
Abstract
Hollow core-shell silica nanoparticles (HCSNs) are being considered as one of the most favorable drug carriers to accomplish targeted drug delivery. In the present study, we developed a simple two-step method, employing polystyrene (PS) nanoparticles (150 ± 20 nm) as a sacrificial template for the synthesis of microporous HCSNs of size 230 ± 30 nm. PS core and the wall structure directing agent cetyl trimethyl ammonium bromide (CTAB) were removed by calcination. Monodispersed spherical HCSNs were synthesized by optimising the parameters like water/ethanol volume ratio, PS/tetraethyl orthosilicate (TEOS) weight ratio, concentration of ammonia, and CTAB. Transmission electron microscopy (TEM) revealed the formation of hollow core-shell structure of silica with tunable thickness from 15 to 30 nm while tailoring the concentration of silica precursor. The results obtained from the cumulative release studies of doxorubicin loaded microporous HCSNs demonstrated the dependence of shell thickness on the controlled drug release behavior. HCSNs with highest shell thickness of 30 nm and lowest surface area of 600 m2/g showed delay in the doxorubicin release, proving their application as a drug carrier in targeted drug delivery systems. The novel concept of application of microporous HCSNs of pore size ~ 1.3 nm with large specific surface area in the field of drug delivery is successful.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Hollow core-shell nanoparticles have attracted attention in the recent years, due to their unique properties like large surface area (Zhao et al. 1998; Zhou et al. 2014), tunable thickness (Jiao et al. 2012), tunable pore diameter (Slowing et al. 2007), low density, high chemical/thermal stability, and good biocompatibility (Liu et al. 2012). They find application in drug delivery, batteries, catalysis, thermal insulation, and sensors (Sandberg et al. 2013; Zhang et al. 2013; Kumar and Himanshu 2014; Li et al. 2015; Wang et al. 2016). Several techniques were reported in the literature to synthesize inorganic hollow core-shell nanoparticles like sacrificial template method (Yuan et al. 2010), galvanic displacement (Li et al. 2010), sol–gel method (Deng et al. 2006), self-assembly technique (Zhang et al. 2009), and surfactant-assisted solvothermal method (He et al. 2004). Among these synthesis methods, sacrificial template method was found to be one of the efficient ways to synthesize hollow structured nanoparticles. Soft templates (vesicles, micelles, emulsions) (Hubert et al. 2000; Che et al. 2003; Nguyen et al. 2014) and hard templates (polymer, carbon, silica) (Gu et al. 2004; Wang et al. 2010; Koike et al. 2013; Han et al. 2014) were the two major types of the templates used during sacrificial template method. The major limitation of soft template method was the formation of irregular shaped particles, which was overcome by the use of hard templates.
Hollow core-shell silica nanoparticles (HCSNs) were found to be nontoxic, highly biocompatible, mechanically stable, and chemically inert (Ogi et al. 2014). Various hard sacrificial templates used for the synthesize of HCSNs were polystyrene (PS) (Yoon et al. 2014), polystyrene methyl acrylic acid, sulfonated polystyrene methyl acrylic acid (Liu et al. 2011), poly(t-butyl acrylate) (Zhou et al. 2014), and SiOH-functionalized PS latexes (Deng et al. 2015). PS nanoparticles are used as efficient sacrificial templates due to the ease of synthesis, monodispersity, spherical shape, and low cost. Emulsion polymerization is one of the methods used to synthesize monodispersed spherical PS particles in nano size range. PS templates could be easily removed by calcination at elevated temperature in order to obtain hollow core-shell nanoparticles (Zhang et al. 2009).
Solid monodispersed silica spheres were synthesized by Stober method via hydrolysis-condensation of silica precursor in water-alcohol medium using ammonia as a catalyst and cetyl trimethyl ammonium bromide (CTAB) as a wall structure directing agent (Stober et al. 1968; Ge et al. 2009). The Stober method was modified to synthesize hollow silica spheres with uniform particle size using polyelectrolytes as template (Nguyen et al. 2014). Use of hollow mesoporous silica nanoparticles has shown sustained drug release behavior and finds potential application in cancer therapy (She et al. 2015). Inorganic drug carriers have high levels of drug encapsulation, better chemical stability, and good biocompatibility compared to organic carriers which are basic requirements of targeted drug delivery systems. Drug release rate has found to depend on parameters like variation in pore size, shell thickness, and surface area of silica nanoparticles (Jiao et al. 2012; Mohamed El-Toni et al. 2012).
There were fewer studies which evaluated the loading and release properties of HCSNs with varied shell thickness and narrow pore size (microporous). In the present research work, we report a simple two-step method to synthesize HCSNs. PS nanoparticles were synthesized in the first step and they were used as sacrificial templates in the second step while synthesizing HCSNs. Effect of parameters like water-ethanol volume ratio, concentration of ammonia, concentration of CTAB, and PS/tetraethyl orthosilicate (TEOS) weight ratio on morphology of HCSNs were analyzed. The microporous HCSNs with tunable shell thickness and hollow core-shell structure found to have high demand in the field of drug delivery. Therefore, loading and release properties of HCSNs were examined using doxorubicin as a model drug.
Materials and methods
Materials
The chemicals used in the present research work were styrene (99%), polyvinyl pyrrolidone (PVP, molecular weight 40,000), TEOS (98%), and doxorubicin purchased from Sigma-Aldrich. Potassium per sulfate (KPS, 98%) and CTAB (98%) were procured from Loba Chemie. Aqueous ammonia solution (25%) and ethanol were purchased from Spectrum Chemicals and Changshu Hongsheng Fine Chemical Co., respectively. All the chemicals were used without further purification. Millipore water was used throughout the experiments.
Synthesis of polystyrene template
Styrene was polymerized by emulsion polymerization in water using KPS as an initiator and PVP as a stabilizer. One hundred twenty-nine micromoles of KPS and 15 μmol of PVP were dissolved in water separately. Of styrene, 43.4 mmols of styrene was taken in 100-mL round bottom flask and PVP solution was added, 1:9 volume ratio of styrene to water was maintained. Reaction mixture was gradually heated to 70 °C and KPS was added. The solution was kept stirring at 70 °C for 24 h in nitrogen atmosphere. The reaction mixture was allowed to cool down to room temperature. PS particles were obtained after centrifugation of reaction mixture at 13,000 rpm for 20 min. Resultant product was subsequently washed with water in order to remove unreacted chemicals. Samples were freeze dried to remove the retained moisture content.
Synthesis of hollow core-shell silica nanoparticles
Synthesized PS template (0.1 g) was dispersed uniformly in water-ethanol mixture by ultrasonication for 30 min. Five percent CTAB solution was added to the mixture and stirred for 2 h. Ammonia was added dropwise to the reaction mixture, followed by addition of TEOS-ethanol mixture (TEOS mixed with equal amount of ethanol). Stirring was continued for 4 h at room temperature. The resulting suspension was aged at room temperature for 24 h. The precipitate was separated by centrifugation at 10,000 rpm for 15 min and washed five times with water. Samples obtained were freeze dried to remove moisture content. Thus, obtained silica-coated PS nanoparticles were calcined at 550 °C for 4 h at a heating rate of 1 °C/min to remove PS core. HCSNs were synthesized by varying parameters like volume ratio of water/ethanol, concentration of ammonia, concentration of CTAB, and weight ratio of PS/TEOS. When one of the parameters was varied, all the remaining parameters were maintained constant.
Loading and release studies
Fifty milligrams of HCSNs was well dispersed in 100 ppm doxorubicin solution by ultrasonication for 1 min. Samples were stored at 4 °C for 48 h. Doxorubicin-loaded silica nanoparticles were separated by centrifugation at 12,000 rpm for 15 min and dispersed in phosphate-buffered saline (PBS) having pH of 7.4. Resultant mixture was gently stirred at 37 °C. To study the release of doxorubicin, 5 mL of solution was extracted at predetermined time intervals and centrifuged at 12,000 rpm for 5 min. The supernatant was analyzed in a UV-visible spectrophotometer at a wavelength of 480 nm and the sample was poured back in order to maintain the constant volume. The loading capacity and entrapment efficiency of doxorubicin were calculated by using the Eqs. (1) and (2), respectively (Yang et al. 2008).
Characterization techniques
Scanning electron microscope (SEM, JEOL-JSM 6380 LA) was used to analyze the surface morphology of synthesized nanoparticles. Samples were gold sputtered prior to visualization in SEM. Hollow core-shell structures of silica nanoparticles were studied using transmission electron microscopy (TEM, JEOL JEM-2100). Before observing in TEM, samples were dispersed in ethanol by ultrasonication and dropped on to the carbon coated copper grid. Nitrogen adsorption/desorption isotherms of the HCSNs were recorded in a volumetric adsorption analyzer (Quantachrome Corporation, NOVA1000). The samples were degassed in vacuum at 450 °C for 8 h and analysis was carried out at 77 K over a range of relative pressures (P/P0). The specific surface area was calculated by using Brunauer–Emmett–Teller (BET) and the pore size distribution was obtained from desorption isotherm curves using Barrett–Joyner–Halenda (BJH) methods. X-ray diffraction pattern of HCSNs was obtained using Rigaku Miniflex-600 diffractometer with Cu Kα radiation, operated at 40 kV and 15 mA. XRD patterns were recorded for 2θ, ranging from 5° to 90° with a scanning step of 0.02°. Fourier transform infrared spectra of the PS, silica-coated PS, and HCSNs were obtained by the KBr pellet technique using FTIR (Bruker Alpha) Thermogravimetric analysis (TGA) was carried out at a heating rate of 10 K/min from the room temperature to 750 °C in an air flow by using TG/TGA instrument (Hitachi-6300). The drug uptake and release were monitored by UV-visible spectrophotometer (Hitachi, U-2900).
Results and discussion
Figure 1 shows the SEM image of PS nanoparticles synthesized by emulsion polymerization. The particles were found to be monodispersed and spherical in shape with an average diameter of 150 ± 20 nm.

SEM image of PS nanoparticles
The effect of variation in water/ethanol volume ratio on the morphology of HCSNs is represented in Fig. 2. The particles were found to be in random shape and highly porous in nature, when water alone was used as the dispersion medium (Fig. 2a). The irregularity in particle size was due to the rapid hydrolysis and condensation of TEOS (Berru et al. 2013). Figure 2b–d shows the variation in morphology of HCSNs as the fraction of water reduced from 4:1 to 1:4, in the water-ethanol mixture. Monodispersed HCSNs with smooth surface and spherical shape were achieved when the volume ratio of water/ethanol was maintained as 1:4 (Fig. 2d). The HCSNs were found to be agglomerated when ethanol alone was used as the dispersion medium (Fig. 2e). Agglomeration was due to the low rate of hydrolysis and condensation of TEOS, which in turn reduced the rate of formation of silica shell on the surface of PS particle (Yuan et al. 2010).

SEM images of HCSNs synthesized by variation of water/ethanol volume ratio. a 100% water. b 4:1. c 3:2. d 1:4. e 100% ethanol (0.6 mL CTAB, 0.1 g TEOS, 50 μL ammonia)
The morphology of silica shell was also influenced by the concentration of ammonia which was used as the catalyst during the synthesis of HCSNs (Stober et al. 1968). At low concentration of ammonia, poor coating of silica on the surface of PS and agglomeration of particles were observed as shown in Fig. 3a, b. Thus, low hydrolysis rate of TEOS would not be preferable to coat silica on the surface of the PS nanoparticles (Yuan et al. 2010). However, as the concentration of ammonia was increased from 20 to 50 μL, uniform coating was observed resulting in the formation of uniform spherical particles (Fig. 3c). Further increasing the concentration of ammonia from 50 to 70 μL, undesirable free silica particles were observed (Fig. 3d). It was due to the heterogeneous nucleation of silica nanoparticles with increased rate of hydrolysis and condensation of TEOS (Park et al. 2009).

SEM images of HCSNs synthesized by variation of ammonia. a 20 μL. b 30 μL. c 50 μL. d 70 μL (1:1 weight ratio of PS/TEOS, 0.6 mL CTAB, 1:4 volume ratio of water/ethanol)
CTAB played a major role in holding silica on the surface of PS nanoparticles. CTAB and TEOS were introduced during condensation step which involved in co-condensation during the synthesis of HCSNs (Trewyn et al. 2007). CTAB coating on PS would introduce positive charge on the surface of the PS particles. Thus, negatively charged silica sol formed from TEOS would bind to the surface of CTAB-coated PS (Niu et al. 2010). When the concentration of CTAB was increased (from 0.2 to 1 mL), PS nanoparticles were coated completely. As a result, reduction in agglomeration between HCSNs was observed as shown in Fig. 4a–c.

SEM images of HCSNs synthesized by variation of CTAB. a 0.2 mL. b 0.6 mL. c 1 mL (1:1 weight ratio of PS/TEOS, 50 μL ammonia, 1:4 volume ratio of water/ethanol)
When the weight ratio of PS/TEOS was maintained as 2:1, formation of solid silica nanoparticles was observed due to the insufficient concentration of silica sol (Fig. 5a). An optimum ratio of 1:1 was maintained in order to synthesize HCSNs (Fig. 5b). When the concentration of TEOS was further increased, formation of undesirable solid silica nanoparticles was observed (Fig. 5c, d), similar to the literature data (Deng et al. 2006; Nguyen et al. 2014).

SEM images of HCSNs synthesized by PS/TEOS weight ratio variation. a 2:1. b 1:1. c 1:2. d 1:4 (0.6 mL CTAB, 50 μL ammonia, 1:4 volume ratio of water/ethanol)
Thickness of silica shell could be modified by varying the concentration of silica precursor (Deng et al. 2006; Yuan et al. 2010). When TEOS was added during the synthesis of HCSNs, initially TEOS hydrolyzed and condensed to form a layer of silica on the surface of CTAB-coated PS template. The silica layer became thicker as more amount of TEOS is condensed (Chen et al. 2013). Thus, HCSNs of varied thickness were synthesized by tailoring the concentration of TEOS, such that the weight ratio of PS/TEOS was maintained in the range of 1:1 to 1:2. Earlier studies have reported the synthesis of mesoporous silica nanoparticles with varied shell thickness by controlling concentration of TEOS. Jiao et al. (2012) showed the variation in shell thickness from 20 to 95 nm, where poly(tert-butylacrylate) (PTBA) was used as a sacrificial template. Mohamed El-Toni et al. (2012) obtained shell thickness range of 41–80 nm while using anionic surfactant as a template. TEM micrographs shown in Fig. 6a–c confirm the formation of hollow core-shell structure of silica nanoparticles of size 230 ± 30 nm. HCSNs were found to be monodispersed and spherical in nature when the weight ratio of PS/TEOS was varied as 1:1, 2:3, and 4:7. In the present study, thickness of the shell was found to increase from 15 to 30 nm with an increase in the amount of TEOS from 1 to 7 g (PS/TEOS weight ratio between 1:1 and 4:7) as reported in Table 1.

TEM micrographs of HCSNs by PS/TEOS variation. a 1:1. b 2:3. c 4:7 (0.6 mL CTAB, 50 μL ammonia, 1:4 volume ratio of water/ethanol)
Typical nitrogen adsorption/desorption isotherms at 77 K and pore size distribution curves for the HCSNs are shown in Figs. 7, 8, and 9. The average specific surface area for the samples T1, T2, and T3 calculated from BET analysis is found in the range of 747.478 to 600.828 m2/g and pore volume calculated by BJH method is observed from 0.917 to 0.768 g/cc (Table 1). The pore size distribution data calculated by the BJH method confirmed the narrow size range from 1.2 to 1.5 nm which is clearly less than the pore sizes reported in the earlier reports. Majority of the previous studies were focused on synthesis of mesoporous silica nanoparticles with pore sizes above 2 nm using various sacrificial templates like PS, anionic surfactant, and amphiphilic block copolymers (Zhang et al. 2009; Mohamed El-Toni et al. 2012; Zhou et al. 2014). In the current study, we found that pore diameters were not significantly affected when the thickness of silica shell was varied. The powder X-ray diffraction (XRD) pattern of the hollow core-shell silica nanoparticles shows characteristic diffraction peak of silica at 23° (Fig. 10) (Nguyen et al. 2014).

Nitrogen adsorption/desorption isotherm and pore size distribution for hollow core-shell silica sample (1:1 weight ratio of PS/TEOS)

Nitrogen adsorption/desorption isotherm and pore size distribution for hollow core-shell silica sample (2:3 weight ratio of PS/TEOS)

Nitrogen adsorption/desorption isotherm and pore size distribution for hollow core-shell silica sample (4:7 weight ratio of PS/TEOS)

X-ray diffraction pattern of silica hollow core-shell nanoparticles
The strong FTIR absorption bands around 2850–3080, 1600, 1500, 750, 700, and 540 cm−1 are the characteristic of PS, as represented in Fig. 11a (Ding et al. 2004; Agrawal et al. 2008). Presence of CTAB in silica-coated PS nanoparticles is confirmed by C-H deformation bands at 1450.2 cm−1 as represented in Fig. 11b (Lee et al. 2008). The peaks due to alkyl groups (in the range of 2800–2900 and 2900–3000 cm−1) disappeared in HCSNs and confirmed the complete removal of PS template by the calcination process (Fig. 11c). Three strong peaks observed at 1089.6, 800.35, and 462.85 cm−1 are from Si-O-Si vibrations confirmed the formation of HCSNs (Nandiyanto et al. 2009; Liu et al. 2012).

FTIR spectra of a PS, b silica-coated PS, and c HCSNs
TGA curves for PS and silica-coated PS exhibited a rapid drop at ∼ 310 °C and plateau > 550 °C as shown in Fig. 12. Weight loss observed at lower temperatures was due to the evaporation of water adsorbed on the surface (˂ 100 °C) (Nguyen et al. 2014). Since there was no weight loss observed in the TGA curve for HCSNs at higher temperature, absence of any remenant PS particles along with silica was confirmed. Approximately twenty-five percent of silica was present in silica-coated PS nanoparticles confirmed by TGA analysis.

TGA analysis of PS, silica-coated PS, and HCSNs
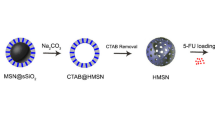
Formation of HCSNs
Scheme of evolution of HCSNs using PS as sacrificial template is represented in Fig. 13. The surface of PS was negatively charged due to the presence of sulfate groups from the initiator KPS (Du and He 2008). CTAB micelles couple with negatively charged PS in the presence of aqueous ammonia solution. CTAB-PS composites were generated via electrostatic interactions between CTA- and PS-, which was similar to the process proposed by Niu et al. (2010). As soon as the TEOS molecules were added, they were hydrolyzed and silica ions started to assemble with CTAB by self-assembly leading to the growth of silica on the surface of the PS particles (Liu et al. 2012; Zhang et al. 2013). PS templates were removed completely by calcination at an elevated temperature of 550 °C which was confirmed by TGA analysis.

Scheme of evolution of HCSNs by sacrificial PS template method
Release studies using doxorubicin
The loading and release properties of HCSNs as drug carriers were explored using doxorubicin an anticancer drug. Doxorubicin-loading capacities of samples T1, T2, and T3 were found to be 1.2, 1.088, and 1.066 wt%, respectively. Variations in loading capacities were low which could be due to the similar core sizes that lead to equal loading of drug (Jiao et al. 2012). Similar values of loading capacities were reported by Bouchoucha et al. (2016). Doxorubicin encapsulation efficiencies of samples T1, T2, and T3 were estimated to be 35.7, 33.01, and 32.27 wt%, respectively. The N2 adsorption-desorption studies revealed that the pore sizes of HCSNs almost remained same (1.2 to 1.5 nm) while the shell thickness was varied (Table 1.), which could be the reason for similar values of encapsulation efficiencies. Thus, doxorubicin was predominantly stored in hollow core and partially adsorbed in the shell. However, slight decrease in encapsulation efficiency could be attributed to the decrease in surface area of HCSN samples T1, T2, and T3 (Table 1). In the present study, moderately higher encapsulation efficiency was observed compared to the earlier studies (Mohamed El-Toni et al. 2012).
Release kinetics showed a high initial dose followed by steady dosage of drug for a prolonged period of time. The rapid leaching of doxorubicin was observed during the initial period, due to the release of molecules coated on the surface and pore entrance (Li et al. 2004; Zhang et al. 2012). Figure 14 shows the cumulative doxorubicin release profile for HCSN samples with different thicknesses (T1, T2, and T3). Doxorubicin release prolonged for 200 min and attained steady state as observed in Fig. 14.

Cumulative doxorubicin release profile for HCSNs at different thicknesses (T1, T2, and T3)
The two major factors influencing the doxorubicin release rate are shell thickness and surface area of HCSNs (Li et al. 2004; Niu et al. 2010). Sample T1 showed the fastest release rate (43.9% after 15 min) with shortest thickness of 15 nm and the lowest microchannel length which permitted the rapid diffusion of doxorubicin into the PBS medium. However, it exhibited largest specific surface area (747.478 m2/g) compared to the other two samples which caused facile leaching of drug molecules from the core. Similarly, variation in release rate for the samples T2 and T3 could be correlated to their shell thickness and surface area. Lowest release rate (34% after 15 min) is observed for the HCSNs with highest shell thickness of 30 nm (sample T3), where the longer microchannel length leads to the slower diffusion rate of the drug molecules into the medium. Moreover, it has lower specific surface area (600.828 m2/g), which inhibited the rapid drug release into to the medium. When the thickness of the silica shell decreases from 30 to 15 nm, doxorubicin diffuses out easily through the porous shell (Jiao et al. 2012). The delays in drug release with an increase in shell thickness and a decrease in surface area are in accordance with the findings of Mohamed El-Toni et al. (2012). Lower drug release rates could be advantageous in delivering therapeutic drug for prolonged period of time by targeted drug delivery system.
Conclusions
HCSNs of size 230 ± 30 nm were successfully synthesized by using PS nanoparticles (150 ± 20 nm) as sacrificial templates. Variation in silica shell thickness from 15 to 30 nm was attained by tailoring the concentration of TEOS. BJH analysis proved that HCSNs were microporous and variation in shell thickness had no significant impact on the pore size (~ 1.3 nm). The percentage cumulative release of doxorubicin was predominately influenced by the specific surface area of HCSNs and the thickness of the shell. Inhibition of rapid drug release was prominent with an increase in the shell thickness from 15 to 30 nm and reduction in specific surface area from 747.47 to 600.82 m2/g. Thus, we anticipate the use of HCSNs as drug carriers would satisfy the demands of targeted drug delivery systems.
References
Agrawal M, Pich A, Gupta S, Zafeiropoulos NE, Simon P, Stamm M (2008) Synthesis of novel tantalum oxide sub-micrometer hollow spheres with tailored shell thickness. Langmuir 24:1013–1018. https://doi.org/10.1021/la702509j
Berru SR, Saniger JM, Flores FJ, Espindola MS (2013) Simple method for the controlled growth of SiO2 spheres. J Mater Sci Eng A 3:237–242. https://doi.org/10.17265/2161-6213/2013.04.004
Bouchoucha M, Cote MF, Gaudreault CR, Fortin MA, Kleitz F (2016) Size-controlled functionalized mesoporous silica nanoparticles for tunable drug release and enhanced anti-tumoral activity. Chem Mater 28:4243–4258. https://doi.org/10.1021/acs.chemmater.6b00877
Che S, Bennett AEG, Yokoi T, Sakamoto K, Kunieda H, Terasaki O, Tatsumi T (2003) A novel anionic surfactant templating route for synthesizing mesoporous silica with unique structure. Nat Mater 2:801–805. https://doi.org/10.1038/nmat1022
Deng TS, Bongard HJ, Marlow F (2015) A one-step method to coat polystyrene particles with an organo-silica shell and their functionalization. Mater Chem Phys 162:548–554. https://doi.org/10.1016/j.matchemphys.2015.06.027
Deng Z, Wu L, Chen M, Zhou S, You B (2006) A novel method for the fabrication of monodisperse hollow silica spheres. Langmuir 22:6403–6407. https://doi.org/10.1021/la060944n
Du X, He J (2008) Facile size-controllable syntheses of highly monodisperse polystyrene nano- and microspheres by polyvinylpyrrolidone-mediated emulsifier-free emulsion polymerization. J Appl Polym Sci 108:1755–1760. https://doi.org/10.1002/app
Ge C, Zhang D, Wang A, Yin H, Ren M, Liu Y, Jiang T, Yu L (2009) Synthesis of porous hollow silica spheres using polystyrene-methyl acrylic acid latex template at different temperatures. J Phys Chem Solids 70:1432–1437. https://doi.org/10.1016/j.jpcs.2009.08.013
Gu S, Kondo T, Konno M (2004) Preparation of silica–polystyrene core–shell particles up to micron sizes. J Colloid Interface Sci 272:314–320. https://doi.org/10.1016/j.jcis.2004.01.056
Han C, Wang S, Wang J, Li M, Deng J, Li H, Wang Y (2014) Controlled synthesis of sustainable n-doped hollow core mesoporous shell carbonaceous nanospheres from biomass. Nano Res 7:1809–1819. https://doi.org/10.1007/s12274-014-0540-x
He T, Chen D, Jiao X (2004) Controlled synthesis of Co3O4 nanoparticles through oriented aggregation. Chem Mater 16:737–743. https://doi.org/10.1021/cm0303033
Hubert DHW, Jung M, Frederik PM, Bomans PHH, Meuldijk J, German AL (2000) Vesicle-directed growth of silica. Adv Mater 12:1286–1290. https://doi.org/10.1002/1521-4095(200009)12:17<1286::AID-ADMA1286>3.0.CO;2-7
Jiao Y, Guo J, Shen S, Chang B, Zhang Y, Jiang X, Yang W (2012) Synthesis of discrete and dispersible hollow mesoporous silica nanoparticles with tailored shell thickness for controlled drug release. J Mater Chem 22:17636–17643. https://doi.org/10.1039/C2JM31821K
Koike N, Ikuno T, Okubo T, Shimojima A (2013) Synthesis of monodisperse organosilica nanoparticles with hollow interiors and porous shells using silica nanospheres as templates. Chem Comm 49:4998–5000. https://doi.org/10.1039/c3cc41904e
Kumar A, Himanshu M (2014) Nanoengineered mesoporous silica nanoparticles for smart delivery of doxorubicin. J Nanoaparticle Res 16:2515–2524. https://doi.org/10.1007/s11051-014-2515-y
Lee CH, Lo LW, Mou CY, Yang CS (2008) Synthesis and characterization of positive-charge functionalized mesoporous silica nanoparticles for oral drug delivery of an anti-inflammatory drug. Adv Funct Mater 18:3283–3292. https://doi.org/10.1002/adfm.200800521
Li B, Yao F, Bae JJ, Chang J, Zamfir MR, Le DT, Pham DT, Yue H, Lee YH (2015) Hollow carbon nanospheres/silicon/alumina core-shell film as an anode for lithium-ion batteries. Sci Rep 5:7659–7657. https://doi.org/10.1038/srep07659
Li H, Ha CS, Kim I (2010) Fabrication of optically tunable silica nanocapsules containing Ag/Au nanostructures by confined galvanic replacement reaction. J Nanopart Res 12:985–992. https://doi.org/10.1007/s11051-009-9650-x
Li ZZ, Wen LX, Shao L, Chen JF (2004) Fabrication of porous hollow silica nanoparticles and their applications in drug release control. J Control Release 98:245–254. https://doi.org/10.1016/j.jconrel.2004.04.019
Liu C, Ge C, Wang A, Yin H, Ren M, Zhang Y, Yu L, Jiang T (2011) Synthesis of porous hollow silica spheres using functionalized polystyrene latex spheres as templates. Korean J Chem Eng 28:1458–1463. https://doi.org/10.1007/s11814-010-0366-5
Liu C, Yin HB, Wang AL, Wu ZA, Wu G, Jiang T, Shen YT, Jiang TS (2012) Size-controlled preparation of hollow silica spheres and glyphosate release. Trans Nonferrous Met Soc China 22:1161–1168. https://doi.org/10.1016/S1003-6326(11)61300-9
Mohamed El-Toni A, Khan A, Abbas Ibrahim M, Puzon Labis J, badr G, Al-Hoshan M, Yin S, Sato T (2012) Synthesis of double mesoporous core-shell silica spheres with tunable core porosity and their drug release and cancer cell apoptosis properties. J Colloid Interface Sci 378:83–92. https://doi.org/10.1016/j.jcis.2012.04.006
Nandiyanto ABD, Kim SG, Iskandar F, Okuyama K (2009) Synthesis of spherical mesoporous silica nanoparticles with nanometer-size controllable pores and outer diameters. Microporous Mesoporous Mater 120:447–453. https://doi.org/10.1016/j.micromeso.2008.12.019
Nguyen AT, Park CW, Kim SH (2014) Synthesis of hollow silica by Stöber method with double polymers as templates. Bull Kor Chem Soc 35:173–176. https://doi.org/10.5012/bkcs.2014.35.1.173
Niu D, Ma Z, Li Y, Shi J (2010) Synthesis of core-shell structured dual-mesoporous silica spheres with tunable pore size and controllable shell thickness. J Am Chem Soc 132:15144–15147. https://doi.org/10.1021/ja1070653
Ogi T, Bayu A, Nandiyanto D, Okuyama K (2014) Nanostructuring strategies in functional fine-particle synthesis towards resource and energy saving applications. Adv Powder Technol 25:3–17. https://doi.org/10.1016/j.apt.2013.11.005
Park I, Ko SH, An YS, Choi KH, Chun H, Lee S, Kim G (2009) Monodisperse polystyrene-silica core-shell particles and silica hollow spheres prepared by the Stober method. J Nanosci Nanotechnol 9:7224–7228. https://doi.org/10.1166/jnn.2009.1636
Sandberg LIC, Gao T, Jelle BP, Gustavsen A (2013) Synthesis of hollow silica nanospheres by sacrificial polystyrene templates for thermal insulation applications. Adv Mater Sci Eng 2013:1–6. https://doi.org/10.1155/2013/483651
Slowing II, Trewyn BG, Giri S, Lin VSY (2007) Mesoporous silica nanoparticles for drug delivery and biosensing applications. Adv Funct Mater 17:1225–1236. https://doi.org/10.1002/adfm.200601191
Stober W, Fink A, Bohn E (1968) Controlled growth of monodisperse silica spheres in the micron size range. J Colloid Interface Sci 26:62–69. https://doi.org/10.1016/0021-9797(68)90272-5
Trewyn BG, Slowing II, Giri S, Chen H, Lin VS (2007) Synthesis and functionalization of a mesoporous silica nanoparticle based on the sol–gel process and applications in controlled release. Acc Chem Res 40:846–853. https://doi.org/10.1021/ar600032u
Wang J, Xu CH, Yao M, Chen J, Xu GJ (2010) A facile route to prepare hierarchical magnetic cobalt–silica hollow nanospheres with tunable shell thickness. J nanoaparticle Res 12:1161–1166. https://doi.org/10.1007/s11051-009-9705-z
Yang J, Lee J, Kang J, Lee K, Suh J-S, Yoon H-G, Huh Y-M, Haam S (2008) Hollow silica nanocontainers as drug delivery vehicles. Langmuir 24(7):3417–3421
Wang Y, Li L, Wang C, Wang T (2016) Facile approach to synthesize uniform Au @ mesoporous SnO2 yolk–shell nanoparticles and their excellent catalytic activity in 4-nitrophenol reduction. J nanoaparticle Res 18:1–11. https://doi.org/10.1007/s11051-015-3307-8
Yoon SY, Park YS, Lee JS (2014) Controlled synthesis of spherical polystyrene beads and their template-assisted manual assembly. Bull Kor Chem Soc 35:2281–2284. https://doi.org/10.5012/bkcs.2014.35.8.2281
Yuan J, Zhou T, Pu H (2010) Nano-sized silica hollow spheres: preparation , mechanism analysis and its water retention property. J Phys Chem Solids 71:1013–1019. https://doi.org/10.1016/j.jpcs.2010.04.004
Zhang J, Rosenholm JM, Gu H (2012) Molecular confinement in fluorescent magnetic mesoporous silica nanoparticles: effect of pore size on multifunctionality. J Chem Phys Phys Chem 13:2016–2019. https://doi.org/10.1002/cphc.201100943
Zhang S, Ren F, Wu W, Zhou J, Xiao X, Sun L, Jiang C (2013) Controllable synthesis of recyclable core–shell ϒ-Fe2O3@SnO2 hollow nanoparticles with enhanced photocatalytic and gas sensing properties. Phys Chem Chem Phys 15:8228–8236. https://doi.org/10.1039/c3cp50925g
Zhang S, Xu L, Liu H, Zhao Y, Zhang Y, Wang Q, Yu Z, Liu Z (2009) A dual template method for synthesizing hollow silica spheres with mesoporous shells. Mater Lett 63:258–259. https://doi.org/10.1016/j.matlet.2008.10.004
Zhao D, Feng J, Huo Q, Melosh N, Fredrickson GH, Chmelka BF, Stucky GD (1998) Triblock copolymer syntheses of mesoporous silica with periodic 50 to 300 angstrom pores. Science 279:548–552. https://doi.org/10.1126/science.279.5350.548
Zhou X, Cheng X, Feng W, Qiu K, Chen L, Nie W, Yin Z, Mo X, Wang H, He C (2014) Synthesis of hollow mesoporous silica nanoparticles with tunable shell thickness and pore size using amphiphilic block copolymers as core templates. Dalt Trans 43:11834–11842. https://doi.org/10.1039/c4dt01138d
Funding
This study was funded by the Council of Scientific and Industrial Research (CSIR), India with grant no. 22/646/13/EMR-II.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Deepika, D., PonnanEttiyappan, J. Synthesis and characterization of microporous hollow core-shell silica nanoparticles (HCSNs) of tunable thickness for controlled release of doxorubicin. J Nanopart Res 20, 187 (2018). https://doi.org/10.1007/s11051-018-4287-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-018-4287-2