
Abstract
Plant molecular breeding is expected to give significant gains in cultivar development through development and utilization of suitable molecular marker systems for genetic diversity analysis, rapid DNA fingerprinting, identification of true hybrids, trait mapping and marker-assisted selection. Transposable elements (TEs) are the most abundant component in a genome and being used as genetic markers in the plant molecular breeding. Here, we review on the high copious transposable element belonging to class-II DNA TEs called “miniature inverted-repeat transposable elements” (MITEs). MITEs are ubiquitous, short and non-autonomous DNA transposable elements which have a tendency to insert into genes and genic regions have paved a way for the development of functional DNA marker systems in plant genomes. This review summarises the characteristics of MITEs, principles and methodologies for development of MITEs based DNA markers, bioinformatics tools and resources for plant MITE discovery and their utilization in crop improvement.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
MITEs are short (~ 100 to 800 bp) non-autonomous truncated versions of autonomous transposable elements belonging to the class-II type of TEs. MITEs possess a pair of conserved terminal inverted repeats (TIRs ≥ 10 bp) that act as receptor sites for the transposase and a pair of short target site duplications (TSDs 2~10 bp) that are located towards the outer end of each TIRs [1]. MITEs initially discovered by Bureau and Wessler [2], during 1992 in maize belonged to Tourist family and another MITE family called Stowaway was identified in Sorghum during 1994. MITEs were later found in several species of plants and animals. Based on sequence similarity, MITEs are primarily classified into families, further grouped into superfamilies with almost identical TSD and TIR sequences shared between MITEs and autonomous transposons capable of encoding transposase enzyme [1, 3]. Through genome sequence analysis in Arabidopsis, the first evidence for such association was reported between a plant MITE family Emigrant with Lemi1 (larger emigrant) transposon which potentially encodes a pogo like transposase was connected to Tc1/mariner transposon superfamily. Apart from being transposed by autonomous DNA transposons belonging to same family, MITEs can also be cross mobilized by DNA transposons belonging distant families [4,5,6]. There are currently seven superfamilies recognized in plant genomes: Tcl/mariner, Pif/Harbinger, hAT, Mutator, CACTA, P-element, and Novosib [1, 7, 8] (Table 1). After discovering MITEs in maize and sorghum genomes, they were reported and characterized in more than 41 plant species including arabidopsis, rice, wheat and groundnut [2, 9,10,11].
During most of host genome evolution period, MITEs remained quiescent. MITEs get activated under natural circumstances when host genome is exposed to severe environmental conditions during domestication. Supporting the theory of "genome shock" proposed by McClintock, that cryptic transposons activate in host genomes upon exposure to stress, as a response of genome to mitigate the danger [12]. Hence, MITEs can amplify and produce more than 1000 copies in plant genomes exposed to extreme genome shocks, as evident in one of the rice cultivars Gimbozu adapted to waterlogging stress through preferential mPing MITEs amplification [13]. Present-day human efforts in plant breeding can also mobilize MITEs through wide hybridization, plant micropropagation techniques such as tissue culture and anther culture and plant chemical mutagen like ethyl methane sulphonate (EMS) and physical mutagen like gamma-ray irradiation [4, 14,15,16,17]. All the above breeding activities seeming to be posing the host plant to experience genomic shock resulting in the MITEs amplification. These examples also indicate that the MITEs in plants are already helping us in generating variability and evolution of host genomes with time and conscious utilization of these elements can further serve us better in crop improvement programmes.
MITEs are exceptionally high in copy number per genome compared to their related autonomous class II transposons and class I transposons [6, 13, 18, 19]. The success of MITEs in reaching such a high copy number in the host genome is because of the generation of stable or neutral mutations due to specifically inserting into the non-translating regions (promoter, intron and untranslated regions), avoiding the exonic region of genes [20]. The short size of MITEs and their ability to escape from being silenced by host surveillance is also considered to be an important reason for a successful transposon burst [6]. Upon their mobilisation by trans-acting TEs, MITEs produce allelic diversity at several loci through insertional polymorphism [21, 22]. MITEs distribution and their abundance in plant genomes play an important role in biosynthetic gene cluster formation through MITEs mediated genomic rearrangement by providing homologous sequences that enable illegitimate recombination and gene relocation [2, 13, 23,24,25]. Therefore, MITE insertional polymorphism can be used as a potential molecular marker system in plant genetics and genomics studies [16, 22, 26,27,28,29,30].
Role of MITEs on regulation of plant genes
The role of MITEs in creating trait variations in plants has been shown by the ample number of studies [31] (Table 2). The wrinkled-seed character, one of the seven traits described by the Mendel to establish fundamental laws of genetics was caused by a transposon-like insertion in a gene encoding starch-branching enzyme (SBEI) by a MITE called hAT element [32]. Other trait variations include, change in the flowering time of maize [33, 34], AltSB, a gene variant in sorghum is responsible for aluminium tolerance [35], ahFAD2B allele is associated with MITE insertion resulted in high oleic acid trait in groundnut [10], change of tuber skin colour in potato [36] and petals colour variation in gentian flowers [37]. A MITE insertion into the gene governing the structure of the glume that encloses and determines rice grain shape has led to slender glume mutant character upon gamma-ray irradiation of Gimbozu rice variety seeds [17, 38]. Enhanced expression of the gene TaHSP16.9-3A due to the insertion of a MITE in its 3′ UTR is attributed for heat tolerance in a wheat cultivar (TAM107) [39]. In the case of maize, MITE insertion in the promotor of the NAC gene (ZmNAC111) is associated with seedling drought tolerance [40]. MITE mediated translational repression was reported for an agronomically important gene Ghd2, that govern grain number, plant height and heading date in rice [41].
MITEs owing to their high copy number and genic preference for insertion [42], have a greater role in the modification of key genes controlling important agricultural traits. The abundance of MITEs in the rice genome and insertional polymorphisms at more than 7000 loci between Oryza sativa subspecies japonica and indica, was attributed for the divergence in gene expression patterns between these subspecies [43]. Recent epigenetic studies in rice reported MITE mediated epigenetic regulation of gene expression is responsible for changes in leaf angle, seed size and subspecies-specific resistance to different pathogens [44, 45]. MITEs have also shown their ability to regulate and control genes involved in abiotic stress responses. These regulatory mechanisms of MITEs will be very useful in understanding and identifying genes involved in abiotic stress response, particularly heat and drought stresses which are essential for the present climate changing situation [20, 39, 40, 45,46,47]. The insertion of TEs into intronic regions of genes can result in mutations that generate new exons. Recently, it is reported that the intronic transposed MITEs in the mulberry genome are frequently associated with alternative splicing due to exonization [48]. The above examples of trait variations associated with MITEs are due to the reprogramming of gene expression mainly by MITE insertional gene inactivation, repression of a nearby gene through methylation, upregulation of the gene by introducing new regulatory information and also involving MITEs mediated miRNA and siRNA pathways. Hence, MITEs can upregulate or downregulate the adjacent genes and the resultant phenotypic change may be associated with domestication and improvement of crop plant species [49].
MITEs based DNA marker systems in plants: principles and methodology
Distinctive characteristics of MITEs such as their abundance, short sequence size (~ 100 to 800 bp), non-autonomous, high frequency of insertion polymorphism and most importantly, preferential insertion of MITEs into the genic region can render them appropriate as an excellent marker system [21, 22, 26,27,28,29,30]. MITE-Display (MD), Inter-MITE polymorphism (IMP) and MITE insertional polymorphism (MIP) are the three prominent MITE based marker systems developed and being used for various molecular plant breeding studies (Table 3). MD is a MITE locus and a restriction site dependent marker system and MIP marker system uses only flanking regions of a MITE for the development of primers to detect presence or absence of a MITE at predetermined genomic region in the host genomes, while TIRs of MITEs are used for designing degenerate primers to amplify the sequence between two MITEs in IMP marker system [50,51,52]. All the three MITE marker systems use MITE locus/loci for detecting sequence variation and generate sequence polymorphism information upon comparing a set of genotypes for that locus.
MITE-display (MD)
MD is a restriction site and polymerase chain reaction (PCR) based marker system. It is similar to amplified fragment length polymorphism (AFLP), MD detects and provides precise information on a large number of MITE insertions in a given plant genotype. MITE insertion at a particular locus can be identified by a ligation-mediated PCR that uses part of the MITE sequence as a template and continues to amplify part of flanking sequence up to a specific restriction site. The amplified PCR product can be analysed in a polyacrylamide gel system. If a MITE insertion is correlated with a particular phenotype and such a co-segregation PCR product can be identified in gel, reamplified, cloned, sequenced and used as a probe to further isolate the tagged gene.
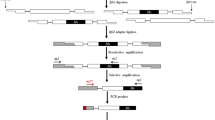
As described in rice [50], the important steps involved in MITE-display are restriction digestion (MseI) of genomic DNA and ligation of AFLP adaptors to the compatible ends. The MITE-containing fragments are then amplified using one AFLP primer and another primer complementary to an internal sequence of the Heartbreak (Hbr) MITE element. As in AFLP, several DNA fragments are then analyzed simultaneously in denaturing polyacrylamide gels (Fig. 1).

Steps involved in MITE-Display marker system
Identification of suitable MITEs for MITE display
MITEs showing high sequence homogeneity within a family indicate that these MITEs are still active or recently active. Such activity by MITEs results in significant insertional polymorphism at several loci within the members of a single plant species. High sequence homogeneity also helps in designing the primers with little or no degeneracy, permitting the use of stringent PCR protocols and leading to reproducible results.
Selection of MITE primers Nested PCR can be used with two sets of primers that recognize conserved regions adjacent to the TIRs. The internal primers confirm that the amplified fragments are truly anchored to the targeted MITE family.
Adapters are made by annealing two oligonucleotides i.e., a restriction enzyme specific sequence with complementary sequences. The sequence of the adapter and the adjacent restriction site serve as primer binding sites for subsequent amplification of the restriction fragments.
PCR conditions The touchdown protocol was used for the very first MITE-Display (Hbr-display) as this protocol increases the specificity of amplification when the primers have different melting temperatures (Tm > 5 °C) [50]. However, a general PCR program can be used based on the length and GC content of the primers and the length of the expected PCR product for any given primer pair.
Pre-selective amplification PCRs are done with the following cycling conditions using a primer complementary to the adapters (MseI + 0) and another primer complementary to an Hbr internal element. A general protocol is programmed to run one cycle of pre-denaturation (94 °C for 5 min), 24 cycles of denaturation (94 °C for 30 s), annealing (59 °C for 30 s) and extension (72 °C for 1 min). Followed by, one cycle of final elongation (72 °C for 5 min).
Selective amplification The 20-fold diluted pre selective amplified reaction is used for selective amplification. The internal primer pair comprising of a primer complementary to the adapter with an overhang (MseI + N) and another primer which is complementary to internal Hbr element is labelled with 33P, designed to anneal within the pre selective amplified product. The following cycling parameters are used for selective amplification [50]. The touch down PCR protocol is programmed to run one cycle of pre-denaturation (94 °C for 5 min), 10 cycles of denaturation (94 °C for 30 s), annealing (starting from 70 °C for 30 s with a decrease of 1 °C/cycle) and extension (72 °C for 1 min). Followed by 27 cycles of denaturation (94 °C for 30 s), annealing (61 °C for 30 s) and extension (72 °C for 1 min). One cycle of final elongation (72 °C for 5 min) was included. The resultant final selective amplified reactions are then analysed in denaturing polyacrylamide gels.
Sometimes, MITE display marker analysis based on denaturing polyacrylamide gels may fail to capture the complete information on MITE insertions when the restriction fragments sequences containing insertions are long enough to produce overlapping bands in the gel profile. To prevent such loss of information and to capture every individual MITE insertion within a genome, the PCR amplified products may be subjected for Next Generation Sequencing (NGS). As the NGS approach gives more precise information compared to the analysis made on denaturing polyacrylamide gels [13]. Recently NGS based MD was attempted to develop high-quality MITEs based marker in Brassica rapa. NGS was integrated with transposon display (TD) analysis to uncover the insertion polymorphism and to develop large-scale MIP markers mediated by recent insertion polymorphism of a high-copy MITE family, BraSto-2 (Bs2) members (IP-Bs2 markers) among Brassica accessions. In this method a degenerate primer developed from the conserved region of Bs2 was used along with the MseI adaptor specific primer to find the insertion polymorphism of Bs2 members among Brassica accessions. Pooled, purified and bar-coded PCR products of Brassica accessions containing MITE flanking sequences were subjected for sequencing with Illumina HiSeq2000 paired-end sequencing platform. Further, the sequences were subjected for in silico mapping on reference genome and accession specific MITE insertion polymorphic sites were identified by eliminating common sites, identified manually based on physical position information. Finally, as compared to conventional gel-based TD analysis NGS based MITE display is more advantageous. As it saves time by preventing multiple rounds of selective amplification in order to amplify most of the MITE family like Bs2 which cannot be amplified/visualized by a single gel analysis. It provides more precise information on MITE insertion polymorphism and it also gives an opportunity to develop large number of MIP markers [53].
Inter-MITE polymorphism (IMP)
IMP is a PCR based marker system. It was developed and utilized in barley for genome mapping and DNA fingerprinting purposes. IMP involves amplification of DNA sequence between two adjacent MITEs as these transposable elements are available in high copy number with insertion preference to the genic region [51]. TIRs of MITEs are most conserved hence these sequences are used for designing degenerate primers (Fig. 2.).

Steps involved in Inter-MITE polymorphism marker system
Identification of suitable MITEs for IMP and primer design The TIR sequences of MITEs families can be aligned to generate a consensus sequence for each family to develop degenerate primers. Outwardly directed oligonucleotide primers can be designed according to the consensus sequences of MITE family TIRs. Such primers are expected to amplify sequence between the two adjacent MITEs within amplifiable distances.
PCR conditions: As per Chang et al. [51], PCR- amplifications were carried out in a final volume of 20 μl containing 2.5 mM of MgCl2, 0.4 mM of dNTPs, 1.0 μM (non-labelled) or 0.1 μM (labelled) of primer and 0.5 units of AmpliTaq DNA polymerase (Perkin-Elmer). The pre-amplification and amplification phases were carried out by setting the conditions for one cycle of pre-denaturation (1 min 30 s at 94 °C), 35 cycles of denaturation (30 s at 94 °C), annealing (45 s at 58 °C or 56 °C), and extension (1 min at 72 °C). In two phases, PCR-amplifications were carried out. Pre-amplification was the first phase and it was done with nonlabelled primers for 35 cycles with 100 ng of DNA template. For the second phase of amplification, an aliquot of 2 μl of the preamplification product was used. The first pre-amplification phase done with nonlabeled primers was meant for the amplification of target sites. The product of pre-amplification was used for the second step of amplification with labelled primers in order to improve the visualization.
MITE insertional polymorphism (MIP)
MITEs by the virtue of their transpositions in host genomes preferably insert into genic regions producing allelic variations for the genes across the genome [18, 54]. This genome-wide insertional polymorphism can be of great importance making them suitable for genome-wide studies, genome analysis, mapping and tagging gene of interest in plants. MIP as a marker system has been developed and utilized in several plant species. In this marker system, the PCR primers are designed flanking to each MITE insertion site across the genome (Fig. 3). These DNA markers were developed based on transposon display, MITEs enriched libraries, in silico analysis and whole genome re-sequencing techniques [16, 29, 55,56,57].

Steps involved in MITE insertional polymorphism marker system
The efficiency of the MIP system over MD and IMP marker systems in capturing sequence variation
MD and IMP marker systems are dependent on a restriction site and a MITE insertion locus respectively in the vicinity of the MITE locus for the successful amplification of the PCR product specific to that MITE locus. Such dependency of MD and IMP marker systems may miss some important MITE insertions but MIP system which is solely dependent upon the individual MITE locus can be more efficient in considering and capturing sequence variation in the host genome created by every individual MITE insertion. As evident by the cases of conversion of MD markers into MIP markers because of ease of marker development, handling and co-dominant nature, MIP system proved to be better suited for QTL mapping studies and marker-assisted selection (MAS) over MD and IMP systems [29, 34]. Furthermore, MITE-display and inter-MITE marker systems being DNA profiling strategies can be effectively employed for rapid fingerprinting and assessment of genetic diversity in a set of genotypes. Unlike MIP, the IMP and MD marker systems do not require host genome sequence information to survey MITEs in plant genomes. It is because of the variations in the approach of primers development in these three marker systems. IMP requires only the sequence information of MITEs for the development of primers. Similarly, MD uses sequence information of a MITE and AFLP adapter sequence for primers development. Whereas MIP marker system utilizes the flanking sequences of MITEs for primers development hence this necessitates the sequence information of host plant genome.
Bioinformatic tools and resources for plant MITEs discovery and annotation
With the advent of sequencing technology and advancement in the computational methods, there is a leap in the genomic sequence databases. In order to discover and annotate MITEs in the plant genomes, several MITEs annotation programs and databases have been developed. Once, MITEs are discovered and characterized in a plant genome, the information on TEs can be catalogued into three types of repositories: TE-centric, genome-centric and polymorphism-centric. These databases can be further used to detect TEs in assembled genomes using ab initio methods [58] (Table 4). MIPS repeat element databases (mips-REdat), a plant repeat database in conjunction with catalogue (mips-REcat) resources are widely used for characterizing and comparative transposon annotation. Currently, the public version mipsREdat_v9.0p consists of 42,000 non-redundant transposon sequences from 44 species covering 20 different genera is available [59].
RepBase Update is used worldwide as a reference collection for masking and annotation of repetitive DNA in genome sequencing projects. It is the most popular TE-centric repository that contains more than 38,000 consensus sequences of TE family and subfamily collected from about 834 species [60]. Dfam is the similar database as that of RepBase, has been used alongside RepeatMasker, which is a widely used database query tool for TEs annotations in eukaryotic genomes [61, 62].
Genomic-centric databases accommodate all the individual TE instances annotated in the reference genomes. P-MITE is one such repository for plant MITEs contains more than 2 Mio. MITE-related sequences of 3527 MITE families, identified from 41 sequenced plant genomes. P-MITE can be used to study the origin and dynamics of MITEs, MITE-derived small RNAs and roles of MITEs on gene and genome evolution [63].
TE insertions of individuals diverging from the annotated reference genome of a particular species are contained in polymorphism-centric repositories. BrassicaTED (Brassica Transposable Elements Database) caters the information on MITEs insertion polymorphism. It has B. rapa and B. oleracea datasets for 6026 members from 20 MITE families apart from information on other miniature transposable elements such as short interspersed elements (SINEs), terminal repeat retrotransposons in miniature (TRIMs) and large retrotransposon derivatives (LARDs) which also do not have transposase coding capacity. BrassicaTED offers to browse structural and positional characteristics for every MITE family. In addition, it has data on 289 MITE insertion polymorphisms from a survey of seven Brassica relatives. It also includes a tool, K BLAST (Karyotype BLAST), for clear visualization of the locations for each member in the B. rapa and B. oleracea pseudo-genome sequences [64].
MITEs are mainly discovered through homology-based and structure-based approaches in the plant genome. Structure-based MITE discovery utilizes defined structure of known MITEs to find putative MITEs, where as sequence similarities are used to identify MITEs in the given plant genome for detecting MITEs with homology-based MITE discovery (Table 5).
For unassembled genomes, raw reads can be used for TE detection with de novo annotation approaches. However, de novo annotation approaches can also be used for assembled genomes for discovering novel TE families that were missing in the TE databases. Following are the popular programs intended to annotate MITEs, detectMITE, MITE Digger, MITE Tracker, MITE-Hunter and MiteFinder. These programs work on different algorithms and operating systems to discover MITEs in a given genome sequence. Each program has its own advantage and disadvantages with respect to the sensitivity, speed and extent of filtering false positives [65,66,67,68,69]. MiteFinder was quick, accurate and more memory efficient compared to detectMITE, MITE Digger and MITE-Hunter remaining while analyzing their relative performance to detect MITEs in the rice genome [67]. However, in a similar test, the MITE Tracker, which is one of the latest programs developed to identify MITEs in plant genomes performed significantly better in terms of processing time and false positive scoring and was the only program able to run with the larger genomes like bread wheat genome as input and discovered 6013 MITE families and allowed the first structural exploration of MITEs in the complete bread wheat genome [65].
Programs or computational tools that allow detection and isolation of polymorphic MITEs can be very useful for efficient DNA marker development for their use in plant molecular breeding. NGS TE Mapper, TE-locate, RelocaTE, TEMP, T-Lex2, PoPoolationTE2, McClintock and PTEMD are the tools that allow detection of TE insertion polymorphisms (TIPs) for a given set of plant genotypes [70,71,72,73,74,75,76]. Recently, around 2957 AhMITE1 specific markers from the whole genome re-sequencing data (WGRS) have been developed using 33 diverse peanut genotypes. This high number of marker development has been credited to the PTEMD software that identified 3546 TIPs from a set of 33 peanut genotypes [55].
Applications and utilization of MITE specific markers
Genetic variability within and between species is key for crop improvement. Accessing molecular genetic diversity may help in understanding evolution, geographical distribution, population structure and genetic relationship in a plant species [77, 78]. Furthermore, the knowledge of the genetic relationship between the inbred lines provides information for the selection of parents and to predict heterosis levels [79,80,81]. Different molecular markers have already been employed in the assessment of plant molecular genetic diversity. But the most commonly used molecular markers like simple sequence repeats (SSR) and single nucleotide polymorphism (SNP) may not sometimes completely cover the diversity existing at the DNA level, mainly due to the transposon mediated intraspecific violation of genetic colinearity and gene structural variations existing in plant species like maize [82,83,84]. In this regard, molecular markers based on TEs can be more efficient in capturing molecular genetic diversity as these elements are involved in evolution and diversification of host genome through their activities like genome expansion by amplification bursts, restructuring of chromosomal regions due to transpositions and creating allelic diversity by insertional polymorphism [85,86,87,88,89,90,91,92,93].
MITE based markers especially, MITE-display and inter MITE marker systems have been used for assessing molecular genetic diversity, relationships and structure of the population in maize, rice, barley, sugarcane, wheat, groundnut, Antirrhinum and Agrostis. The level of polymorphism for MITE based markers in these studies was high and the estimated diversity was similar and congruent with known relationships among the genotypes studied, suggesting that MITE specific markers can be useful for plant molecular genetic diversity assessment [11, 34, 94,95,96,97,98,99,100,101]. Hence, these markers were proposed to use along with other molecular markers for genotyping and relationships studies in maize [102]. Recently, Transposon-based genetic diversity assessment was made in wild and cultivated barley using both class I and class II Transposons with three types of marker systems: IMP (Inter-MITE Polymorphism) and IRAP (Inter-Retrotransposon Amplified Polymorphism). The mean polymorphic information content (PIC) and discrimination power (D) values for IMP and IRAP markers was found to be similar in wild and cultivated species of barley [90].
True hybrid identification by MIP marker system
MIP, a PCR based co-dominant marker system has the power to discriminate heterozygote by generating DNA amplicons from both insertion and deletion for a given locus. This ability of hybrid discrimination was used for identifying true hybrids upon crossing two inbreds or diverse parental genotypes. About 15 MITEs based markers discriminated two laboratory inbred lines 165E and Sippe 50 and twelve commercial varieties of snapdragon [101]. However, DcMaster transposon display markers were also used for hybrid seed purity testing in carrot but the heterozygote discriminatory capacity was confined for only a subset of total markers [98]. Hence, co-dominant MIP marker system can be comparatively better true hybrid identification marker system. Although MIP is a co-dominant marker system, sometimes it may fail to identify true hybrids particularly in polyploid plant species like groundnut due to the presence of homeologus genomic regions and in such cases, MIP markers in homeologus genomic regions may be avoided for true hybrid identification [56].
MITE markers for trait mapping
MITE markers have been used for mapping and saturating the previously developed genetic maps in several plant species. Heartbreak (Hbr) MITE-Display was employed in maize to map 100 recombinant inbred lines derived from a cross between the maize inbreds B73 × Mo17. HBr markers when added to the maize RFLP genetic map, the total length was 1092 cM and map length was increased by 150 cM and also reduced the overall distance between markers [26]. Barfly MITE based IMP markers system was used to saturate RFLP map by mapping a total of 88 IMP markers in barley using doubled-haploid segregating population between Hordeum vulgare and Hordeum spontaneum [50].
As MITE-Display and Inter-MITE markers systems being tedious techniques compared to MIP marker systems, hence, some workers converted these markers into simple PCR markers to exploit MIP. MIP marker system can be more efficient because of its simplicity of marker development, ease of use and co-dominant nature, it has been used for mapping and trait association studies in several plant species. In rice, mPing-SCAR markers were developed from MITE-Display data and they were used for QTL mapping using RIL population derived from a japonica intraspecific cross between Nipponbare X Gimbozu EG4. As compared to SSR markers (7.00%) mPing-SCAR markers (82.30%) showed significantly high polymorphism and it was possible to get better genomic coverage [29]. In another study, 33 MIP PCR based markers were developed from MITE-Display generated information. Furthermore, these markers were used for association mapping in a panel of 367 maize lines to understand MITEs contribution to the phenotypic variation resulting in the identification of a marker ZmV1-9, highly significantly associated with the delayed male flowering [34].
A MIP marker system called Arachis hypogaea transposable element (AhTE) markers developed based on one of the MITE families, i.e., AhMITE1 in groundnut genome, it is being used extensively in groundnut evolutionary studies and construction of linkage maps as a sole marker system as well as in conjunction with other markers systems [16, 55, 56, 103,104,105,106,107,108]. The utility of AhTE Markers for genetic and genomic studies in groundnut (Arachis hypogaea L.) was recently analysed by estimating population genetic parameters among mutants and recombinant inbred lines (RILs). Association analysis and trait mapping with AhTE markers in different groundnut populations have identified MITE markers significantly associated with traits like productivity, foliar disease resistance and oil quality. Furthermore, upon validation, these markers may be advocated for utilization in groundnut improvement through MAS [27, 85, 109,110,111].
All the three MITE based marker systems (MD, IMP and MIP) are being used in mapping and marker-trait association studies as a sole marker system or conjunction with other marker systems and these markers not only saturated the maps by adding additional markers to the previously developed maps but also they were able to map and associate new traits with MITE specific markers.
Conclusion
Transposition activities of MITEs in the plant genomes can produce a wide spectrum of variation in plants both at genotypic and phenotypic levels, which can be useful for crop plant adaptation to the environment or any trait variation that improve yield and quality traits of crop species for sustainable production in this climate changing era. As already witnessed by the utilities of well-known MITEs based marker systems in different plant species like mPing-rice, Heartbreaker-maize, Barfly-barley and AhMITE-groundnut and so far, MITEs have been discovered and characterized in about more than 41 plant species. MITEs by their virtue of abundance, genic preference, short size and gene regulation mechanisms make them suitable for using them as a genetic molecular marker system. Hence, MITEs based molecular markers can be of choice for overall crop improvement through molecular plant breeding. The wide range of phenotypic or trait variations generated by MITEs indicates their ability to be an excellent molecular marker system for genetic and molecular studies in plants. The latest trends in molecular plant breeding are the MAS and genomics, which are based on the development of different molecular marker systems and using them in mapping and tagging several important genes in plants [112, 113]. Molecular markers developed based on MITEs are emerging as a potential marker system in plant species, as these markers are user-friendly, informative and highly reproducible compared to other commonly used SSR-based marker systems. The utility of MITE specific markers in genetics and genomic studies have already been shown in major crop plant species like maize, rice, wheat and groundnut [11, 16, 26, 27, 29].
With the availability of genome sequences of new species, we understand that the plant genome is made up of TEs and particularly MITEs that are widely distributed in the genome and also enriched in the genic region with high copy numbers [20, 24, 114]. Available TE databases and bioinformatics resources for plant MITEs annotation are handy in discovery, identification and classification of MITEs and this helps in the development of existing type (MD, IMP and MIP) of MITE based markers in newly genome sequenced plant species or already existing but MITEs unexploited plant species. Further, there is also scope for integrating MITEs with other marker systems/sequences of any genes to evolve new kind of MITE based marker systems, as a couple of examples cited here. MITE/RGA is a modified MITE Display marker system. It uses a combination of primers specific to MITEs and primers based on resistant gene analogs (RGAs). This marker system provided a unique opportunity for mapping and identification of candidate genes conferring resistance to grey leaf spot in maize. Compared to MITE display, this technique was more cost-effective, less time consuming as it does not require restriction digestion of plant genomic DNA and ligation of primer adaptors [115]. Another marker system called MITE based intron length polymorphism, which is also a simple and cost-effective PCR marker system was developed for its utilization in carrot molecular breeding [57]. Recently, the bibliometric analysis of plant molecular markers showed the predominance of SNPs and SSRs marker systems and also identified US, China, India, France, and Germany as the top countries to use DNA markers for molecular studies [116]. Like SSR markers, which are labelled as markers of choice, MITE based MIP marker system is also co-dominant, highly polymorphic and easily visualized in agarose gel electrophoresis compared to SSR [29]. With these added advantages, MIP markers may be integrated with SSRs or they can be used as a sole marker system for the routine needs of plant molecular studies. MITEs in rice can create a significantly very high rate of new insertions i.e., ~ 40 per plant per segregating generation as compared to a relatively low rate of new SNPs formation per generation per plant [13, 117]. Hence, this high rate of insertional polymorphisms created by dynamic MITEs activity resulting in beneficial adaptive trait variations arising due to exposure of plant genome to the present climate-changing situation allows us to utilize them as effective marker systems in plant molecular breeding.
References
Feschotte C, Zhang X, Wessler SR (2002) Miniature inverted-repeat transposable elements and their relationship to established DNA transposons. In: Mobile DNA II American Society of Microbiology 1147–1158.
Bureau TE, Wessler SR (1992) Tourist: a large family of small inverted repeat elements frequently associated with maize genes. Plant Cell 4:1283–1294. https://doi.org/10.2307/3869414
Feschotte C, Mouchès C (2000) Evidence that a family of miniature inverted-repeat transposable elements (MITEs) from the Arabidopsis thaliana genome has arisen from a pogo- like DNA transposon. Mol Biol Evol 17:730–737. https://doi.org/10.1093/oxfordjournals.molbev.a026351
Jiang N, Bao Z, Zhang X et al (2003) An active DNA transposon family in rice. Nature 421:163–167. https://doi.org/10.1038/nature01214
Yang G, Nagel DH, Feschotte C (2009) Tuned for transposition: Molecular determinants underlying the hyperactivity of a stowaway MITE. Science 325:1391–1394. https://doi.org/10.1126/science.1175688
Chen J, Lu L, Benjamin J et al (2019) Tracking the origin of two genetic components associated with transposable element bursts in domesticated rice. Nat Commun 10:641. https://doi.org/10.1038/s41467-019-08451-3
Fattash I, Rooke R, Wong A et al (2013) Miniature inverted-repeat transposable elements: discovery, distribution, and activity. Genome 56:475–486. https://doi.org/10.1139/gen-2012-0174
Jiang N, Feschotte C, Zhang X, Wessler SR (2004) Using rice to understand the origin and amplification of miniature inverted repeat transposable elements (MITEs). Curr Opin Plant Biol 7:115–119
Casacuberta E, Casacuberta JM, Puigdomènech P, Monfort A (1998) Presence of miniature inverted-repeat transposable elements (MITEs) in the genome of Arabidopsis thaliana: characterisation of the Emigrant family of elements. Plant J 16:79–85. https://doi.org/10.1046/j.1365-313X.1998.00267.x
Patel M, Jung S, Moore K et al (2004) High-oleate peanut mutants result from a MITE insertion into the FAD2 gene. Theor Appl Genet 108:1492–1502. https://doi.org/10.1007/s00122-004-1590-3
Yaakov B, Ceylan E, Domb K, Kashkush K (2012) Marker utility of miniature inverted-repeat transposable elements for wheat biodiversity and evolution. Theor Appl Genet 124:1365–1373. https://doi.org/10.1007/s00122-012-1793-y
McClintock B (1984) The significance of responses of the genome to challenge. Science 226:792–801. https://doi.org/10.1126/science.15739260
Naito K, Zhang F, Tsukiyama T et al (2009) Unexpected consequences of a sudden and massive transposon amplification on rice gene expression. Nature 461:1130–1134. https://doi.org/10.1038/nature08479
Shan X, Liu Z, Dong Z et al (2005) Mobilization of the active MITE transposons mPing and Pong in rice by introgression from wild rice (Zizania latifolia Griseb.). Mol Biol Evol. 22:976–990. https://doi.org/10.1093/molbev/msi082
Kikuchi K, Terauchi K, Wada M, Hirano H-Y (2003) The plant MITE mPing is mobilized in anther culture. Nature 421:167–170. https://doi.org/10.1038/nature01218
Shirasawa K, Hirakawa H, Tabata S et al (2012) Characterization of active miniature inverted-repeat transposable elements in the peanut genome. Theor Appl Genet 124:1429–1438. https://doi.org/10.1007/s00122-012-1798-6
Nakazaki T, Okumoto Y, Horibata A et al (2003) Mobilization of a transposon in the rice genome. Nature 421:170–172. https://doi.org/10.1038/nature01219
Zhang Q, Arbuckle J, Wessler SR (2000) Recent, extensive, and preferential insertion of members of the miniature inverted-repeat transposable element family Heartbreaker into genic regions of maize. Proc Natl Acad Sci 97:1160–1165. https://doi.org/10.1073/pnas.97.3.1160
Turcotte K, Srinivasan S, Bureau T (2001) Survey of transposable elements from rice genomic sequences. Plant J 25:169–179
Naito K, Cho E, Yang G et al (2006) Dramatic amplification of a rice transposable element during recent domestication. Proc Natl Acad Sci 103:17620–17625. https://doi.org/10.1073/pnas.0605421103
Liu Y, Tahir UlQamar M, Feng JW et al (2019) Comparative analysis of miniature inverted-repeat transposable elements (MITEs) and long terminal repeat (LTR) retrotransposons in six Citrus species. BMC Plant Biol 19:140. https://doi.org/10.1186/s12870-019-1757-3
Sampath P, Yang T-J (2014) Miniature inverted-repeat transposable elements (MITEs) as valuable genomic resources for the evolution and breeding for Brassica crops. Plant Breed Biotech 2:322–333. https://doi.org/10.9787/PBB.2014.2.4.322
Boutanaev AM, Osbourn AE (2018) Multigenome analysis implicates miniature inverted-repeat transposable elements (MITEs) in metabolic diversification in eudicots. Proc Natl Acad Sci 115:E6650–E6658. https://doi.org/10.1073/pnas.1721318115
Guo C, Spinelli M, Ye C et al (2017) Genome-wide comparative analysis of miniature inverted repeat transposable elements in 19 Arabidopsis thaliana ecotype accessions. Sci Rep 7:1–12. https://doi.org/10.1038/s41598-017-02855-1
Jiang N (2001) Insertion preference of maize and rice miniature inverted repeat transposable elements as revealed by the analysis of nested elements. Plant Cell Online 13:2553–2564. https://doi.org/10.1105/tpc.13.112553
Casa AM, Brouwer C, Nagel A et al (2000) The MITE family Heartbreaker (Hbr): molecular markers in maize. Proc Natl Acad Sci 97:10083–10089. https://doi.org/10.1073/pnas.97.18.10083
Hake AA, Shirasawa K, Yadawad A et al (2017) Identification of transposable element markers associated with yield and quality traits from an association panel of independent mutants in peanut (Arachis hypogaea L.). Euphytica 213:283. https://doi.org/10.1007/s10681-017-2070-6
Komori T, Nitta N (2003) High frequency of sequence polymorphism in rice MITEs and application to efficient development of PCR-based markers. Breed Sci 53:85–92. https://doi.org/10.1270/jsbbs.53.85
Monden Y, Naito K, Okumoto Y et al (2009) High potential of a transposon mPing as a marker system in japonica x japonica cross in rice. DNA Res 16:131–140. https://doi.org/10.1093/dnares/dsp004
Hake AA, Bhat RS (2017) Utility of AhTE markers for genetic and genomic studies in groundnut (Arachis hypogaea L.). Int J Curr Microbiol Appl Sci 6:2051–2060
Vitte C, Fustier M-A, Alix K, Tenaillon MI (2014) The bright side of transposons in crop evolution. Brief Funct Genom 13:276–295. https://doi.org/10.1093/bfgp/elu002
Bhattacharyya MK, Smith AM, Ellis THN et al (1990) The wrinkled-seed character of pea described by Mendel is caused by a transposon-like insertion in a gene encoding starch-branching enzyme. Cell 60:115–122. https://doi.org/10.1016/0092-8674(90)90721-P
Castelletti S, Tuberosa R, Pindo M, Salvi S (2014) A MITE transposon insertion is associated with differential methylation at the maize flowering time QTL Vgt1. G3 4:805–812. https://doi.org/10.1534/g3.114.010686
Zerjal T, Rousselet A, Mhiri C et al (2012) Maize genetic diversity and association mapping using transposable element insertion polymorphisms. Theor Appl Genet 124:1521–1537. https://doi.org/10.1007/s00122-012-1807-9
Magalhaes JV, Liu J, Guimarães CT et al (2007) A gene in the multidrug and toxic compound extrusion (MATE) family confers aluminum tolerance in sorghum. Nat Genet. 39:1156. https://doi.org/10.1038/ng2074
Momose M, Abe Y, Ozeki Y (2010) Miniature inverted-repeat transposable elements of Stowaway are active in potato. Genetics 186:59–66. https://doi.org/10.1534/genetics.110.117606
Nishihara M, Hikage T, Yamada E, Nakatsuka T (2011) A single-base substitution suppresses flower color mutation caused by a novel miniature inverted-repeat transposable element in gentian. Mol Genet Genomics 286:371–382. https://doi.org/10.1007/s00438-011-0652-x
Teraishi M, Okumoto Y, Hirochika H et al (1999) Identification of a mutable slender glume gene in rice (Oryza sativa L.). Mol Gen Genet 261:487–494. https://doi.org/10.1007/s004380050992
Li J, Wang Z, Peng H, Liu Z (2014) A MITE insertion into the 3′-UTR regulates the transcription of TaHSP16. 9 in common wheat. Crop J 2:381–387. https://doi.org/10.1016/j.cj.2014.07.001
Mao H, Wang H, Liu S et al (2015) A transposable element in a NAC gene is associated with drought tolerance in maize seedlings. Nat Commun 6:8326. https://doi.org/10.1038/ncomms9326
Shen J, Liu J, Xie K et al (2017) Translational repression by a miniature inverted-repeat transposable element in the 3′ untranslated region. Nat Commun 8:14651. https://doi.org/10.1038/ncomms14651
Lu C, Chen J, Zhang Y et al (2012) Miniature inverted-repeat transposable elements (MITEs) have been accumulated through amplification bursts and play important roles in gene expression and species diversity in Oryza sativa. MolBiolEvol 29:1005–1017. https://doi.org/10.1093/molbev/msr282
Chen J, Lu C, Zhang Y, Kuang H (2012) Miniature inverted-repeat transposable elements (MITEs) in rice were originated and amplified predominantly after the divergence of Oryza and Brachypodium and contributed considerable diversity to the species. Mob Genet Elements 2:127–132. https://doi.org/10.4161/mge.20773
Zhang H, Tao Z, Hong H et al (2016) Transposon-derived small RNA is responsible for modified function of WRKY45 locus. Nat Plants 2:16016. https://doi.org/10.1038/NPLANTS.2016.16
Xianwei S, Zhang X, Sun J, Cao X (2015) Epigenetic mutation of RAV6 affects leaf angle and seed size in rice. Plant Physiol 169:2118–2128. https://doi.org/10.1104/pp.15.00836
Wei L, Gu L, Song X et al (2014) Dicer-like 3 produces transposable element-associated 24-nt siRNAs that control agricultural traits in rice. Proc Natl Acad Sci USA 111:3877–3882. https://doi.org/10.1073/pnas.1318131111
Yan Y, Zhang Y, Yang K et al (2011) Small RNAs from MITE-derived stem-loop precursors regulate abscisic acid signaling and abiotic stress responses in rice. Plant J 65:820–828. https://doi.org/10.1111/j.1365-313X.2010.04467.x
Xin Y, Ma B, Xiang Z et al (2019) Amplification of miniature inverted-repeat transposable elements and the associated impact on gene regulation and alternative splicing in mulberry (Morusno tabilis). Mob DNA 10:27. https://doi.org/10.1186/s13100-019-0169-0
Vaschetto LM (2016) Miniature inverted-repeat transposable elements (MITEs) and their effects on the regulation of major genes in cereal grass genomes. Mol Breed 36:30. https://doi.org/10.1007/s11032-016-0440-8
Casa AM, Nagel A, Wessler SR (2004) MITE display. Methods MolBiol 260:175–188. https://doi.org/10.1385/1-59259-755-6:175
Chang R-Y, O’Donoughue LS, Bureau TE (2001) Inter-MITE polymorphisms (IMP): a high throughput transposon-based genome mapping and fingerprinting approach. Theor Appl Genet 102:773–781. https://doi.org/10.1007/s001220051709
Sampath P, Lee SC, Lee J et al (2013) Characterization of a new high copy stowaway family MITE, BRAMI-1 in Brassica genome. BMC Plant Biol 13:56. https://doi.org/10.1186/1471-2229-13-56
Perumal S, Waminal N, Lee J et al (2016) Next-generation sequencing based transposon display to detect high-throughput insertion polymorphism markers in brassica. Plant Breed Biotechnol 4:285–296
Casacuberta JM, Santiago N (2003) Plant LTR-retrotransposons and MITEs: control of transposition and impact on the evolution of plant genes and genomes. Gene 311:1–11. https://doi.org/10.1016/S0378-1119(03)00557-2
Gayathri M, Shirasawa K, Varshney RK et al (2018) Development of AhMITE1 markers through genome-wide analysis in peanut (Arachis hypogaea L.). BMC Res Notes 11:8–13. https://doi.org/10.1186/s13104-017-3121-8
Shirasawa K, Koilkonda P, Aoki K et al (2012) Insilico polymorphism analysis for the development of simple sequence repeat and transposon markers and construction of linkage map in cultivated peanut. BMC Plant Biol 12:80. https://doi.org/10.1186/1471-2229-12-80
Stelmach K, Macko-Podgórni A, Machaj G, Grzebelus D (2017) Miniature inverted repeat transposable element insertions provide a source of intron length polymorphism markers in the carrot (Daucuscarota L.). Front Plant Sci 8:725. https://doi.org/10.3389/FPLS.2017.00725
Goerner-Potvin P, Bourque G (2018) Computational tools to unmask transposable elements. Nat Rev Genet 19:688–704. https://doi.org/10.1038/s41576-018-0050-x
Nussbaumer T, Martis MM, Roessner SK et al (2012) MIPS PlantsDB: a database framework for comparative plant genome research. Nucleic Acids Res 41:D1144–D1151. https://doi.org/10.1093/nar/gks1153
Bao W, Kojima KK, Kohany O (2015) Repbase update, a database of repetitive elements in eukaryotic genomes. Mob DNA 6:11. https://doi.org/10.1186/s13100-015-0041-9
Hubley R, Finn RD, Clements J et al (2016) The Dfam database of repetitive DNA families. Nucleic Acids Res 44:D81–D89. https://doi.org/10.1093/nar/gkv1272
Wheeler TJ, Clements J, Eddy SR et al (2013) Dfam: a database of repetitive DNA based on profile hidden Markov models. Nucleic Acids Res 41:D70–D82. https://doi.org/10.1093/nar/gks1265
Chen J, Hu Q, Zhang Y et al (2014) P-MITE: a database for plant miniature inverted-repeat transposable elements. Nucleic Acids Res 42:D1176–1181. https://doi.org/10.1093/nar/gkt1000
Murukarthick J, Sampath P, Lee S et al (2014) BrassicaTED - a public database for utilization of miniature transposable elements in Brassica species. BMC Res Notes 7:379. https://doi.org/10.1186/1756-0500-7-379
Crescente JM, Zavallo D, Helguera M, Vanzetti LS (2018) MITE Tracker: an accurate approach to identify miniature inverted-repeat transposable elements in large genomes. BMC Bioinform 19:348. https://doi.org/10.1186/s12859-018-2376-y
Han Y, Wessler SR (2010) MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res 38:e199–e199. https://doi.org/10.1093/nar/gkq862
Hu J, Zheng Y, Shang X (2017) MiteFinder: A fast approach to identify miniature inverted-repeat transposable elements on a genome-wide scale. IEEE International Conference on Bioinformatics and Biomedicine: 164–168. https://doi.org/10.1109/BIBM.2017.8217644
Yang G (2013) MITE Digger, an efficient and accurate algorithm for genome wide discovery of miniature inverted repeat transposable elements. BMC Bioinform 14:186. https://doi.org/10.1186/1471-2105-14-186
Ye C, Ji G, Liang C (2016) Detect MITE: a novel approach to detect miniature inverted repeat transposable elements in genomes. Sci Rep 6:19688. https://doi.org/10.1038/srep19688
Fiston-Lavier AS, Barrón MG, Petrov DA, González J (2015) T-lex2: Genotyping, frequency estimation and re-annotation of transposable elements using single or pooled next-generation sequencing data. Nucleic Acids Res 43:e22–e22. https://doi.org/10.1093/nar/gku1250
Kang H, Zhu D, Lin R et al (2016) A novel method for identifying polymorphic transposable elements via scanning of high-throughput short reads. DNA Res 23:241–251. https://doi.org/10.1093/dnares/dsw011
Kofler R, Gómez-Sánchez D, Schlötterer C (2016) PoPoolationTE2: comparative population genomics of transposable elements using pool-seq. MolBiolEvol 33:2759–2764. https://doi.org/10.1093/molbev/msw137
Linheiro RS, Bergman CM (2012) Whole genome resequencing reveals natural target site preferences of transposable elements in Drosophila melanogaster. PLoS ONE 7:e30008. https://doi.org/10.1371/journal.pone.0030008
Nelson MG, Linheiro RS, Bergman CM (2017) McClintock: an integrated pipeline for detecting transposable element insertions in Whole-Genome Shotgun sequencing data. Genes Genomes Genet 7:2763–2778. https://doi.org/10.1534/g3.117.043893
Platzer A, Nizhynska V, Long Q (2012) TE-locate: a tool to locate and group transposable element occurrences using paired-end next-generation sequencing data. Biology 1:395–410. https://doi.org/10.3390/biology1020395
Zhuang J, Wang J, Theurkauf W, Weng Z (2014) TEMP: A computational method for analyzing transposable element polymorphism in populations. Nucleic Acids Res 42:6826–6838. https://doi.org/10.1093/nar/gku323
Glaszmann JC, Kilian B, Upadhyaya HD, Varshney RK (2010) Accessing genetic diversity for crop improvement. CurrOpin Plant Biol 13:167–173. https://doi.org/10.1016/j.pbi.2010.01.004
Govindaraj M, Vetriventhan M, Srinivasan M (2015) Importance of genetic diversity assessment in crop plants and its recent advances: an overview of its analytical perspectives. Genet Res Int. https://doi.org/10.1155/2015/431487
Dudley J, Maroof M, Rufener G (1992) Molecular marker information and selection of parents in corn breeding programs. Crop Sci 32:301–304. https://doi.org/10.2135/cropsci1992.0011183X003200020002x
Xing N, Fan C, Zhou Y (2014) Parental selection of hybrid breeding based on maternal and paternal inheritance of traits in rapeseed (Brassica napus L.). PLoS ONE 9:e103165. https://doi.org/10.1371/journal.pone.0103165
Soni SK, Tiwari S, Newmah JT et al (2018) Prediction of hybrid performance in crop plants : molecular and recent approaches. Int J Curr Microbiol Appl Sci 7:98–108
Sun S, Zhou Y, Chen J et al (2018) Extensive intraspecific gene order and gene structural variations between Mo17 and other maize genomes. Nat Genet 50:1289. https://doi.org/10.1038/s41588-018-0182-0
Buckler ES, Gaut BS, McMullen MD (2006) Molecular and functional diversity of maize. Curr Opin Plant Biol 9:172–176
Fu H, Dooner HK (2002) Intraspecific violation of genetic colinearity and its implications in maize. Proc Natl Acad Sci USA 99:9573–9578. https://doi.org/10.1073/pnas.132259199
Hake AA, Shirasawa K, Yadawad A et al (2018) Genome-wide structural mutations among the lines resulting from genetic instability in peanut (Arachis hypogaea L.). Plant Gene 13:1–7. https://doi.org/10.1016/j.plgene2017.11.001
Jiang N, Bao Z, Zhang X et al (2004) Pack-MULE transposable elements mediate gene evolution in plants. Nature 431:569. https://doi.org/10.1038/nature02953
Keidar-Friedman D, Bariah I, Kashkush K (2018) Genome-wide analyses of miniature inverted-repeat transposable elements reveals new insights into the evolution of the Triticum-Aegilops group. PLoS ONE 13:e0204972. https://doi.org/10.1371/journal.pone.0204972
Lai J, Li Y, Messing J, Dooner HK (2005) Gene movement by Helitron transposons contributes to the haplotype variability of maize. Proc Natl Acad Sci USA 102:9068–9073. https://doi.org/10.1073/pnas.0502923102
Morgante M, Brunner S, Pea G et al (2005) Gene duplication and exon shuffling by helitron-like transposons generate intraspecies diversity in maize. Nat Genet 37:997. https://doi.org/10.1038/ng1615
Singh S, Nandha PS, Singh J (2017) Transposon-based genetic diversity assessment in wild and cultivated barley. Crop J 5:296–304. https://doi.org/10.1016/j.cj.2017.01.003
Tang Y, Ma X, Zhao S et al (2019) Identification of an active miniature inverted-repeat transposable element in rice. Plant J 98:639–653. https://doi.org/10.1111/tpj.14260
Wessler SR, Nagel A, Casa A (2008) Miniature inverted repeat transposable elements help create genomic diversity in maize and rice. Rice Genet 4:107–116
Wicker T, Gundlach H, Spannagl M et al (2018) Impact of transposable elements on genome structure and evolution in bread wheat. Genome Biol 19:103. https://doi.org/10.1186/s13059-018-1479-0
Amundsen K, Warnke S (2011) Species relationships in the genus Agrostis based on flow Cytometry and MITE-display molecular markers. Crop Sci 51:1224–1231. https://doi.org/10.2135/cropsci2010.09.0512
Hong SM, Kwon SJ, Oh CS (2006) Diversity analysis of Japonica Rice using MITE-transposon display. Korean J Crop Sci 51:259–268. https://doi.org/10.1093/molbev/msr282
Kavar T, Meglic V, Rozman L (2007) Diversity of Slovenian maize (Zea mays) populations by Hbr (MITE) markers and morphological traits. Rus J Genet 43:989–995. https://doi.org/10.1134/S1022795407090049
Lyons M, Cardle L, Rostoks N et al (2008) Isolation, analysis and marker utility of novel miniature inverted repeat transposable elements from the barley genome. Mol Genet Genomics 280:275–285. https://doi.org/10.1007/s00438-008-0363-0
Macko A, Grzebelus D (2008) DcMaster transposon display markers as a tool for diversity evaluation of carrot breeding materials and for hybrid seed purity testing. J Appl Genet 49:33–39. https://doi.org/10.1007/BF03195246
Nakayama S (2012) Inter-MITE polymorphisms of a newly identified MITE show relationships among sugarcane (Saccharum) species. Genet Resour Crop Evol 59:1389–1396. https://doi.org/10.1007/s10722-011-9766-6
Park KC, Kim NH, Cho YS et al (2003) Genetic variations of AA genome Oryza species measured by MITE-AFLP. Theor Appl Genet 107:203–209. https://doi.org/10.1007/s00122-003-1252-x
Weiss J, Mallona I, Gomez-di-Marco P (2012) Genotyping Antirrhinum commercial varieties using miniature inverted-repeat transposable elements (MITEs). SciHortic (Amsterdam) 144:161–167. https://doi.org/10.1016/j.scienta.2012.06.040
Casa AM, Mitchell SE, Smith OS et al (2002) Evaluation of Hbr (MITE) markers for assessment of genetic relationships among maize (Zea mays L.) inbred lines. Theor Appl Genet 104:104–110. https://doi.org/10.1007/s001220200012
Bhat RS, Patil VU, Chandrashekar TM et al (2008) Recovering flanking sequence tags of a miniature inverted-repeat transposable element by thermal asymmetric interlaced-PCR in peanut. Curr Sci 95:452–453
Gowda MVC, Bhat RS, Motagi BN et al (2010) Association of high-frequency origin of late leaf spot resistant mutants with AhMITE1 transposition in peanut. Plant Breed 129:567–569. https://doi.org/10.1111/j.1439-0523.2009.01704.x
Gowda MVC, Bhat RS, Sujay V et al (2011) Characterization of AhMITE1 transposition and its association with the mutational and evolutionary origin of botanical types in peanut (Arachis spp.). Plant Syst Evol 291:153–158. https://doi.org/10.1007/s00606-010-0373-3
Kolekar RM, Sujay V, Shirasawa K et al (2016) QTL mapping for late leaf spot and rust resistance using an improved genetic map and extensive phenotypic data on a recombinant inbred line population in peanut (Arachis hypogaea L.). Euphytica 209:147–156. https://doi.org/10.1007/s10681-016-1651-0
Leal-Bertioli SCM, Cavalcante U, Gouvea EG et al (2015) G3 5:1403–1413. https://doi.org/10.1534/g3.115.018796
Mondal S, Hande P, Badigannavar AM (2014) Identification of transposable element markers for a rust (Puccinia arachidis Speg.) resistance gene in cultivated peanut. J Phytopathol 162:548–552. https://doi.org/10.1111/jph.12220
Venkatesh (2014) Association analysis for taxonomic traits using transposon specific markers (AhMITE1) in a mutant population of groundnut. MSc. Dissertation University of Agricultural Sciences, Dharwad, Karnataka, India
Kamble VM (2014) Association analysis for yield related and foliar disease resistance using transposon specific markers in a mutant population of groundnut. University of Agricultural Sciences, Dharwad, Karnataka
Venkatesh BN, Vijaykumar A, Motagi B, Bhat R (2019) Single marker analysis using transposon specific markers (AhMITE1) for yield, foliar disease resistance and oil quality in a mutant population of groundnut (Arachis hypogaea L.). Int J Curr Microbiol Appl Sci 8:2376–2385
Nadeem MA, Nawaz MA, Shahid MQ et al (2018) DNA molecular markers in plant breeding: current status and recent advancements in genomic selection and genome editing. Biotechnol Biotechnol Equip 32:261–285. https://doi.org/10.1080/13102818.2017.1400401
Yuan Y, Bayer PE, Batley J, Edwards D (2017) Improvements in genomic technologies: application to crop genomics. Trends Biotechnol 35:547–558
Liu J, Guyot R, Ming R (2018) Transposable Elements in the Pineapple Genome. Genetics and Genomics of Pineapple, 155–165. Springer, Cham.
Lambert CA (2001) A novel marker technique: using miniature inverted-repeat transposable elements (MITEs) in combination with resistant gene analogues (RGAs). Doctoral dissertation, Stellenbosch University.
Garrido-Cardenas JA, Mesa-Valle C, Manzano-Agugliaro F (2018) Trends in plant research using molecular markers. Planta 247:543–557
Grzebelus D (2018) The functional impact of transposable elements on the diversity of plant genomes. Diversity 10:18. https://doi.org/10.3390/d10020018
Bureau TE, Wessler SR (1994) Stowaway: a new family of inverted repeat elements associated with the genes of both monocotyledonous and dicotyledonous plants. Plant Cell Online 6:907–916. https://doi.org/10.1105/tpc.6.6.907
Surzycki SA, Belknap WR (1999) Characterization of repetitive DNA elements in Arabidopsis. J MolEvol 48:684–691. https://doi.org/10.1007/PL00006512
Charrier B, Foucher F, Kondorosi E et al (1999) Bigfoot: a new family of MITE elements characterized from the Medicago genus. Plant J 18:431–441. https://doi.org/10.1046/j.1365-313X.1999.00469.x
Gierlb A, Saedler H, Peterson PA (1989) Maize transposable elements. Annu Rev Genet 23:71–85
Smit. AFA, Hubley R, Green P (2013–2015) RepeatMasker Open-4.0 https://repeatmasker.org.
Robb SMS, Lu L, Valencia E et al (2013) The use of relocate and unassembled short reads to produce high-resolution snapshots of transposable element generated diversity in rice. G3 3:949–957. https://doi.org/10.1534/g3.112.005348
Acknowledgements
We thank H. L. Nadaf, S. A. Desai, Sampath Perumal, Spoorthi Nayak, Basavaraj Bagewadi and Ravikumar Hosmani for proofreading the manuscript, providing helpful comments and suggestions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Venkatesh, Nandini, B. Miniature inverted-repeat transposable elements (MITEs), derived insertional polymorphism as a tool of marker systems for molecular plant breeding. Mol Biol Rep 47, 3155–3167 (2020). https://doi.org/10.1007/s11033-020-05365-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-020-05365-y