
Abstract
Diabetes and other lifestyle disorders have been recognized as the leading cause of morbidity and mortality globally. Nuclear factor kappa B (NF-κB) is a major factor involved in the early pathobiology of diabetes and studies reveal that hyperglycemic conditions in body leads to NF-κB mediated activation of several cytokines, chemokines and inflammatory molecules. NF-κB family comprises of certain DNA-binding protein factors that elicit the transcription of pro-inflammatory molecules. Various studies have identified NF-κB as a promising target for diabetic management. Probiotics have been proposed as bio-therapeutic agents for treatment of inflammatory disorders and many other chronic clinical stages. The precise mechanisms by which probiotics acts is yet to be fully understood, however research findings have indicated their role in NF-κB modulation. The current review highlights NF-κB as a bio-therapeutic target for probable management of type 2 diabetes through probiotic intervention.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetes mellitus is one of the leading global health emergencies, affecting all major sectors of the society, creating huge burden on global health and economy [1, 2]. As per International Diabetes Federation (IDF) Diabetes Atlas latest update, 425 million people are suffering from diabetes globally, and if the trends continue un-checked, the figure is expected to cross 629 million mark by 2045 [1]. Type 1 diabetes is characterized by autoimmune mediated destruction of pancreatic beta cells; while type 2 diabetes, the more prevalent form is defined by progressive loss of beta cells, disturbed insulin secretion and resistance to insulin [3, 4]. It is a complex metabolic disorder known to be mediated by oxidative stress led hyperglycemia [5]. Several other risk factors such as, sedentary lifestyle, genetic pre-disposition, epigenetic changes, and altered gut microbiota are associated with diabetes [2, 6]. However, pancreatic β cells dysfunction or death leading to hampered insulin secretion remains the most prominent factor for development of both type 1 and type 2 diabetes [7]. Healthy β-cells synthesize, store and secrete insulin in response to glucose, nutrients, hormones and nervous stimuli [8]. Proper functioning of β cells is vital for regulation of glucose levels and management of metabolic energy. There are multiple events responsible for apoptosis of β cells in both types of diabetes [9,10,11].
Apart from β cell apoptosis, other factors leading to death of β cells involves nuclear factor kappa B (NF-κB) mediated cytokine induced cell death. NF-κB levels are frequently observed to be elevated in diabetic patients. A recent study indicated involvement of common gene variants of NF-κB in diabetes and renal function impairment, thereby showing the association of NF-κB 1 variants in type 2 diabetic patients [12]. In another recent study it was observed that β cell de-differentiation and impaired insulin secretion which eventually leads to β cell death were promoted by NF-κB signalling pathway. Thus, modulating NF-κB signalling pathway can efficiently prevent the β cell death and provide regulated therapeutic agents for management of type 2 diabetes [13]. The transcription factor NF-κB poses major threat to health and activity of β cells and could be a potential therapeutic target for management of diabetes. Specific roles of NF-κB in type 2 diabetes and role of commensal gut microbiota and probiotics in its management has been reviewed in successive sections.
Evidently the human gut harbours a complex community of over 100 trillion bacterial cells belonging to over 1000 bacterial species [14]. Several studies have shown an association of these gut microorganisms with conditions like allergies, intestinal inflammatory diseases, cancer, diabetes, cardiovascular diseases, non-alcoholic fatty liver disease, and dyslipidaemia [2, 15,16,17,18]. Similarly, several researchers have observed that modulation of intestinal microbiota by beneficial microbes (probiotics) may facilitate the management of a number of clinical conditions [15, 19, 20]. Probiotics as defined by FAO/WHO are "live microorganisms which when administered in adequate amounts confer a health benefit on the host" [21]. Probiotics benefit the host by maintaining a healthier gut microbiota, immunomodulation and other mechanisms, and are frequently involved in host-microbe cross-talk. This cross-talk is by various secreted and non-secreted bacterial factors and cell signalling is an essential part of this interaction. Role of different cell signalling pathway in type 2 diabetes is now well established and probiotics are known to influence a vast array of host cell signalling events [22]. Given the general beneficial effect of probiotics on host health, it could prove useful in prophylactic and/or therapeutic strategies against diabetes. Hence, this review appraises modulation of NF-κB as possible intervention for management of diabetes through probiotic intervention. Our aim is to bring together the current status and possible role of probiotics in the prevention and management of diabetes through intercession of NF-κB signalling pathway.
Nuclear factor kappa B
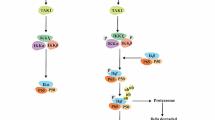
For better understanding of the topic, it is imperative to have basic knowledge of function and regulation of NF-κB, a nuclear transcription factor found in almost all cell types [23] (Fig. 1). NF-κB performs an essential role in myriad aspects of human health including the development of both innate as well as adaptive immunity, through control over as much as 150 genes involved in a variety of cellular processes [24,25,26]. Five members of the NF-κB family including p50, p52, p65 (RelA), c-Rel, and RelB form different combinations of homo- and heterodimers with different DNA binding specificities and transactivation potential [27, 28]. Heterodimer, p50/ RelA or p52 is the commonly found active form of NF-κB. Each member of the NF-κB family has a conserved N-terminal region called the Rel-homology domain, which contains the dimerization, nuclear localization, and DNA binding domains [27, 29]. Under healthy state, inactive forms of NF-κB complexes are sequestered in the cytoplasm via non-covalent interaction with inhibitory protein, IκBα.

Regulatory circuit of NF-κB: In response to multiple triggering factors IKK phosphorylates IkBα, which is subsequently ubiquitinated and degraded by proteasome. The free NF-κB hetero-dimer translocates to the nucleus and binds to its DNA binding site
In response to multiple triggering factors, including cytokines, viral and bacterial pathogens, inflammation and stress-inducing agents, the inactive cytoplasmic NF-κB/IκBα complex gets phosphorylated on conserved serine residues in the N-terminal portion of IκBα, followed by ubiquitinylation and proteosomal degradation resulting in breakage of non-covalent interactions and activation of NF-κB. This phosphorylation process is mediated by a multimeric IκB kinase (IKK) complex [30]. Active NF-κB translocates to the nucleus, where it binds to its DNA binding site in the promoter or enhancer regions of specific genes. Level of transcription of individual genes and the amount of transcribed product is regulated by several factors including the composition of NF-κB dimers, nature of activating stimulus and the number of consensus sites in the target genes [31, 32]. Although NF-κB is responsible for mediating immune response and homeostasis, it is better known for its pro-inflammatory potential and serves as potential target during drug therapy against infections. Excessive activation of NF-κB leads to the over-production of pro-inflammatory cytokines and chemokines resulting in chronic inflammation. NF-κB dysregulation has been widely associated with many clinical conditions, including diabetes [33].
Role of NF-κB in diabetes
NF-κB plays a mediator in development of diabetes and related complications. Activation of NF-κB is capable of triggering either pro-inflammatory or anti-inflammatory cascade [12, 34, 35]; however, in terms of β cells, the activity is reported to be predominantly pro-apoptotic [36]. NF-κB activity is suppressed in healthy β cells, however, upon oxidative stress and inflammation, NF-κB gets activated and translocate to the nucleus. Production of reactive oxygen species (ROS) intermediates play a critical role in signalling of autoimmune/inflammatory response, mediated through NF-κB. NF-κB regulates the expression of several genes involved in dysfunction and death of β cells [37]. One such study that involved the identification of novel diabetes candidate genes, gave direct evidence of role of IKK/NF-κB activation in triggering β cell death in type 1 diabetes. Recently, exhaustive bioinformatics analyses were carried out to identify core genes and pathways involved in development of type 2 diabetes [38, 39]. Role of genes viz. major histocompatibility complex class I/II and signalling pathways viz. tumor necrosis factor, cyclic adenosine monophosphate, and peroxisome proliferators-activated receptor signalling pathway in β cells death were also identified [39]. Similarly, Luo and co-workers identified the role of NF-κB and NLRP3 inflammasome activation in the development of diabetes and diabetic cardiomyopathy in a rat model. The study also revealed that activation of NLRP3 inflammasome was through NF-κB mediated pathway in high glucose treated H9c2 cells [40].
Inhibition of NF-κB has been shown to improve insulin sensitivity [41] and prevent apoptosis in both human islets [42] and in alloxan induced diabetic mice models [43]. NF-κBp50 subunit knockout conferred resistance towards streptozotocin induced diabetes in mice models [44]. Role of c-Rel and p50/p105 sub unit has also been suggested in streptozotocin induced diabetes [45, 46]. Many in vitro and in vivo studies have shown that the inhibition of NF-κB pathway provides protection against cytokine induced apoptosis of pancreatic β cells [47,48,49].
The role of NF-κB and its associated target genes is very well documented in the pathogenesis of insulin resistance and type 2 diabetes. Two independent studies, one using the selective transgenic expression and the other using IKKβ knockout in the liver [50, 51] offered adequate evidence to support the key role of NF-κB in development of insulin resistance. Over expression of IKKβ led to the activation of NF-κB, which mimics the effects of high fat diet or obesity induced insulin resistance in experimental mice model. Tumor necrosis factor α (TNF α), a pro-inflammatory cytokine, one of the best characterized inducer of NF-κB is also known to induce insulin resistance through serine phosphorylation of insulin receptor substrate 1 (IRS1). Members of TNF family induce rapid transcription of genes involved in cell survival, inflammation, proliferation and differentiation, mediated through activation of NF-κB [52]. Furthermore, there are various evidences that TNF α is highly induced in the adipose tissues of obese human and animal subjects, while the neutralization of TNF α can reverse insulin resistance, indicating towards involvement of NF-κB activation in insulin resistance [53]. The type and level of polymorphism in NF-κB gene determines the level of complications. For instance, a study conducted to investigate the correlation between NF-κB gene polymorphism and their susceptibility to diabetic nephropathy revealed that a NF-κB1 gene, -94 ATG insertion/deletion polymorphism in Asian Indian subjects with previous history of diabetes mellitus might be associated with an increased risk of nephropathy development [54]. Romzova and co-workers reported increase in homozygous AA genotype of IκBα (NF-κB inhibitor) gene in human subjects with type 2 diabetes suggesting role of NF-κB polymorphism in pathogenesis of type 2 diabetes [55]. Variation in the NF-κB1 was independently responsible as a risk factor for the development of type 2 diabetes in elderly Caucasian subjects [12]. Chronic exposure to glucose and free fatty acids also induces β cell apoptosis. Although high glucose does not induce NF-κB, indicating that the glucose induced β cell apoptosis is primarily independent of NF-κB. However, few studies linked higher glucose concentration to aggravated NF-κB expression in pancreatic cells [56].
Although, majority of studies have demonstrated apoptotic effect of NF-κB in pancreatic β cells, it has been earlier reported that NF-κB has both protective and destructive effects which depends on pathophysiology and on the tissue. Blockage or knockout of NF-κB gene resulted in reduced expression of insulin secretion pathway genes and marginal decrease in the count of endocrine cells in adult pancreas [57]. In another such study, A20 was identified as an NF-kB dependent antiapoptotic gene in β cells that protected β cells from TNF induced apoptosis [58].
NF-κB is involved in the expression of GLUT2 (Glucose transporter 2), which contributes to insulin secretion by β cells [59]. Reports generated from various cell and animal studies suggest that an inhibition of GLUT2 transcription factor might have certain deleterious effects, ultimately leading to the development of insulin resistance and type 2 diabetes [34]. The appropriate control and regulation of NF-κB activity, by means of gene modification as well as pharmacological strategies would provide a potential approach for the management of NF-κB related human diseases including diabetes. Keeping this in mind, several research groups have explored different compounds for modulating NF-κB signalling in different clinical conditions including diabetes. Free phenolic extracts from cereal grains, and plumbagin, a vitamin K3 analogue inhibited Lipopolysaccharide (LPS) induced NF-κB under in vitro cell line conditions [60, 61]. Lycopene inhibited promoter binding activity of NF-κB and intracellular ROS production in human hepatoma Hep 1 cells [62]. Pyrrolidine dithiocarbamate, an anti-oxidant is known to inhibit DNA binding and nuclear translocation of NF-κB in neurons. However, DDTC, another NF-κB inhibiting thiocarbamate has been shown to have negative health effects [63]. NF-κB modulation by probiotic strains can be a safe, dietary intervention for possible management of diabetes.
Management of NF-κB by probiotics
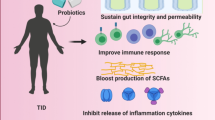
Substantial evidence proves that certain probiotic strains can modulate immune response exerting metabolic changes in the host [59]. Potential sites where probiotics and their metabolites can influence NF-κB are depicted in Fig. 2. In particular, the NF-kB signalling can be modulated by probiotics and their active biological molecules at different sites with probiotic induced effects reported on inhibition of different processes viz. induction through TLRs, transcriptional activation, phosphorylation, ubiquitination and proteasomal degradation of IkBα, nuclear translocation, and DNA binding of the p50/p65 isoforms (Table 1).

Schematic illustration of signalling pathways involved in activation of NF-κB and their probable regulation by probiotics: Probiotic mediated regulation of NF-κB can take place at several steps viz. inactivation of IKK, inhibition of IkBα phosphorylation and ubiquitination, inhibition of IkBα degrading proteasomes, suppression of translocation of NF-κB dimer to the nucleus and transcription inhibition
Various pathogens interact with different toll like receptors (TLRs) to activate NF-κB leading to inflammation. Probiotic bacteria such as L. casei suppresses the Shigella flexneri induced transcription of inflammatory cytokines, adhesion molecules and chemokines in intestinal epithelial cells by inhibiting NF-κB activation [64]. Several other studies supported NF-κB down-regulating potential of L. reuteri, L. rhamnosus GG (LGG), B. infantis and L. salivarius expressed through suppressed TNF α or Salmonella Typhimurium induced IL 8 gene expression and secretion by intestinal epithelial cells [65,66,67]. Inhibition of pathogen recognition associated TLRs is one of the mechanism identified for inhibiting NF-κB [68, 69]. Among other, down regulation of the transcriptional activity of NF-κB via targeting NF-κB signalling pathway is the most reported mechanism. Different steps in NF-κB signalling pathway can act as potential targets for anti-inflammatory probiotics and several other commensals to weaken its transcription. In this line, Kaci et al. (2011) documented the NF-κB suppressing activity of commensal Streptococcus salivarius K12 in human intestinal epithelial cells. Cell free supernatants (< 3 kDa fraction) of S. salivarius and S. vestibularis strains markedly inhibited TNF α induced NF-κB activation in different in vitro models viz. THP-1 reporter cells, Caco-2/kB-seap-7 cells suggesting role of an active microbial metabolite modulating the inflammatory response [70]. In another similar study, S. salivarius K12 was reported to attenuate NF-κB activation, suggesting preventive role of this bacterium in inflammation [71]. Bifidobacterium lactis was observed to suppress NF-κB activation in TNF α, IL 1β and LPS induced HT-29 cells [72]. In contrast, conditioned media from bifidobacteria were reported to stimulate NF-κB in HT-29 cells, while inhibiting the same in Caco-2 cells co-cultured with pro-inflammatory cytokines and bifidobacteria conditioned media. However, under similar conditions TNF α induced NF-κB activation was restrained in the two cell lines only by Colinsella aerofasciens [73]. In another study, LGG pre-feeding prevented TNF α induced intestinal activation of NF-κB [74]. A study carried out by Johnson-Henry et al. (2005) demonstrated probiotics induced downregulation of pro-inflammatory Th1 response with a simultaneous shift towards an improved Th2 response in mice infected with Citrobacter rodentium [75]. This response was attributed to downregulation of NF-κB mediated pathways by probiotic L. helveticus R0052 and L. rhamnosus R0011. Recently, peptides belonging to microbial anti-inflammatory molecule (MEM) secreted by Faecalibacterium prausnitzii were shown to inhibit NF-κB under in vitro epithelial cell culture model and displayed anti-inflammatory properties in colitis model. In transgenic mice models, MEM administration inhibited Th1, Th2 and Th17 immune response through mechanism affecting NF-κB activation [76]. Pre-incubation with yeasts induced NF-κB mediated downregulation in expression of pro-inflammatory chemokines [77]. Another study which evaluated the suppression of Salmonella enteric infection in mice, showed an association of probiotic bacteria with reduced mRNA expression of a group of genes (RelB, Myd88, IKKα, Jun, Irak2) regulated through NF-κB signal transduction pathway as a part of cytokine response [78].
Effects of probiotics on NF-κB transcriptional activity in the nucleus is modulated through PPAR gamma dependent pathway in a strain and dose dependent manner [59]. PPAR gamma forms complex with nuclear RelA and enhances its nuclear export, thereby diminishing NF-κB transcription [79]. B. thetaiotaomicron induced nuclear export of the RelA subunit of NF-κB associated with PPAR. In another study, L. crispatus M247 was able to produce hydrogen peroxide which acts as a signal transducing molecule thereby helping in the activation of PPAR gamma leading to suppression in NF-κB activity. Other non-hydrogen peroxide producing Lactobacillus strains were unable to activate the PPAR gamma mechanism [80].
Presence of reactive oxygen species was also observed to suppress degradation of IκBα [81]. Role of probiotics in modulating inflammatory responses by inducing local generation of ROS has been reported [82]. ROS can oxidize and inactivate key regulatory enzymes. In one such study, LGG induced ROS exhibited increased oxidation of the Ubc12 enzyme in intestinal epithelia. Ubc12 is responsible for the ubiquitination of the inhibitory molecule IκBα, therefore IκBα is not targeted for proteasomal degradation keeping NF-κB inactive in the cytosol. Pre-treatment of Caco-2 cells with LGG inhibited nuclear translocation of the NF-κB p65 resulting in decreased production of TNF α [83]. L. reuteri was shown to inhibit IκB phosphorylation in the intestine during LPS exposure which in turn inhibited the translocation of free NF-κB to the nucleus, thereby inhibiting the later anti-inflammatory response [84]. L. paracasei attenuated the LPS induced secretion of TNF α and IL 1β, concurrently with or before LPS challenge and the effect was due to decrease in IκB phosphorylation and NF-κB nuclear translocation [69]. Further investigators proposed the role of various probiotics which acts after the subsequent NF-κB translocation into the nucleus and is preceded by proteolytic degradation of IκBα. To elucidate whether probiotics could suppress NF-κB activation, inhibition of IκBα degradation was tested in human myeloid leukemia‐derived cells and results indicated that the probiotic could suppresses TNF induced IκBα degradation [85].
Activation of cytosolic NF-κB is followed by its translocation inside the nucleus. Blockage or inhibition of NF-κB nuclear translocation is another intervention point where probiotics can act. In order to study the effect of microbial metabolites on nuclear translocation of active NF-κB, THP-1 cells were stimulated with LPS in presence or absence of Streptococcus thermophilus and B. breve conditioned medium filtrate fractions (< 3 kDa), which significantly inhibited nuclear translocation of active NF-κB subunits [86]. Lactococcus lactis subsp. cremoris strains also inhibited NF-κB nuclear translocation in RAW264.7 cells along with notable suppression in expression of TNF-α, IFN-γ, IL-6, iNOS, and MIP-2 [87]. In another study, pre-treatment of HT-29 cells with LGG attenuated LPS induced NF-κB nuclear translocation along with blockage of LPS induced IκBα degradation [77]. LGG reduced the nuclear translocation of NF-κB by reducing the p65 subunit, necessary for the nuclear translocation [88]. L. reuteri was found to block nuclear translocation of RelA by preventing IκBα degradation in response to TNF stimulation [89]. Another study showed that L. casei suppresses S. flexneri induced transcription of inflammatory chemokines, cytokines and various adhesion molecules by manipulating the ubiquitin pathway to stabilise IκBα and thereby inhibit NF-κB nuclear translocation [23]. The activation of NF-κB was inhibited, thereby inhibiting p65 nuclear translocation and reversal of IκBα degradation when the Raw264.7 cells were treated with L. casei 3260 [90].
In another study binding of p50/p65 isoforms of NF-κB in presence of pro-inflammatory stimulus and bacterial conditioned media was studied. Pre-treatment of intestinal epithelial cells and macrophages (RAW 264.7) with conditioned media from several different Gram positive and Gram negative commensal bacteria, followed by pro-inflammatory stimulation with TNF α inhibited the binding of the p50/p65 subunits. The study depicted that pre-treatment with conditioned medium inhibits the chymotrypsin like activity of the proteasome responsible for release and activation of p50/p65 subunit of NF-κB, thus inhibiting its DNA binding activity [91]. Probiotic treatment was also shown to reduce NF-kB binding activity in high fat diet fed mice [92]. Many studies revealed that the binding activity of NF-κB was limited due to blocking of degradation of IκBα subunit which is a key step in the activation of NF-κB [91]. L. reuteri secretes various factors promoting Bcl-2 and Bcl-xL (anti-apoptotic protein) production in human myeloid leukemia derived cells by inhibiting NF-κB activation. These secreted proteins inhibit NF-κB activation through inhibition of IκB Ubiquitination [85].
Probiotics Lactobacillus strains are known to prevent damage from inflammatory response during autoimmune diseases as well as bacterial infections. In one such study, L. brevis G-101 was shown to inhibit phosphorylation of both Akt (alpha serine/threonine protein kinase) and IRAK1 (Interleukin-1 receptor-associated kinase-1) via the traditional MyD88 pathway, preventing the activation of NF-κB [93]. Investigators studied the role of intracellular events of anti-proliferative activity of L. plantarum JSA22 through the signalling cascade involving an overall decrease in NF-κB activation in colon fibroblast cells when stimulated with S. Typhimurium. The study indicated that L. plantarum JSA22 promotes intestinal epithelial cells survival through inhibition of Akt factor, which is pro-apoptotic in nature, through the inactivation of p38. The phosphorylation levels of Akt and p38 were estimated with or without probiotics. A significant decrease was observed in both the proteins under study when the host cells were infected with L. plantarum JSA22 or even L. rhamnosus GG [94]. In contrast, LGG and their soluble factors (p75 and p40) were reported to prevent epithelial cell apoptosis through activating anti-apoptotic Akt and inhibiting pro-apoptotic p38/MAPK [59].
The effect of eukaryotic probiotic Saccharomyces boulardii on NF-κB DNA binding was studied and mechanism of IκBα degradation was observed. The expression of NF-κB regulated gene was evaluated by transient transfection of THP-1 cells with a NF-κB responsive luciferase reporter gene. S. boulardii inhibited IκBα degradation and reduced both NF-κB DNA binding and NF-κB reporter gene up-regulation in LPS stimulated THP-1 cells. S. boulardii also exerts an anti-inflammatory effect that blocks NF-κB activation in intestinal epithelial cells and monocytes [95]. β-glucan from Saccharomyces cerevisiae was reported to induce sheep β-defensin 1 expression in ovine ruminal epithelial cells mediated through activation of NF-κB. β-defensins play a key role in innate and adaptive immunity [96].
The efficacy of probiotics against diabetes has also been proven in experimental in vivo models. For instance, Bifidobacterium spp. reduced blood glucose levels and increased the expressions of insulin receptor β, insulin receptor substrate 1, protein kinase B (Akt/PKB). Increased Akt suppress IκBα degradation in adipose tissue of diabetic mice [97]. Authors also reported that feeding of probiotic induced the adiponectin expression and decreased both macrophage chemoattractant protein-1 (MCP-1) and interleukin 6 (IL 6) expression in the test organism. An in vivo study showed that B. infantis was responsible for the generation and function of Treg to suppress LPS induced NF-κB activation [98]. Furthermore, the implication of probiotic action has also been reported through production of short chain fatty acids (SCFA) in large intestine. SCFA are established as histone deacetylase inhibitors and affects the expression of various genes, which are directly and indirectly involved in glucose metabolism and pathogenesis of diabetes [99]. For example, probiotic generated SCFA, such as butyrate is reported to downregulate NF-κB activation through blocking cullin-1 neddylation, a critical step in the ubiquitination system which leads to NF-κB suppression [100]. Orally administered probiotic cocktail consisting of L. acidophilus, L. plantarum, B. lactis and B. breve reduced colonic expression of NF-κB, TLR-4 and iNOS in dextran sulfate sodium (DSS) induced acute colitis mice model [101].
Probiotics also have potential to enhance the immunocompetence to prevent spontaneous autoimmune response in diabetic subjects. Probiotic containing kefir improved the phagocytic capacity of peritoneal macrophages and increased concentration of IL 10, TNF α, IL 17 and IL 1β in diabetic mice challenged with LPS [102]. Likewise, Bernini et al. [103] studied the effects of B. lactis HN019 on inflammatory state and nitro-oxidative stress in patients with and without the metabolic syndrome. The study revealed that probiotic intervention decreased homocysteine, hydroperoxides, IL 6 levels and increased adiponectin and nitric oxide metabolites in metabolic syndrome group. The mechanism behind could involve the attenuation of pro-inflammatory Th1 and Th17 cytokines and generation of regulatory T cells that produce IL 10 like cytokines in the process of immune tolerance [104, 105]. Intestinal gut microbiota is known to regulate Th17 cell homeostasis and govern the outcome of metabolic disorders [106]. Moreover, changes in the level of NF-κB expression by T lymphocyte cells have a direct impact on the differentiation and activation of T helper and T regulatory lymphocytes. The application of probiotics could change the expression of NF-κB in immunopositive cells leading to impact on physiological disease outcomes [107].
Another key mechanism involves modulation of gut microbiota to improve insulin sensitivity in diabetic condition. The composition of gut microflora has direct impact on energy metabolism, immunity, inflammation and metabolic dysfunction. Altered microbial population enhance gut permeability and activates LPS induced downstream signalling of MAPK, JNK and P38 molecules which eventually activates NF-κB in epithelial cells, immune cells and metabolically active tissues [97]. Probiotics have proven efficacy in the treatment of dysbiosis and its complications. The transplantation of butyrate producing probiotic Faecalibacterium prausnitzii has been proposed to improve symptoms of metabolic syndrome like obesity and diabetes using such mechanism [108]. Besides, other probiotic species such as, L. rhamnosus, L. acidophilus and B. bifidum have also been reported to influence gut microbiota, intestinal permeability and insulin sensitivity in mice subjected to high fat diet [109].
In contrast to the available reports supporting the NF-κB inhibitory potential of probiotic strains, few reports documented NF-κB induction by probiotics. The ability of L. plantarum JSA22 to activate the innate response via the NF-κB dependent manner was evaluated based on the assessment of NF-κB nuclear translocation. The results indicated that co-infection of cells with L. plantarum JSA22 and S. Typhimurium, significantly induced NF-κB dependent gene activation in intestinal epithelial cells [94]. In a co-culture model (intestinal epithelial cells and macrophages) of the undeveloped small intestine, members of Lactobacillus spp. influenced NF-kB p65 nuclear translocation in both intestinal epithelial cells and underlying macrophages in a strain dependent manner. LGG and PCS 20 strains significantly increased NF-kB p65 translocation; however no significant induction was reported with PCS 26. This nuclear translocation was linked to the ability of commensal microbiome to train the early immune system against pathogens [110].
Apart from their role in prevention of diabetes, probiotics also play a major role in improvement of metabolic diseases including alcoholic fatty liver disease (AFLD) and non-alcoholic fatty liver disease (NAFLD). Hepatic fat accumulation is associated with hepatic insulin resistance in obesity and type 2 diabetes. Increasing evidence suggest that intervention of probiotics could reduce the risk of metabolic syndrome associated NAFLD. One such study conducted by Li et al. documented VSL#3 mediated inhibition of TNF α, thereby leading to an improvement in NAFLD in ob/ob mice models. The results were also consistent with patients of nonalcoholic steatohepatitis (NASH), where enhanced β oxidation of fatty acids was reported in hepatic cells. It was proposed earlier that VSL#3 might tend to normalize the abnormalities in fatty acid β oxidation in ob/ob mice, which may further improve NASH in this mice model. On evaluation of the effects of probiotic VSL#3 therapy on peroxisomal and mitochondrial fatty acid oxidation, results showed that VSL#3 and/or anti TNF antibodies restored the hepatic fatty acid β-oxidation levels towards normal [111]. Furthermore, probiotic B. adolescentis had protective effect on high-fat diet induced NAFLD mice model through reduction of expression of MyD88 mRNA and activation of nuclear factor NF-κB [112].
Prospects/conclusion
Growing burden of diabetes and sub-optimal performance of available treatment strategies highlights the requirement for novel therapeutic strategies. Pancreatic beta cells play a central role in diabetes pathogenesis. Preserving beta cells via management of NF-κB can be a promising strategy for the management of diabetes. The correlation established between gut microbiota and NF-kB supports that the probiotic mediated NF-kB targeted therapy can be explored for possible management of diabetes and other related metabolic and inflammation driven disorders. Many Lactobacillus strains have been shown to regulate NF-κB expression under in vitro and in vivo conditions. Emerging leads from available data reflects NF-κB as a promising biotherapeutic target against diabetes, which can be modulated with dietary intervention involving probiotics. Selective probiotics strains may possibly be harnessed to regulate NF-κB for maintaining health and protecting against diabetes.
Abbreviations
- NF-κB:
-
Nuclear factor kappa B
- LAB:
-
Lactic acid bacteria
- IDF:
-
International diabetes federation
References
Cho N, Shaw JE, Karuranga S, Huang Y, da Rocha Fernandes JD, Ohlrogge AW, Malanda B (2018) IDF Diabetes Atlas: global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pract 138:271–281
Panwar H, Rashmi HM, Batish VK, Grover S, (2013) Probiotics as potential biotherapeutics in the management of type 2 diabetes–prospects and perspectives. Diabetes Metab Res Rev 29(2):103–112
Salsali A, Nathan M (2006) A review of types 1 and 2 diabetes mellitus and their treatment with insulin. Am J Ther 13(4):349–361
Ndisang JF, Vannacci A, Rastogi S (2017) Insulin resistance, type 1 and type 2 diabetes, and related complications. J Diabetes Res. https://doi.org/10.1155/2017/1478294
Jayachandran M, Vinayagam R, Ambati RR, Xu B, Chung SSM, (2018) Guava leaf extract diminishes hyperglycemia and oxidative stress, prevents β-cell death, inhibits inflammation, and regulates NF-κB signaling pathway in STZ induced diabetic rats. BioMed Res Int. https://doi.org/10.1155/2018/4601649
Lyssenko V, Laakso M (2013) Genetic screening for the risk of type 2 diabetes: worthless or valuable? Diabetes Care 36:S120–S126
Baeyens L, Lemper M, Staels W, De Groef S, De Leu N, Heremans Y et al (2018) (Re) generating human beta cells: status, pitfalls, and perspectives. Physiol Rev 98(3):1143–1167
Kulkarni RN (2004) The islet β-cell. Int J Biochem Cell Biol 36(3):365–371
Cnop M, Welsh N, Jonas JC, Jörns A, Lenzen S, Eizirik DL, (2005) Mechanisms of pancreatic β-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes 54(suppl 2):S97–S107
Nogueira TC, Paula FM, Villate O, Colli ML, Moura RF, Cunha DA et al (2013) GLIS3, a susceptibility gene for type 1 and type 2 diabetes, modulates pancreatic beta cell apoptosis via regulation of a splice variant of the BH3-only protein Bim. PLoS Genet. https://doi.org/10.1371/journal.pgen.1003532
Ardestani A, Paroni F, Azizi Z, Kaur S, Khobragade V, Yuan T et al (2014) MST1 is a key regulator of beta cell apoptosis and dysfunction in diabetes. Nat Med 20(4):385
Coto E, Díaz-Corte C, Tranche S, Gómez J, Alonso B, Iglesias S et al (2018) Gene variants in the NF-KB pathway (NFKB1, NFKBIA, NFKBIZ) and their association with type 2 diabetes and impaired renal function. Hum Immunol 79(6):494–498
Chen H, Zhou W, Ruan Y, Yang L, Xu N, Chen R et al (2018) Reversal of angiotensin ll-induced β-cell dedifferentiation via inhibition of NF-κb signaling. Mol Med 24(1):43
Guinane CM, Cotter PD (2013) Role of the gut microbiota in health and chronic gastrointestinal disease: understanding a hidden metabolic organ. Therap Adv Gastroenterol 6(4):295–308
Gomes AC, Bueno AA, de Souza RGM, Mota JF (2014) Gut microbiota, probiotics and diabetes. Nutr J 13(1):60
Holmes E, Li JV, Athanasiou T, Ashrafian H, Nicholson JK, (2011) Understanding the role of gut microbiome–host metabolic signal disruption in health and disease. Trends Microbiol 19(7):349–359
Elshaghabee F, Rokana N, Panwar H, Heller KJ, Schrezenmeir J (2019) Probiotics for dietary management of non-alcoholic fatty liver disease. Environ Chem Lett. https://doi.org/10.1007/s10311-019-00896-8
Thakur N, Rokana N, Panwar H (2016) Probiotics: selection criteria, safety and role in health and disease. J Innov Biol 3(1):259–270
Floch MH, Montrose DC (2005) Use of probiotics in humans: an analysis of the literature. Gastroenterol Clin 34(3):547–570
Panwar H, Calderwood D, Grant IR, Grover S, Green BD (2014) Lactobacillus strains isolated from infant faeces possess potent inhibitory activity against intestinal alpha- and beta-glucosidases suggesting anti-diabetic potential. Eur J Nutr 53(7):1465–1474
Hill C, Guarner F, Reid G, Gibson GR, Merenstein DJ, Pot B, Morelli L, Canani RB, Flint HJ, Salminen S, Calder PC (2014) Expert consensus document: the International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat Rev Gastroenterol Hepatol 11(8):506
Llewellyn A, Foey A (2017) Probiotic modulation of innate cell pathogen sensing and signalling events. Nutrients 9(10):1156
Yan F, Polk DB (2010) Probiotics: progress toward novel therapies for intestinal diseases. Curr Opin Gastroenterol 26(2):95
Ghosh S, May MJ, Kopp EB (1998) NF-κB and Rel proteins: evolutionarily conserved mediators of immune responses. Ann Rev Immunol 16(1):225–260
Pahl HL (1999) Activators and target genes of Rel/NF-κB transcription factors. Oncogene. https://doi.org/10.1038/sj.onc.1203239
Tang C, Zhu G (2019) Classic and novel signaling pathways involved cancer: targeting the NF-κB and Syk signaling pathways. Curr Stem Cell Res Ther 14(3):219–225
Li Q, Verma IM (2002) NF-κB regulation in the immune system. Nat Rev Immunol 2(10):725
Senftleben U, Karin M (2002) The Ikk/nf-κb pathway. Crit Care Med 30(1):S18–S26
Silverman N, Maniatis T (2001) NF-κB signaling pathways in mammalian and insect innate immunity. Genes Dev 15(18):2321–2342
Chen ZJ, Parent L, Maniatis T (1996) Site-specific phosphorylation of IκBα by a novel ubiquitination-dependent protein kinase activity. Cell 84(6):853–862
Karin M, Ben-Neriah Y (2000) Phosphorylation meets ubiquitination: the control of NF-κB activity. Ann Rev Immunol 18(1):621–663
Zhou LZH, Johnson AP, Rando TA (2001) NFκB and AP-1 mediate transcriptional responses to oxidative stress in skeletal muscle cells. Free Radic Biol Med 31(11):1405–1416
Serasanambati M, Chilakapati SR (2016) Function of nuclear factor kappa B (NF-kB) in human diseases-a review. South Indian J Biol Sci 2(4):368–387
Patel S, Santani D (2009) Role of NF-κB in the pathogenesis of diabetes and its associated complications. Pharmacol Rep 61(4):595–603
Arakelyan A, Nersisyan L, Poghosyan D, Khondkaryan L, Hakobyan A, Löffler-Wirth H et al (2017) Autoimmunity and autoinflammation: a systems view on signaling pathway dysregulation profiles. PLoS ONE. https://doi.org/10.1371/journal.pone.0187572
Eizirik DL, Mandrup-Poulsen T (2001) A choice of death–the signal-transduction of immune-mediated beta-cell apoptosis. Diabetologia 44(12):2115–2133
Cardozo AK, Heimberg H, Heremans Y, Leeman R, Kutlu B, Kruhøffer M et al (2001) A comprehensive analysis of cytokine-induced and nuclear factor-κB-dependent genes in primary rat pancreatic β-cells. J Biol Chem 276(52):48879–48886
Ding L, Fan L, Xu X, Fu J, Xue Y (2019) Identification of core genes and pathways in type 2 diabetes mellitus by bioinformatics analysis. Mol Med Rep 20:2597–2608
Li L, Pan Z, Yang S, Shan W, Yang Y (2018) Identification of key gene pathways and coexpression networks of islets in human type 2 diabetes. Diabetes Metab Syndr Obes 11:553–563
Luo B, Huang F, Liu Y, Liang Y, Wei Z, Ke H, Zeng Z, Huang W, He Y (2017) NLRP3 inflammasome as a molecular marker in diabetic cardiomyopathy. Front physiol 8:519
Zhang N, Valentine JM, Zhou Y, Li ME, Zhang Y, Bhattacharya A, Walsh ME, Fisher KE, Austad SN, Osmulski P, Gaczynska M, Shoelson SE, Remmen HV, Chen HI, Chen Y, Liang H, Musi N (2017) Sustained NFkB inhibition improves insulin sensitivity but is detrimental to muscle health. Aging Cell 16:847–858
Giannoukakis N, Mi Z, Rudert WA, Gambotto A, Trucco M, Robbins P (2000) Prevention of beta cell dysfunction and apoptosis activation in human islets by adenoviral gene transfer of the insulin-like growth factor I. Gene Thera 7(23):2015
Ho E, Quan N, Tsai YH, Lai W, Bray TM (2001) Dietary zinc supplementation inhibits NFκB activation and protects against chemically induced diabetes in CD1 mice. Exp Biol Med 226(2):103–111
Mabley JG, Hasko G, Liaudet L, Soriano F, Southan GJ, Salzman AL et al (2002) NFkappaB1 (p50)-deficient mice are not susceptible to multiple low-dose streptozotocin-induced diabetes. J Endocrinol 173(3):457–464
Lamhamedi-Cherradi SE, Zheng S, Hilliard BA, Xu L, Sun J, Alsheadat S et al (2003) Transcriptional regulation of type I diabetes by NF-κB. J Immunol 171(9):4886–4892
Kumar D, Robertson S, Burns KD (2004) Evidence of apoptosis in human diabetic kidney. Mol Cell Biochem 259(1–2):67–70
Song MY, Jeong GS, Kwon KB, Ka SO, Jang HY, Park JW et al (2010) Sulfuretin protects against cytokine-induced beta-cell damage and prevents streptozotocin-induced diabetes. Exp Mol Med 42(9):628–638
Collier JJ, Burke SJ, Eisenhauer ME, Lu D, Sapp RC, Frydman CJ et al (2011) Pancreatic β-cell death in response to pro-inflammatory cytokines is distinct from genuine apoptosis. PLoS ONE. https://doi.org/10.1371/journal.pone.0022485
Burke SJ, Lu D, Sparer TE, Masi T, Goff MR, Karlstad MD et al (2013) NF-κB and STAT1 control CXCL1 and CXCL2 gene transcription. Am J Physiol Endocrinol Metab 306(2):E131–E149
Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM et al (2005) IKK-β links inflammation to obesity-induced insulin resistance. Nat Med 11(2):191
Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J et al (2005) Local and systemic insulin resistance resulting from hepatic activation of IKK-β and NF-κB. Nat Med 11(2):183
Hayden MS, Ghosh S (2014) Regulation of NF-kB by TNF family cytokines. Semin Immunol 26(3):253–266
Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM (1995) Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest 95(5):2409–2415
Gautam A, Gupta S, Mehndiratta M, Sharma M, Singh K, Kalra OP, Agarwal S, Gambhir JK (2017) Association of NFKB1 gene polymorphism (rs28362491) with levels of inflammatory biomarkers and susceptibility to diabetic nephropathy in Asian Indians. World J Diabetes 8(2):66
Romzova M, Hohenadel D, Kolostova K, Pinterova D, Fojtikova M, Ruzickova S, Dostal C, Bosak V, Rychlik I, Cerna M (2006) NFκB and its inhibitor IκB in relation to type 2 diabetes and its microvascular and atherosclerotic complications. Hum Immunol 67(9):706–713
Cernea S, Dobreanu M (2013) Diabetes and beta cell function: from mechanisms to evaluation and clinical implications. Biochem Med 23(3):266–280
Norlin S, Ahlgren U, Edlund H (2005) Nuclear factor-κB activity in β-cells is required for glucose-stimulated insulin secretion. Diabetes 54(1):125–132
Liuwantara D, Elliot M, Smith MW, Yam AO, Walters SN, Marino E, McShea A, Grey ST (2006) Nuclear factor- κB regulates β cell death. Diabetes 55(9):2491–2501
Madsen KL (2012) Enhancement of epithelial barrier function by probiotics. J Epithel Biol Pharmacol 5(1):55–59
Hole AS, Grimmer S, Naterstad K, Jensen MR, Paur I, Johansen SG, Balstad TR, Blomhoff R, Sahlstrom S (2009) Activation and inhibition of nuclear factor kappa B activity by cereal extracts: role of dietary phenolic acids. J Agric Food Chem 57:9481–9488
Wang T, Wu F, Jin Z, Zhai Z, Wang Y, Tu B, Yan W, Tang T (2014) Plumbagin inhibits LPS-induced inflammation through the inactivation of the nuclear factor-kappa B and mitogen activated protein kinase signaling pathways in RAW 264.7 cells. Food Chem Toxicol 64:177–183
Huang CS, Fan YE, Lin CY, Hu ML (2007) Lycopene inhibits matrix metalloproteinase-9 expression and down-regulates the binding activity of nuclear factor-kappa B and stimulatory protein-1. J Nutr Biochem 18(7):449–456
Nurmi A, Vartiainen N, Pihlaja R, Goldsteins G, Yrjanheikki J, Koistinaho J (2004) Pyrrolidine dithiocarbamate inhibits translocation of nuclear factor kappa-B in neurons and protects against brain ischaemia with a wide therapeutic time window. J Neurochem 91(3):755–765
Tien MT, Girardin SE, Regnault B, Le Bourhis L, Dillies MA, Coppée JY et al (2006) Anti-inflammatory effect of Lactobacillus casei on Shigella-infected human intestinal epithelial cells. J Immunol 176(2):1228–1237
Ma D, Forsythe P, Bienenstock J (2004) Live Lactobacillus reuteri is essential for the inhibitory effect on tumor necrosis factor alpha-induced interleukin-8 expression. Infect Immun 72(9):5308–5314
Zhang L, Li N, Caicedo R, Neu J (2005) Alive and dead Lactobacillus rhamnosus GG decrease tumor necrosis factor-α–induced interleukin-8 production in caco-2 cells. J Nutr 135(7):1752–1756
O'hara AM, O'regan P, Fanning Á, O'mahony C, MacSharry J, Lyons A, et al (2006) Functional modulation of human intestinal epithelial cell responses by Bifidobacterium infantis and Lactobacillus salivarius. Immunology 118(2):202–215
Bu X, Lian X, Wang Y, Luo C, Tao S, Liao Y, Yang J, Chen A, Yang Y (2019) Dietary yeast culture modulates immune response related to TLR2-MyD88-NF-kβ signaling pathway, antioxidant capability and disease resistance against Aeromonas hydrophila for Ussuri catfish (Pseudobagrus ussuriensis). Fish Shellfish Immunol 84:711–718
Sun KY, Xu DH, Xie C, Plummer S, Tang J, Yang XF et al (2017) Lactobacillus paracasei modulates LPS-induced inflammatory cytokine release by monocyte-macrophages via the up-regulation of negative regulators of NF-kappaB signaling in a TLR2-dependent manner. Cytokine 92:1–11
Kaci G, Lakhdari O, Doré J, Ehrlich SD, Renault P, Blottière HM et al (2011) Inhibition of the NF-κB pathway in human intestinal epithelial cells by commensal Streptococcus salivarius. Appl Environ Microbiol 77(13):4681–4684
Cosseau C, Devine DA, Dullaghan E, Gardy JL, Chikatamarla A, Gellatly S et al (2008) The commensal Streptococcus salivarius K12 downregulates the innate immune responses of human epithelial cells and promotes host-microbe homeostasis. Infect Immun 76(9):4163–4175
Kim SW, Kim HM, Yang KM, Kim SA, Kim SK, An MJ et al (2010) Bifidobacterium lactis inhibits NF-κB in intestinal epithelial cells and prevents acute colitis and colitis-associated colon cancer in mice. Inflamm Bowel Dis 16(9):1514–1525
Lakhdari O, Tap J, Béguet-Crespel F, Le Roux K, De Wouters T, Cultrone A et al (2011) Identification of NF-κB modulation capabilities within human intestinal commensal bacteria. BioMed Res Int. https://doi.org/10.1155/2011/282356
Lin PW, Myers LE, Ray L, Song SC, Nasr TR, Berardinelli AJ et al (2009) Lactobacillus rhamnosus blocks inflammatory signaling in vivo via reactive oxygen species generation. Free Rad Biol Med 47(8):1205–1211
Johnson-Henry KC, Nadjafi M, Avitzur Y, Mitchell DJ, Ngan BY, Galindo-Mata E et al (2005) Amelioration of the effects of Citrobacter rodentium infection in mice by pretreatment with probiotics. J Infect Dis 191(12):2106–2117
Breyner NM, Michon C, de Sousa CS, Vilas Boas PB, Chain F, Azevedo VA, Langella P, Chatel JM (2017) Microbial anti-inflammatory molecule (MAM) from Faecalibacterium prausnitzii shows a protective effect on DNBS and DSS-induced colitis model in mice through inhibition of NF-κB pathway. Front Microbiol. https://doi.org/10.3389/fmicb.2017.00114
Lee SK, Yang KM, Cheon JH, Kim TI, Kim WH (2012) Anti-inflammatory mechanism of Lactobacillus rhamnosus GG in lipopolysaccharide-stimulated HT-29 cell. Korean J Gastroenterol 60(2):86–93
Wagner RD, Johnson SJ (2017) Probiotic bacteria prevent Salmonella–induced suppression of lymphoproliferation in mice by an immunomodulatory mechanism. BMC Microbiol 17(1):77
Kelly D, Campbell JI, King TP, Grant G, Jansson EA, Coutts AG et al (2004) Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of PPAR-gamma and RelA. Nat Immunol 5:104–112
Voltan S, Martines D, Elli M, Brun P, Longo S, Porzionato A et al (2008) Lactobacillus crispatus M247-derived H2O2 acts as a signal transducing molecule activating peroxisome proliferator activated receptor-γ in the intestinal mucosa. Gastroenterol 135(4):1216–1227
Volpe C, Villar-Delfino PH, Dos Anjos P, Nogueira-Machado JA (2018) Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis 9(2):119
Kumar A, Wu H, Collier-Hyams LS, Hansen JM, Li T, Yamoah K et al (2007) Commensal bacteria modulate cullin-dependent signaling via generation of reactive oxygen species. EMBO J 26(21):4457–4466
Donato KA, Gareau MG, Wang YJJ, Sherman PM (2010) Lactobacillus rhamnosus GG attenuates interferon-γ and tumour necrosis factor-α-induced barrier dysfunction and pro-inflammatory signalling. Microbiol 156(11):3288–3297
Liu Y, Fatheree NY, Mangalat N, Rhoads JM (2012) Lactobacillus reuteri strains reduce incidence and severity of experimental necrotizing enterocolitis via modulation of TLR4 and NFκB signaling in the intestine. Am J Physiol Heart Circ Physiol 302(6):G608–G617
Iyer C, Kosters A, Sethi G, Kunnumakkara AB, Aggarwal BB, Versalovic J, (2008) Probiotic Lactobacillus reuteri promotes TNF-induced apoptosis in human myeloid leukemia-derived cells by modulation of NF-κB and MAPK signalling. Cell Microbiol 10(7):1442–1452
Menard S, Candalh C, Bambou JC, Terpend K, Cerf-Bensussan N, Heyman M (2004) Lactic acid bacteria secrete metabolites retaining anti-inflammatory properties after intestinal transport. Gut 53(6):821–828
Nishitani Y, Tanoue T, Yamada K, Ishida T, Yoshida M, Azuma T et al (2009) Lactococcus lactis subsp. cremoris FC alleviates symptoms of colitis induced by dextran sulfate sodium in mice. Int Immunopharmacol 9(12):1444–1451
Versalovic J, Iyer C, Ping Lin Y, Huang Y, Dobrogosz W (2008) Commensal-derived probiotics as anti-inflammatory agents. Microbial Ecol Health Dis 20(2):86–93
Rahman MM, McFadden G (2011) Modulation of NF-κB signalling by microbial pathogens. Nat Rev Microbiol 9(4):291
Lee JM, Hwang KT, Jun WJ, Park CS, Lee MY (2008) Antiinflammatory effect of lactic acid bacteria: inhibition of cyclooxygenase-2 by suppressing nuclear factor-kappaB in Raw 264.7 macrophage cells. J Microbiol Biotechnol 18(10):1683–1688
Petrof EO, Claud EC, Sun J, Abramova T, Guo Y, Waypa TS et al (2009) Bacteria-free solution derived from Lactobacillus plantarum inhibits multiple NF-kappaB pathways and inhibits proteasome function. Inflamm Bowel Dis 15(10):1537–1547
Ma X, Hua J, Li Z (2008) Probiotics improve high fat diet-induced hepatic steatosis and insulin resistance by increasing hepatic NKT cells. J Hepatol 49:821–830
Jang SE, Hyam SR, Han MJ, Kim SY, Lee BG, Kim DH (2013) Lactobacillus brevis G-101 ameliorates colitis in mice by inhibiting NF-κB, MAPK and AKT pathways and by polarizing M1 macrophages to M2-like macrophages. J Appl Microbiol 115(3):888–896
Eom JS, Song J, Choi HS (2015) Protective effects of a novel probiotic strain of Lactobacillus plantarum JSA22 from traditional fermented soybean food against infection by Salmonella enterica serovar Typhimurium. J Microbiol Biotechnol 25(4):479–491
Sougioultzis S, Simeonidis S, Bhaskar KR, Chen X, Anton PM, Keates S et al (2006) Saccharomyces boulardii produces a soluble anti-inflammatory factor that inhibits NF-κB-mediated IL-8 gene expression. Biochem Biophys Res Commun 343(1):69–76
Zhang M, Jin X, Yang YF (2019) β-Glucan from Saccharomyces cerevisiae induces SBD-1 production in ovine ruminal epithelial cells via the dectin-1–Syk–NF-κB signaling pathway. Cell Signal 53:304–315
Le TK, Hosaka T, Nguyen TT, Kassu A, Dang TO, Tran HB, Pham TP, Tran QB, Le TH, Da Pham X (2015) Bifidobacterium species lower serum glucose, increase expressions of insulin signaling proteins, and improve adipokine profile in diabetic mice. Biomed Res 36(1):63–70
O'Mahony C, Scully P, O'Mahony D, Murphy S, O'Brien F, Lyons A et al (2008) Commensal-induced regulatory T cells mediate protection against pathogen-stimulated NF-κB activation. PLoS Pathog. https://doi.org/10.1371/journal.ppat.1000112
Khan S, Maremanda KP, Jena G. (2019) Butyrate, a short-chain fatty acid and histone deacetylases inhibitor: nutritional, physiological, and pharmacological aspects in diabetes. In: Handbook of nutrition, diet, and epigenetics Cham: Springer. 793–807
Kumar A, Wu H, Collier-Hyams LS, Kwon YM, Hanson JM, Neish AS, (2009) The bacterial fermentation product butyrate influences epithelial signaling via reactive oxygen species-mediated changes in cullin-1 neddylation. J Immunol 182(1):538–546
Toumi R, Soufli I, Rafa H, Belkhelfa M, Biad A, Touil-Boukoffa C (2014) Probiotic bacteria lactobacillus and bifidobacterium attenuate inflammation in dextran sulfate sodium-induced experimental colitis in mice. Int J Immunopathol Pharmacol 27(4):615–627
Maciel FR, Punaro GR, Rodrigues AM, Bogsan CS, Rogero MM, Oliveira MN, Mouro MG, Higa EM (2016) Immunomodulation and nitric oxide restoration by a probiotic and its activity in gut and peritoneal macrophages in diabetic rats. Clin Nutr 35(5):1066–1072
Bernini LJ, Simão ANC, de Souza CH, Alfieri DF, Segura LG, Costa GN, Dichi I (2018) Effect of Bifidobacterium lactis HN019 on inflammatory markers and oxidative stress in subjects with and without the metabolic syndrome. Br J Nutr 120(6):645–652
Calcinaro F, Dionisi S, Marinaro M, Candeloro P, Bonato V, Marzotti S, Corneli RB, Ferretti E, Gulino A, Grasso F, De Simone C (2005) Oral probiotic administration induces interleukin-10 production and prevents spontaneous autoimmune diabetes in the non-obese diabetic mouse. Diabetologia 48(8):1565–1575
Lavasani S, Dzhambazov B, Nouri M, Fåk F, Buske S, Molin G, Thorlacius H, Alenfall J, Jeppsson B, Weström B (2010) A novel probiotic mixture exerts a therapeutic effect on experimental autoimmune encephalomyelitis mediated by IL-10 producing regulatory T cells. PLoS ONE. https://doi.org/10.1371/journal.pone.0009009
Garidou L, Pomie C, Klopp P, Waget A, Charpentier J, Aloulou M, Giry A, Serino M, Stenman L, Lahtinen S, Dray C, Iacovoni JS, Courtney M, Collet X, Amar J, Servant F, Lelouvier B, Valet P, Eberl G, Fazilleau N, Douin-Echinard V, Heymes C, Burcelin R (2015) The gut microbiota regulates intestinal CD4 T cells expressing RORγt and controls metabolic disease. Cell Metab 22(1):100–112
Topol I, Kamyshny A (2013) Study of expression of TLR2, TLR4 and transckription factor NF-kB structures of galt of rats in the conditions of the chronic social stress and modulation of structure of intestinal microflora. Georgian Med News 12(225):115–120
Ganesan K, Chung SK, Vanamala J, Xu B (2018) Causal relationship between diet-induced gut microbiota changes and diabetes: a novel strategy to transplant Faecalibacterium prausnitzii in preventing diabetes. Int J Mol 19(12):3720
Bagarolli RA, Tobar N, Oliveira AG, Araújo TG, Carvalho BM, Rocha GZ, Vecina JF, Calisto K, Guadagnini D, Prada PO, Santos A (2017) Probiotics modulate gut microbiota and improve insulin sensitivity in DIO mice. J Nutr Biochem 50:16–25
Trapecar M, Goropevsek A, Gorenjak M, Gradisnik L, Rupnik MS (2014) A co-culture model of the developing small intestine offers new insight in the early immunomodulation of enterocytes and macrophages by Lactobacillus spp. through STAT1 and NF-kB p65 Translocation. PLoS ONE. https://doi.org/10.1371/journal.pone.0086297
Li Z, Yang S, Lin H, Huang J, Watkins PA, Moser AB et al (2003) Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatol 37(2):343–435
Reichold A, Brenner SA, Spruss A, Förster-Fromme K, Bergheim I, Bischoff SC (2014) Bifidobacterium adolescentis protects from the development of nonalcoholic steatohepatitis in a mouse model. J Nutr Biochem 25(2):118–125
Funding
This study did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bhardwaj, R., Singh, B.P., Sandhu, N. et al. Probiotic mediated NF-κB regulation for prospective management of type 2 diabetes. Mol Biol Rep 47, 2301–2313 (2020). https://doi.org/10.1007/s11033-020-05254-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-020-05254-4