mixtures were blended with NPs in the same ratios of nanoparticles and drugs. The final washing stages were carried out by a mixture of DI water:ethanol (60:40, v/v) for both compounds. The amounts of DOX and CUR in supernatants were analyzed by the HPLC–UV method. The calibration curves were plotted in the range of 250 ng/mL and 10 μg/mL in DI water and ethanol for both compounds. Entrapment efficiency (EE) and drug loading (DL) were calculated using Eqs. (1) and (2) as follows:
Drug release study
A prerequisite for an accurate evaluation of the release profile of NPs is to ensure the sink conditions being in the release medium. To accomplish this purpose, the final drug concentration in the release medium should be at least three times less than its measured saturation concentration in that medium (Phillips et al. 2012). The required volume to achieve sink conditions can be calculated according to the following equations:
To evaluate the release profile of DOX, HMSN-DOX, and HMSN-DOX-CUR, nanoparticles were dispersed in the appropriate volume of PBS (pH = 7.4). The suspension was stirred under constant shaking conditions (150 RPM) at 37 °C. Two hundred microliter of aliquot was taken out at specific time intervals (1, 2, 3, 4, 5, 6, and 24 h), and was then replaced with the same volume of fresh medium. The samples were centrifuged at 7000 RPM for 10 min. The released amounts of DOX in each sampling time were measured by the HPLC method. All release procedures were performed in triplicate.
Depending on the loaded amount of curcumin, an appropriate amount of HMSN-CUR or HMSN-DOX-CUR delivery systems was separately distributed in albumin 3% (pH = 7.4) to evaluate the release profile of the compound. The suspension was stirred under a 150 RPM shaking condition at 37 °C. The samples (200 µL) were collected immediately, and then every hour, in the first 6 h subsequently, the last sampling was collected 24 h after starting the release study. The same volume of fresh medium was replaced after each sampling time point. To assure complete solubility of CUR, 200 µL of methanol was added to the sample; then, 20 µL of perchloric acid (7.56 M) was added to the mixture to precipitate the albumin before CUR analysis. All samples were centrifuged at 15,000 RPM for 10 min, and 20 µL of the supernatant was separated for analysis.
Statistical analysis
In this section, all data are shown as the mean ± standard deviation (SD) of the experiments performed at least three times. Statistical comparisons were carried out using Student’s t test. P value < 0.05 was significant, and F1 (difference factor) and F2 (similarity factor) were used to analyze the release profiles of each drug in different systems.
Results and discussion
HMSN characterization
The hydrodynamic sizes of dSiO2 NPs and HMSNs were 139.5 ± 5 nm and 206.8 ± 10 nm, and the zeta potentials were − 45.8 ± 1 mV and − 28.8 ± 2 mV respectively (Table 1). The TEM images showed spherical nanoparticles with a total diameter of 150 nm and an outer shell of 30 nm, with a relatively smooth surface and uniform size distribution (Fig. 1b).
The FT-IR spectrum of HMSNs was quite simple and well-allocated. C-H stretching vibration peaks at 2928 cm−1 and C-H deformation vibration peaks at about 1480 cm−1 and 1569 cm−1 were assigned to BTESPD which was integrated in HMSN preparation. The broad absorption peaks at 3432 cm−1 and 1637 cm−1 were attributed to the stretching vibration of the silanol groups. Transmission peaks at about 1086 cm−1, 796 cm−1, and 466 cm−1 were ascribed to the characteristic stretching vibrations of the Si–O bond (Fig. 2a). TGA analysis of NPs demonstrated that HMSNs had a weight loss of c.a. 27.65% from 25 to 600 °C under nitrogen flow (Fig. 2b). This was attributed to the presence of organic compounds in nanoparticles. The nitrogen adsorption–desorption isotherm obtained from the HMSNs indicated that the existence of a gap between the adsorption and desorption branch and their sigmoid curves proved the existence of these nanoparticles (Fig. 2c). The BET diagram data displayed the surface area of HMSNs, which was 305.72 ± 8.1 m2/g (Table 2). The high surface area is one of the major features of HMSNs. As shown in Table 2, the BJH diagram also illustrated the total pore area (ap,BJH), total pore volume (vp,BJH), and pore diameter (rp,BJH) were 157.71 ± 6.7 m2/g, 0.48 ± 0.2 cm3/g, and 1.22 ± 0.4 nm, respectively.

a FT-IR spectra, b thermogravimetric analysis (TGA), and c nitrogen adsorption–desorption isotherm analysis of HMSNs
Figure 3a and Table 3 demonstrate approximate atomic densities in HMSNs via EDXS detector. As shown (Fig. 3b), the presence of sulfur (S) and silicon (Si) atoms in HMSNs was verified by the EDXS spectrum.

a The EDXS spectrum and b presence of sulfur (S) and silicon (Si) atoms in HMSNs
Entrapment efficiency and drug loading of NPs
The EE of DOX in HMSN-DOX NPs were 89.87 ± 4.64%, 90.14 ± 3.26%, and 70.77 ± 3.58% for the NPs-to-drug ratios of 2:1, 1:1, and 1:2, respectively (Fig. 4a). As shown in Fig. 4a, after adding CUR to that delivery system, the EE changed to 81.01 ± 4.12%, 80.67 ± 3.92%, and 60.14 ± 4.68% for DOX in HMSN-DOX-CUR NPs with the mentioned ratios of the nanoparticles to the compound, respectively. The results of the drug loading evaluation of NPs as illustrated in Fig. 4b were 29.96 ± 1.55%, 45.07 ± 1.63%, and 46.18 ± 2.39% for DOX in HMSN-DOX NPs with 2:1, 1:1, and 1:2 nanoparticle-to-drug ratios respectively. In addition, this parameter changed to 20.67 ± 0.64%, 27 ± 1.28%, and 25.75 ± 1.87% in the nanoparticle-to-drug ratios of 2:1, 1:1, and 1:2 in HMSN-DOX-CUR NPs, respectively.

a Entrapment efficiency and b loading capacity of DOX in HMSN-DOX NPs and HMSN-DOX-CUR NPs in nanoparticle-to-drug ratios 2:1, 1:1, and 1:2. c Entrapment efficiency and d loading capacity of CUR in HMSN-CUR NPs and HMSN-DOX-CUR NPs in nanoparticle-to-drug ratios 2:1, 1:1, and 1:2. Comparison of e entrapment efficiency and f loading capacity of DOX and CUR in HMSN-DOX NPs, HMSN-CUR NPs, and HMSN-DOX-CUR NPs in nanoparticle-to-drug ratios 1:2
As shown in Fig. 4c, the EE of CUR in HMSN-CUR NPs in the nanoparticle-to-drug ratios of 2:1, 1:1, and 1:2 were 95.15 ± 4.56%, 95.82 ± 3.12%, and 95.06 ± 3.29%, respectively. The parameter was calculated to be about 95.46 ± 3.48%, 95.48 ± 3.02%, and 94.85 ± 3.78% for CUR in HMSN-DOX-CUR NPs in the similar mentioned ratios, respectively. Although no significant variation was observed in the EE of CUR in different nanoparticle-to-drug ratios, the loading parameter was calculated to be about 31.72 ± 1.52%, 47.91 ± 1.56%, and 63.37 ± 2.19% for CUR in HMSN-CUR NPs with the nanoparticle-to-drug ratios of 2:1, 1:1, and 1:2 (Fig. 4d.). Moreover, the CUR loading capacities at the similar mentioned ratios in HMSN-DOX-CUR NPs were about 23.87 ± 0.87%, 31.83 ± 1.01%, and 37.94 ± 1.51%, respectively.
As the primary goal of the present study was to investigate probable differences in loading capacity and the release profiles of curcumin (a hydrophobic compound) and DOX (a hydrophilic drug) in HMSN NPs, the release study was only completed for nano-drug delivery systems with a nanoparticle-to-drug ratio of 1:2 which demonstrated the highest differences in loading capacity of the two compounds.
As can be seen in Fig. 4e, the results of the EE assessment could be indicating a greater tendency of nanoparticles to entrap more lipophilic compounds, where this parameter was calculated to be about 92.41 ± 4.29% and 65.6 ± 12.44% for HMSN-CUR NPs and HMSN-DOX NPs respectively (Pvalue < 0.001). In addition, this parameter was calculated in co-delivery systems where the EE of CUR and DOX in DOX-CUR NPs was determined as 94.49 ± 5.4% and 4.43 ± 15.54%, respectively (Pvalue < 0.001). On the other hand, although co-delivery of the compounds did not affect the EE of CUR (92.41 ± 4.29% vs 92.41 ± 4.29% for HMSN-CUR NPs and HMSN-DOX-CUR NPs respectively), the parameter significantly changed for DOX where the EE was calculated to be 65.6 ± 12.44% for HMSN-DOX NPs and 46.43 ± 15.54% for HMSN-DOX-CUR respectively (Pvalue < 0.05) (Fig. 4e).
Furthermore, as displayed in Fig. 4f, the loading results were in accordance with EE observations. The drug loading of NPs was higher for CUR in comparison to that for DOX, where the parameter was 61.57 ± 2.82% and 37.51 ± 2.07% for HMSN-CUR NPs and HMSN- DOX NPs, respectively (Pvalue < 0.001). Although the loading capacity was decreased for both compounds in a co-delivery system, the observed reduction for DOX was more noticeable, where CUR and DOX loading in HMSN-DOX-CUR NPs changed to 43.73 ± 8.30% and 18.57 ± 6.22%, respectively (Pvalue < 0.001).
In vitro release study
The release profile is recognized as an important parameter for evaluating the safety and efficiency of the product and is therefore utilized as a valid predictor of in vivo behavior of both traditional and novel dosage forms (Amann et al. 2010; Buch et al. 2010; Souza et al. 2014). In addition, although cellular studies may show the cytotoxicity of drug-loaded nanoparticles on cancerous cells, the release profile in a simulated plasma environment may reject their efficacy because a high percentage of compounds is released from nanoparticles before reaching target cells. Therefore, it can be said that release studies have a significant role among in vitro studies (D’Souza 2014).
To ensure an accurate release test, the sink condition is recognized as one of the essential requirements which could be more challenging for highly hydrophobic drugs. Therefore, several release mediums including PBS, SLS 0.1%, SLS 0.5%, and albumin 3% were examined for the highly lipophilic compound CUR in this study (Log P = 3.62–4.12).
It is worth considering that the powder form of a solid compound should be used to determine the saturation concentration because even a small amount of a co-solvent could considerably change the calculated parameter. This finding has been carefully discussed by Abouelmagd et al. (2015), showing particularly different solubilities for paclitaxel in different mediums when using the same amount of compound in solid form or when dissolved in DMSO.
The calculated saturation concentration of CUR was 0.9, 8, 32, and 8 μg/mL in PBS, SLS 0.1%, SLS 0.5%, and albumin 3%, respectively. Although PBS is used in most release studies, it was not appropriate for this investigation because large volumes of the medium (about 2 L) should be applied to maintain the sink condition for the loading amount of the compound in the delivery systems, a factor which could reduce the accuracy of the compounds’ analysis, especially at early sampling time points. Despite the suitable observed saturation concentrations, the SLS-containing mediums were not applied in this study due to the complications during the HPLC analysis of the collected samples. Furthermore, the surfactant-containing mediums are more recommended in release studies of oral delivery systems (Rahman et al. 2009). Albumin-containing solutions appear to be preferred for release tests, especially for vascular delivery systems, as albumin can be extremely helpful in establishing sink conditions due to the high protein binding capacity. It also makes approximately similar conditions to the actual plasma environment. Therefore, a PBS solution containing 3% of albumin (pH = 7.4) was selected as the final CUR release medium. In this regard, the appropriate release volume was estimated to be about 600 mL according to the loaded amount of the compound in each delivery system and the saturation concentration of CUR in this medium (Eq. 4).
At the same time, as the saturation concentration for DOX was calculated to be about 20 μg/mL in PBS at pH = 7.4, this solution was selected as the simplest and most convenient medium applied for the release test of this compound and the suitable volume for the release medium was determined according to this saturation concentration.
The release profiles of DOX from the two studied NPs (HMSN-DOX NPs and HMSN-DOX-CUR) are depicted in Fig. 5a. The statistical analysis demonstrated no significant difference in the amount of DOX released from the delivery systems (HMSN-DOX NPs and HMSN-DOX-CUR NPs), where approximately about 9.96 ± 3.22% and 8.38 ± 0.51% of the encapsulated amount of DOX were released from HMSN-DOX NPs and HMSN-DOX-CUR NPs until 6 h after starting the release study respectively (F1 = 24.25, F2 = 81.64). It seems that the designed nanoparticles do not tend to release more DOX because only about 10.36 ± 0.7% and 13.06 ± 0.26% of the encapsulated compound were quantified in the medium 24 h after the study from HMSN-DOX NPs and HMSN-DOX-CUR NPs respectively (F1 = 24.54, F2 = 79.74).

a The DOX release from HMSN-DOX NPs and HMSN-DOX-CUR NPs after 24 h in phosphate buffer saline (PBS, pH = 7.4). b CUR release from HMSN-CUR NPs and HMSN-DOX-CUR NPs after 24 h in albumin 3% (pH = 7.4)
As shown in Fig. 5b, about 92.04 ± 3.89% and 95.53 ± 0.29% of CUR were released from HMSN-CUR NPs and HMSN-DOX-CUR NPs after 6 h, respectively. As approximately more than 90% of the compound was released from both delivery systems in the early sampling time points, no statistical evaluation was performed to compare the release profiles. Unexpectedly, the continuation of the release test for up to 24 h showed a significant reduction in the rate of release, where the amounts of CUR measured at this time point were about 62.89 ± 4.79% and 67.14 ± 4.12% from HMSN-CUR NPs and HMSN-DOX-CUR NPs respectively. Since almost all loaded CUR was released in the early sampling time points, and also due to the reduction of the compound with a similar slope in both studied delivery systems, the observed decrease could be explained by the instability of CUR in the physiologic and acidic pHs. To examine this hypothesis, all samples collected in the first 6 h of the release study which were injected into the HPLC immediately after collection were re-analyzed after 24 h. As shown in Figs. S2-a and S2-b, the results approved the instability of the CUR in the release medium.
As shown in Fig. 5a and b, the co-delivery of the two compounds cannot affect the dissolution process of drugs, where the release profiles of each compound were similar in both designed delivery systems with no statistical differences.
The most important finding of the study seems to be that although the release profiles of DOX (Fig. 5a), as a hydrophilic compound, suggested that HMSNs can be used as a potential drug delivery system candidate for cancer therapy, these NPs cannot be used as a suitable cancer therapy candidate for hydrophobic compounds.
In other words, illustrating all the characteristics, including high loading capacity and low burst release as well as releasing only a small percentage of DOX (less than 14%) within 24 h of the release study, can suggest the ability of NPs as a potential drug delivery system candidate for cancer therapy. Zhai et al. proved that HMSNs were degradable inside the cytoplasm and lysosomes of human umbilical vein endothelial cells (Zhai et al. 2012). Besides, various studies have shown that MSNs degrade in different cells after becoming hollow (Croissant et al. 2017). On the other hand, the absence and non-functionality of lymphatic vessels, high vascular density caused by angiogenesis, and large gaps between endothelial cells of blood vessels in tumors lead to the enhanced permeability and retention (EPR) effect, which causes considerable accumulation of nanocarriers in tumor cells rather than others (Danhier et al. 2010; Fang et al. 2011). Therefore, since the nano-system releases a small amount of DOX in the simulated plasma model, it seems that a high percentage of DOX will be released in tumor cells. Obviously, further investigations will be required to confirm the efficacy of this nano-system in cancer therapy if it aims to be used for this purpose.
However, the NPs demonstrated completely different release characteristics when delivering a lipophilic compound (CUR), where more than 90% of the compound was released in very early sampling time points in the simulated plasma model (Fig. 5b). Furthermore, taking out more samples during the first hour of the release study proved that almost all loaded CUR has been released in the first sampling time point (15 min). The observed properties, despite the high loading capacity, cannot serve these NPs as a suitable candidate for cancer therapy of lipophilic compounds. Similarly, Hillerström et al. (2014) demonstrated that 90% of ibuprofen was released from mesoporous silica nanoparticles to the solution after 200 s. In addition, Zhang et al. (2015), Sun et al. (2017), Reddy et al. (2019), and He et al. (2020) loaded DOX and CUR into T-DNA-NCs (DNA-hybrid-gated photothermal mesoporous silica nanoparticles), MSN-Pep (peptide-decorated mesoporous silica nanoparticle), UCNP@mSiO2 (up-conversion nanoparticles coated with porous silica and functionalized with an amine), and SP-FS-USMSN (spiropyran and fluorinated silane–modified ultrasmall mesoporous silica nanoparticles), respectively. In these studies, DOX and CUR had been released less than 30% in the medium at pH = 7.4 without any intervention. However, it seems that the method of analysis and sink condition should be validated in these studies.
From another point of view, the independent release profiles of DOX and CUR in co-delivery systems could confirm a bi-structural characteristic of HMSNs, similar to nano-emulsion and nanoliposome suggested in previous studies, allowing for the loading of hydrophobic and hydrophobic agents in different structures of these NPs (Chen et al. 2012). It is proposed that the large interior hollow of HMSNs could act out as a reservoir for hydrophobic agents, while the shell as the reservoir for hydrophilic agents (Chen et al. 2012). In addition, Li et al. (2013) claimed that their synthesized mesoporous silica nanospheres are composed of a hydrophobic core and a hydrophilic shell.
In this study, the lack of burst release of DOX increases the probability of electrostatic bonding between DOX and silanol groups contrary to the findings of others regarding the entrapment of hydrophilic drugs in HMSN shells. Besides, the very rapid release rate of CUR, even at the early sampling time points, increases the probability of this lipophilic agent loading on the surface of the shell instead of loading in the interior hole of NPs.
To investigate this hypothesis, HMSNs-NH2 in which the silanol groups were replaced with -NH2 (especially at the surface of the shell) were synthesized, after which the CUR was loaded in synthesized NPs. As shown in Figs. S3-a and S3-b, the amounts of encapsulated and loaded CUR in HMSNs-NH2 NPs were significantly reduced compared to HMSNs, which may confirm the reduction in lipophilicity of the surface of NPs leading to lower entrapment of a lipophilic agent as CUR. However, it should be noted that to fully confirm the loading site of different compounds, especially with different lipophilicities, more experiments must be performed.
Conclusion
The result of this study suggests that HMSNs can be used as a suitable candidate system for the delivery of hydrophilic drugs in cancer therapies, thanks to their ability to prevent premature drug release from NPs. On the other hand, the release study of CUR as a hydrophobic drug model suggests that the considered NPs may not be a suitable candidate for the delivery of such drugs in cancer therapies because of the rapid release of the compounds in a simulated plasma model. It seems that the appropriate drug selection for each designed nano-system is necessary to predict whether the nano-drug system can be used as a potential candidate in cancer therapy or not. For example, this study showed that the lipophilicity of the selected drug model might be a significant characteristic for the application prediction of a designed drug delivery system. Therefore, studies about the generalization of the results on delivery systems for investigating other drugs are relevant to be considered as future works.
References
Abdelaal HM, Shaikjee A (2020) Microwave-based fast synthesis of clear-cut hollow spheres with mesoporous wall of silica nanoparticles as excellent drug delivery vehicles. J Nanoparticle Res 22:1–9. https://doi.org/10.1007/s11051-020-04918-3
Abouelmagd SA, Sun B, Chang AC, Ku YJ, Yeo Y (2015) Release kinetics study of poorly water-soluble drugs from nanoparticles: are we doing it right? Mol Pharm 12:997–1003. https://doi.org/10.1021/mp500817h
Akhter MH, Rizwanullah M, Ahmad J, Ahsan MJ, Mujtaba MA, Amin S (2018) Nanocarriers in advanced drug targeting: setting novel paradigm in cancer therapeutics. Artif Cells Nanomed Biotechnol 46:873–884. https://doi.org/10.1080/21691401.2017.1366333
Alrushaid S, Sayre CL, Yáñez JA, Forrest ML, Senadheera SN, Burczynski FJ, Löbenberg R, Davies NM (2017) Pharmacokinetic and toxicodynamic characterization of a novel doxorubicin derivative. Pharmaceutics 9.https://doi.org/10.3390/pharmaceutics9030035
Amann LC, Gandal MJ, Lin R, Liang Y, Siegel SJ (2010) In vitro-in vivo correlations of scalable PLGA-risperidone implants for the treatment of schizophrenia. Pharm Res 27:1730–1737. https://doi.org/10.1007/s11095-010-0152-4
Bharti C, Gulati N, Nagaich U, Pal A (2015) Mesoporous silica nanoparticles in target drug delivery system: a review. Int J Pharm Investig 5:124–133. https://doi.org/10.4103/2230-973x.160844
Buch P, Holm P, Thomassen JQ, Scherer D, Branscheid R, Kolb U, Langguth P (2010) IVIVC for fenofibrate immediate release tablets using solubility and permeability as in vitro predictors for pharmacokinetics. J Pharm Sci 99:4427–4436. https://doi.org/10.1002/jps.22148
Chen F, Hong H, Shi S, Goel S, Valdovinos HF, Hernandez R, Theuer CP, Barnhart TE, Cai W (2014) Engineering of hollow mesoporous silica nanoparticles for remarkably enhanced tumor active targeting efficacy. Sci Rep 4.https://doi.org/10.1038/srep05080
Chen Y, Gao Y, Chen H, Zeng D, Li Y, Zheng Y, Li F, Ji X, Wang X, Chen F, He Q, Zhang L, Shi J (2012) Engineering inorganic nanoemulsions/nanoliposomes by fluoride-silica chemistry for efficient delivery/co-delivery of hydrophobic agents. Adv Funct Mater 22:1586–1597. https://doi.org/10.1002/adfm.201102052
Croissant JG, Fatieiev Y, Khashab NM (2017) Degradability and clearance of silicon, organosilica, silsesquioxane, silica mixed oxide, and mesoporous silica nanoparticles. Adv Mater 29:1604634. https://doi.org/10.1002/ADMA.201604634
Danhier F, Fabienne O, Préat V (2010) To exploit the tumor microenvironment: passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Control Release 148:135–146. https://doi.org/10.1016/J.JCONREL.2010.08.027
Deepika D, PonnanEttiyappan JB (2018) Synthesis and characterization of microporous hollow core-shell silica nanoparticles (HCSNs) of tunable thickness for controlled release of doxorubicin. J Nanoparticle Res 20:1–15. https://doi.org/10.1007/s11051-018-4287-2
D’Souza S (2014) A review of in vitro drug release test methods for nano-sized dosage forms. Adv Pharm 2014:1–12. https://doi.org/10.1155/2014/304757
Fang J, Nakamura H, Maeda H (2011) The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev 63:136–151. https://doi.org/10.1016/J.ADDR.2010.04.009
He Y, Shao L, Usman I, Hu Y, Pan A, Liang S, Xu H (2020) A pH-responsive dissociable mesoporous silica-based nanoplatform enabling efficient dual-drug co-delivery and rapid clearance for cancer therapy. Biomater Sci 8:3418–3429. https://doi.org/10.1039/d0bm00204f
Hillerström A, Andersson M, Samuelsson J, Van Stam J (2014) Solvent strategies for loading and release in mesoporous silica. Colloids Interface Sci Commun 3:5–8. https://doi.org/10.1016/j.colcom.2015.01.001
Huang YW, Wu CH, Aronstam RS (2010) Toxicity of transition metal oxide nanoparticles: recent insights from in vitro studies. Materials (Basel) 3:4842–4859. https://doi.org/10.3390/ma3104842
Karavasili C, Andreadis DA, Katsamenis OL, Panteris E, Anastasiadou P, Kakazanis Z, Zoumpourlis V, Markopoulou CK, Koutsopoulos S, Vizirianakis IS, Fatouros DG (2019) Synergistic antitumor potency of a self-assembling peptide hydrogel for the local co-delivery of doxorubicin and curcumin in the treatment of head and neck cancer. Mol Pharm 16:2326–2341. https://doi.org/10.1021/acs.molpharmaceut.8b01221
Li S, Jiao X, Yang H (2013) Hydrophobic core/hydrophilic shell structured mesoporous silica nanospheres: enhanced adsorption of organic compounds from water. Langmuir 29:1228–1237. https://doi.org/10.1021/la303733w
Liu JL, Liu T, Pan J, Liu S, Max Lu GQ (2018) Advances in multicompartment mesoporous silica micro/nanoparticles or theranostic applications. Annu Rev Chem Biomol Eng 9:389–411
Maghsoudnia N, Eftekhari RB, Sohi AN, Zamzami A, Dorkoosh FA (2020) Application of nano-based systems for drug delivery and targeting: a review. J Nanoparticle Res 22:1–41. https://doi.org/10.1007/s11051-020-04959-8
Mistiran AF, Dzarr AA, Gan SH (2010) HPLC method development and validation for simultaneous detection of arabinoside-C and doxorubicin. Toxicol Mech Methods 20:472–481. https://doi.org/10.3109/15376516.2010.503246
Narayan R, Nayak UY, Raichur AM, Garg S (2018) Mesoporous silica nanoparticles: a comprehensive review on synthesis and recent advances. Pharmaceutics 10.https://doi.org/10.3390/pharmaceutics10030118
Nelson KM, Dahlin JL, Bisson J, Graham J, Pauli GF, Walters MA (2017) The essential medicinal chemistry of curcumin. J Med Chem 60:1620–1637. https://doi.org/10.1021/acs.jmedchem.6b00975
Phillips DJ, Pygall SR, Cooper VB, Mann JC (2012) Overcoming sink limitations in dissolution testing: a review of traditional methods and the potential utility of biphasic systems. J Pharm Pharmacol 64:1549–1559. https://doi.org/10.1111/j.2042-7158.2012.01523.x
Rahman SMH, Telny TC, Ravi TK, Kuppusamy S (2009) Role of surfactant and pH in dissolution of curcumin. Indian J Pharm Sci 71:139–142. https://doi.org/10.4103/0250-474X.54280
Reddy KL, Sharma PK, Singh A, Kumar A, Shankar KR, Singh Y, Garg N, Krishnan V (2019) Amine-functionalized, porous silica-coated NaYF 4:Yb/Er upconversion nanophosphors for efficient delivery of doxorubicin and curcumin. Mater Sci Eng C 96:86–95. https://doi.org/10.1016/j.msec.2018.11.007
Souza SD’, Faraj JA, Giovagnoli S, Deluca PP (2014) IVIVC from long acting olanzapine microspheres. Int J Biomater 2014.https://doi.org/10.1155/2014/407065
Sun X, Luo Y, Huang L, Yu BY, Tian J (2017) A peptide-decorated and curcumin-loaded mesoporous silica nanomedicine for effectively overcoming multidrug resistance in cancer cells. RSC Adv 7:16401–16409. https://doi.org/10.1039/c7ra01128h
Vallet-Regí M, Colilla M, Izquierdo-Barba I, Manzano M (2018) Mesoporous silica nanoparticles for drug delivery: current insights. Molecules 23.https://doi.org/10.3390/molecules23010047
Yen F-L, Wu T-H, Tzeng C-W, Lin L-T, Lin C-C (2010) Curcumin nanoparticles improve the physicochemical properties of curcumin and effectively enhance its antioxidant and antihepatoma activities. J Agric Food Chem 58:7376–7382. https://doi.org/10.1021/jf100135h
Zhai W, He C, Wu L, Zhou Y, Chen H, Chang J, Zhang H (2012) Degradation of hollow mesoporous silica nanoparticles in human umbilical vein endothelial cells. J Biomed Mater Res B Appl Biomater 100:1397–1403. https://doi.org/10.1002/JBM.B.32711
Zhang Y, Hou Z, Ge Y, Deng K, Liu B, Li X, Li Q, Cheng Z, Ma P, Li C, Lin J (2015) DNA-hybrid-gated photothermal mesoporous silica nanoparticles for NIR-responsive and aptamer-targeted drug delivery. ACS Appl Mater Interfaces 7:20696–20706. https://doi.org/10.1021/acsami.5b05522
Zhang Y, Yang C, Wang W, Liu J, Liu Q, Huang F, Chu L, Gao H, Li C, Kong D, Liu Q, Liu J (2016) Co-delivery of doxorubicin and curcumin by pH-sensitive prodrug nanoparticle for combination therapy of cancer. Sci Rep 6:1–12. https://doi.org/10.1038/srep21225
Zhao X, Chen Q, Liu W, Li Y, Tang H, Liu X, Yang X (2014) Codelivery of doxorubicin and curcumin with lipid nanoparticles results in improved efficacy of chemotherapy in liver cancer. Int J Nanomed 10:257–270. https://doi.org/10.2147/IJN.S73322
Zhou Y, Quan G, Wu Q, Zhang X, Niu B, Wu B, Huang Y, Pan X, Wu C (2018) Mesoporous silica nanoparticles for drug and gene delivery. Acta Pharm Sin B 8:165–177. https://doi.org/10.1016/j.apsb.2018.01.007
Acknowledgements
We thank the Nanotechnology Research Center, Faculty of Pharmacy, Tehran University of Medical Science for financial support.
Funding
This study was funded by the grant from Tehran University of Medical Science (No. 43747).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kabiri, F., Mirfakhraee, S., Ardakani, Y.H. et al. Hollow mesoporous silica nanoparticles for co-delivery of hydrophobic and hydrophilic molecules: mechanism of drug loading and release. J Nanopart Res 23, 226 (2021). https://doi.org/10.1007/s11051-021-05332-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-021-05332-z