
Abstract
PTEN (Phosphatase and tensin homolog deleted on chromosome ten) is a tumor suppressor that is frequently mutated in most human cancers. PTEN is a lipid and protein phosphatase that antagonizes PI3K/AKT pathway through lipid phosphatase activity at the plasma membrane. More recent studies showed that, in addition to the putative role of PTEN as a PI(3,4,5)P3 3-phosphatase, it is a PI(3,4)P2 3-phosphatase during stimulation of class I PI3K signaling pathway by growth factor. Although PTEN tumor suppressor function via it’s lipid phosphatase activity occurs primarily in the plasma membrane, it can also be found in the nucleus, in cytoplasmic organelles and extracellular space. PTEN has also shown phosphatase independent functions in the nucleus. PTEN can exit from the cell through exosomal export or secretion and has a tumor suppressor function in adjacent cells. PTEN has a critical role in growth, the cell cycle, protein synthesis, survival, DNA repair and migration. Understanding the regulation of PTEN function, activity, stability, localization and its dysregulation outcomes and also the intracellular and extracellular role of PTEN and paracrine role of PTEN-L in tumor cells as an exogenous therapeutic agent can help to improve clinical conceptualization and treatment of cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
PTEN was first identified in 1997 as a tumor suppressor gene on human chromosome 10q23. High susceptibility of PTEN gene to mutation and loss of its normal function is frequently found in a variety of cancers [1, 2]. PTEN is a dual-specificity phosphatase that has both protein phosphatase and lipid phosphatase activity [3, 4]. On the one hand, PTEN as a tumor suppressor is capable of dephosphorylation of the lipid second messenger PIP3 (phosphatidylinositol(3,4,5)-trisphosphate) and creates PIP2 (phosphatidylinositol(4,5)bi-phosphate). PTEN inhibits the PI3K/AKT signaling pathway by Hydrolyzing PIP3 to PIP2 and prevents PIP3 membrane recruitment and stimulation of AKT [5]. Therefore, loss of PTEN phosphatase activity leads to activating cell survival, growth, and proliferation [6, 7]. On the other hand, PTEN protein phosphatase activity is demonstrated and can dephosphorylate phosphopeptides at tyrosine, serine, and threonine sites [8]. PTEN prevents cellular migration and controls cell adhesion by protein phosphatase activity and interacts with FAK and Shc (Src-homologous collagen) [9, 10]. Secretion of hepatitis C virus particles in the liver is regulated by protein phosphatase activity of PTEN, most likely through regulation of cholesterol metabolism [11]. Additionally, the lipid phosphatase activity of PTEN is promoted by its auto-dephosphorylation at serine and/or threonine residues through its protein phosphatase activity [12, 13]. Despite cytosol, PTEN can also be found in specific cellular compartments and is involved in PI3K/AKT-independent activities [14]. Nuclear PTEN plays an important role in chromosome stability, DNA repair and apoptosis by phosphatase-independent tumor suppressive functions [4, 15]. In addition, recent evidence suggests that PTEN is able to exit from cell to intercellular space [16, 17]. Expression level, stability and enzymatic activity of PTEN are important and are regulated by transcriptional, post-translational and protein–protein interactions [18] .
PTEN structure
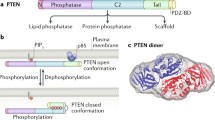
PTEN has nine exons and 1209 nucleotides that encode for a 403-amino acid protein [19]. PTEN protein structure consists of two main functional domains: a phosphatase domain, a C2 domain and three structural domains: an N-terminal PBD (phosphatidylinositol-4,5-bisphosphate-binding domain), a C terminal tail (C-tail), and a PDZ-B [20]. N-terminal PIP2-binding domain plays a role in both cellular localization and catalytic activity of PTEN [21, 22]. The amino acid sequence of PTEN contains the active site sequence motif HCxxGxxR which is a landmark of protein tyrosine and phosphatase (PTPase) superfamily [2]. Amino acids 1–185 form the N-terminal phosphatase domain that is shown with the catalytic core [23]. The N-terminal region of PTEN is homologous to auxilin and tensin. Auxilin is well known for its roles in the uncoating of clathrin-coated vesicles. Tensin as a focal adhesion protein binds to actin filaments through its actin-binding domains [24, 25]. Amino acids 186–403 form the C-terminal domain contains C2 domain (amino acids 186–351) and the C-terminal tail. PTEN C2 domain has the ability to bind to membrane phospholipid. C2 domain found in many protein structures is involved in membrane localization and binding to phospholipid bilayer [23]. The PTEN C2 domain binds to phospholipid without the canonical loops which is necessary for binding Ca2+. This procedure, unlike other signaling proteins that possess C2 domain, binds to the membrane in Ca2+ independent manner [20]. C-terminal tail contains the PDZ domain which has an essential role in protein–protein interaction [26] (Fig. 1).

Structure of PTEN
The PI3K/PTEN/AKT pathway
PI3K phosphorylates phosphatidylinositol (4,5)-bisphosphate to phosphatidylinositol (3,4,5)-trisphosphate in response to growth factor stimulation by binding to tyrosine kinase receptors and Ras at the plasma membrane [27]. Some of the proteins with pleckstrin homology domain, such as AKT are activated in reaction to second messenger PIP3. AKT activation stimulates survival, growth, and proliferation [28, 29]. PI3K/AKT pathway is negatively regulated by PTEN as a tumor suppressor through dephosphorylation of PIP3. PTEN antagonizes PI3K/AKT signaling pathway and its function loss leads to increasing PI3K/AKT pathway activity. As a consequence, PTEN alterations can induce tumorigenesis and other disease [30] .Vivanco et al demonstrated that in addition to AKT, the JNK (Jun-N-terminal kinase) also could be stimulated in response to growth factors as a PI3K effector. AKT and JNK are complementary signals with the parallel function in PIP3-driven tumorigenesis. JNK is a PTEN-regulated pathway and its activity is increased in PTEN null cells compared to PTEN positive cells in an AKT-independent manner. Thus, clinically JNK inhibitors accompanied by AKT inhibitors may provide more potent therapeutic effects on PTEN null cancer cells [31].
PTEN as a PI(3,4)P2 phosphatase
The main function of PTEN has been associated with its tumor suppressor ability through dephosphorylation of phosphatidylinositol (3,4,5)-trisphosphate (PIP3) and inhibition of AKT activity. However, recent studies demonstrated that PTEN can also dephosphorylate phosphatidylinositol (3,4)-bisphosphate (PIP2) during stimulation of class I PI3K signaling pathway by growth factor [32]. Loss of PTEN like INPP4B alone has no noticeable effect on PI(3,4)P2 accumulation. But, PTEN loss together with INPP4B loss has significant effects on PI(3,4)P2 accumulation upon epidermal growth factor (EGF) stimulation that leads to hyperactivation of AKT. In contrary with the previous study which showed that an increase in PI(3,4)P2 level leads to a reduction in phosphorylation of AKT. Therefore, in addition to the putative role of PTEN as a PI(3,4,5)P3 3-phosphatase, it is a PI(3,4)P2 3-phosphatase, suggesting that PTEN together with INPP4B regulates PI(3,4)P2 levels within EGF stimulation, and they compensate each other [33]. Likely, PTEN has a wide effect on the biology of cells by controlling of PI(3,4)P2 levels. PI(3,4)P2 and PI(3,4,5)P3 bind AKT with a similar affinity. Distortion in class I PI3K signaling pathway will occur as a result of accumulations of a large amount of PI(3,4)P2 through a quantitative effect on common PI(3,4)P2 and PI(3,4,5)P3 effectors activation such as AKT [34]. Previous studies have shown that PI(3,4)P2 also has a main role in many endocytic processes, membrane ruffling and invadopodia formation through activation of specific PI(3,4)P2 effectors [34, 35]. Effects of PI(3,4)P2 accumulations are context dependent [32], however, in the PTEN-dependent tumor, possible involvements and effects of PI(3,4)P2-specific processes demand more investigation.
Genetic alteration, transcriptional and posttranscriptional regulation of PTEN
Various genetic alterations and molecular mechanisms can imply the loss of PTEN function partially or completely in many types of cancer [36,37,38]. PTEN can be lost or inactivated by complete Allelic losses, point mutations or truncation mutations. Epigenetic alteration also causes PTEN silencing through hypermethylation or mutation of PTEN promoter region [18, 23]. Numerous genes can positively or negatively control PTEN gene expression in a variety of cell types. Positive regulators contain early growth response protein 1 (EGR-1) [39], peroxisome proliferator activated receptor γ (PPARγ) [40], P53 [41], ATF2 [42] and Myc [43]. NFκB [44], c-Jun [45], HES-1 [43], and TGFβ signaling [46] negatively regulate PTEN transcription. EGR-1, an initial member of the transcription factors has been shown to affect PTEN gene expression, upregulates PTEN transcription at the beginning of the apoptotic pathway by binding to PTEN promoter in reaction to Insulin-like growth factor-2 (IGF-2) stimulation or radiation [39, 47]. Insulin-like growth factor-1 (IGF-1) affects PTEN mRNA upregulation through EGR1 phosphorylation by binding to IGF-1R and activation of AKT. This leads to activation and migration of EGR1 to the nucleolus. EGR1 sumoylation occurs through an ARF-dependent mechanism in the nucleolus. It is proved that the deletion of EGR1 or ARF in tumor cells leads to the reduction of PTEN [48]. A previous study demonstrated that IGF-1 could suppress PTEN’s phosphorylation, resulting in the upregulation of cell proliferation and invasiveness [49]. p53 regulates PTEN gene transcription by binding to PTEN and form a regulatory complex between PTEN and p53 [41, 50]. PTEN gene expression can also be upregulated by activation of PPARγ which works as an anti-inflammatory and anti-tumor transcription factor through binding to two sites of PTEN promoter, PPAR response element 1 (PPRE1) and PPAR response element 2 (PPRE2) [40]. Resistin, a cytokine involved in inflammatory and insulin resistance, activates p38 MAPK pathway in aortic vascular endothelial cells. p38 activation results in phosphorylation and activation of ATF2, then ATF2 binds to PTEN promoter and increases PTEN expression [42]. MKK4 can repress PTEN transcription by activation of NF-kB, a transcriptional suppressor of PTEN [44]. The proto-oncogene c-JUN, one of the AP-1 family of the transcription factors, promotes resistance to apoptosis and progresses cancer via binding to a variant AP-1 site found in the 5′ upstream sequences of PTEN promoter and suppress PTEN transcription [45]. Despite PTEN mutation occurring rarely in pancreatic cancers, TGF-β could suppress PTEN expression [46]. Ecotropic virus integration site 1 (EVI-1) has an important role in hematopoietic stem cells proliferation from bone marrow cells. EVI-1 can also suppress PTEN transcription via binding directly to the PTEN promoter. Overexpression of EVI-1 has been reported, especially in acute myeloid leukemia [51]. NOTCH1 regulatory function on PTEN transcription depends on the cellular context and tissue specificity could be both negative and positive. Active NOTCH1 might increase PTEN expression through interacting with MYC [43] and CBF-1 transcription factor [52, 53] and decrease in PTEN expression through mechanisms involving the HES-1 transcription factor [43]. However, these results propose that transcriptional regulation of PTEN has a complex network of agents as tumor suppressors or oncogenes with a feedback loop that could affect PTEN protein levels concomitant with alteration of Akt activation [18]. miRNAs are small noncoding single-stranded RNAs (20–25 nucleotide) that modulate gene expression. miRNAs could post-transcriptionally repress gene expression through base-pairing with target mRNAs [54,55,56]. It has been demonstrated that miRNAs could affect PTEN expression through PTEN mRNA silencing and protein level reduction in multiple human cancers [57, 58] (Fig. 2). A large number of miRNAs involved in PTEN expression have been discovered, but Researches are continuing to find the new miRNAs. Some of these newly discovered miRNA still have unclear functions. Direct miRNAs targeting might be a major step toward cancer treatment.

The conformational regulation of PTEN. Phosphorylation of the C-tail domain of PTEN promotes an interaction between this acidic tail and the C2 domain, and this conformation (‘closed’ conformation) masks the membrane binding of PTEN. In the ‘open’ conformation, the basic N terminus binds to the acidic surface of the membrane and the PDZ—binding domain interacts with the PDZ domain—containing proteins in the membrane
Various post-translational modifications consist of phosphorylation [26], oxidation [59], acetylation [60], ubiquitination [61] and SUMOylation [62] can regulate PTEN activity and function (Fig. 3).

Post-translational modifications of PTEN. PTEN is subject to several post-translational modifications including phosphorylation, oxidation, acetylation, ubiquitination and SUMOylation. Phosphorylation of multiple sites on the C-terminal region of PTEN affects protein stability, phosphatase activity and protein–protein interactions. Oxidation of PTEN at Cys124 leads to the formation of a disulfide bond with Cys71 resulting in decreased PTEN activity. PTEN is also acetylated at Lys125 and Lys128 by PCAF and at Lys402 by CBP. Ubiquitination of PTEN at Lys13 and Lys 289 by NEDD4-1, XIAP, and WWP2 regulates PTEN stability and cellular localization. PTEN SUMOylation at K254 and K266 is critical for PTEN tumor suppressive functions
Phosphorylation of PTEN
Phosphorylation of C2 domain and C-terminal tail of PTEN regulate PTEN activity and modulate its function. Phosphorylation of PTEN mainly at Thr366, Ser370 and a cluster containing Ser380, Thr382, Thr383 and Ser385 in the C-terminal tail can lead to C-terminal tail interacting with the N-terminal C2 and phosphatase domains [26, 63]. During phosphorylation, the conformational change of PTEN to “closed” state, is associated with deactivity of PTEN and increased protein stability, (Fig. 4). Mutation of these phosphorylation sites to the nonphosphorylatable alanine leads to the conformational change of PTEN to “open” state and consequently, increase in membrane affinity, catalytic activity, more instability and rapid degradation of PTEN [26]. It was suggested that the lipid phosphatase function of PTEN can be controlled by its auto-inhibitory mechanism through phosphorylation of the C-terminal tail. In fact, normal biological activities of PTEN have a balance between phosphorylation and dephosphorylation of PTEN [64]. Previous evidence shows that PTEN can be phosphorylated by several kinases. Casein kinase 2 (CK2) plays a main role in the phosphorylation of PTEN [65] at Ser370, Ser380, Thr382, Thr383, and Ser385 [63]. Glycogen synthase kinase-3β (GSK3β) can phosphorylate PTEN at Ser362 and Thr366. Phosphorylation of PTEN by GSK3β, as part of the negative feedback loop of the PI3K signaling pathway, can control PTEN and PI3K activity [63, 66, 67]. Interestingly, phosphorylation at Ser370 by CK2 leads to enhanced phosphorylation of Thr366 by GSK3, which suggests phosphorylation at specific sites can be prime phosphorylation at distant sites [64, 65]. Ataxia telangiectasia mutated (ATM) can phosphorylate SUMOylated PTEN at T398 in genotoxic stress [68]. PTEN can be inactivated through phosphorylation on Ser385 by Liver kinase B1 (LKB1) [69]. PTEN can also be translocated to the membrane by the RhoA-associated kinase (ROCK) through phosphorylation at Thr223, Ser229, Thr319 and Thr321 in the C2 domain in chemoattractant stimulated leukocytes by unknown mechanisms [70]. Unexpectedly, PTEN is inactivated by p110, catalytic subunit of PI3K kinase, via a pathway involving RhoA and ROCK which decrease the activity of PTEN and increase tyrosine phosphorylation of PTEN [71] A Src family tyrosine kinase, FRK (Fyn-related kinase also known as RAK), targets PTEN on Tyr336 and promotes the phosphorylation and stability of PTEN through preventing PTEN from binding to the E3 ubiquitin ligase NEDD4-1 (neural precursor cell expressed developmentally downregulated protein 41) and protecting PTEN from polyubiquitination and proteasomal degradation [61, 71, 72]. The site of phosphorylation and identity of the kinase, play an important role in PTEN activity and stability. Therefore, phosphorylation of PTEN’s C2 domain increases PTEN’s membrane affinity and decreases PTEN degradation, whereas phosphorylation of the C-tail domain changes PTEN’s conformation and increases PTEN stability but reduces its activity and PTEN’s membrane targeting. GLTSCR2 (glioma tumor suppressor candidate region 2 also known as PICT-1), is capable of enhancing PTEN stability through phosphorylation of PTEN at Ser380 in C-terminal [73]. Previous studies demonstrated that down regulated GLTSCR2 by RNA interference increases PTEN degradation by the proteasome and reduced PTEN phosphorylation and stability in MCF7 cells [60, 74].

Transcriptional regulation of PTEN. Transcription factors that positively regulate PTEN gene expression (blue ovals) include EGR-1, PPARγ, MYC, p53 and ATF2. Transcription factors that negatively regulate PTEN messenger RNA (mRNA) levels (purple ovals) include NFκB, c-JUN, HES-1, CBF-1, TGFB and EVI-1. NOTCH1 may be able to activate or repress PTEN transcription depending on the cellular context. CBF-1 serves as a switch for PTEN regulation by Notch. In the presence of Notch, CBF-1 becomes an activator of PTEN transcription. On the other hand, NOTCH1 activation has also been demonstrated to repress PTEN transcription through the HES-1 transcription factor. miRNAs could be affected PTEN expression trough PTEN mRNA silencing and protein levels reduction and miR-21 was identified as the first microRNA to regulate the expression of PTEN. (Color figure online)
Oxidation and acetylation of PTEN
PTEN by having a high reactive catalytic site cysteine has catalytic activity as a protein tyrosine phosphatase which is sensitive to oxidation [75, 76]. Reactive oxygen species (ROS) can reversibly oxidize cysteine124 and decrease PTEN phosphatase activity by creating a link between Cys124 and Cys71 through the disulfide bond [59, 77]. Reversible cysteine oxidation by hydrogen peroxide (H2O2) can inactivate PTEN. Thioredoxin reduces the H2O2-oxidized cysteine residues and inactivation of PTEN that occurs following oxidation [78]. Also, direct interaction between PTEN and peroxiredoxin I (PRDX1) prevents to forming the disulfide bond. Indeed, PRDX1 protects PTEN from oxidation by forming PTEN–PRDX1 complex results in preventing to PTEN inactivation [79]. Thioredoxin-interacting protein (TXNIP) is an endogenous inhibitor of thioredoxin that modulates thioredoxin activity and subsequently reactivates oxidized PTEN and antagonize the PI3K/AKT signaling pathway [80]. PTEN inactivation by oxidative stress can indirectly occur through regulation of PTEN interaction proteins. Parkinson protein 7 (PARK7, DJ-1) binds PTEN under oxidative stress conditions resulting in inhibiting its activity and an increase in AKT activation [81]. Therefore, the increased levels of intracellular ROS in various tumor cells can cause oxidation-driven inactivation of PTEN resulting in activation of the PI3K/AKT signaling pathway. Treatment with ROS scavengers can enhance PTEN activity in T cell acute lymphoblastic leukemia cells.
Previous researches showed that acetylation can regulate the catalytic activity of PTEN. Acetylation of PTEN at Lys125 and 128 in the catalytic cleft in response to growth factors occurs through interaction between the histone acetyltransferase PCAF (p300/CREB-binding protein (CBP)-associated factor also called KAT2B) and PTEN which reduces PTEN catalytic activity and enhances AKT phosphorylation [60]. Acetylation of the PDZ binding domain of PTEN at Lys402 by CREB-binding protein can also regulate PTEN activity through increased communication and binding PDZ domain related proteins to PTEN [82, 83]. Reversely, PTEN can be deacetylated by the histone deacetylase sirtuin SIRT1 [84, 85].
Ubiquitylation of PTEN
PTEN downregulation through the ubiquitin/proteasome pathway is another mechanism that can also affect PTEN protein levels. Ubiquitylation of PTEN at Lys13 and 289 sites by NEDD4-1, which is the first identified E3 ubiquitin ligase involved in PTEN ubiquitylation, can help in degradation of PTEN, nuclear-cytoplasmic shuttling of PTEN and inhibition of phosphatase activity. Polyubiquitylation of PTEN by NEDD4-1 results in degradation and missing its tumor-suppressor activity, however, monoubiquitylation of PTEN causes nuclear import, genomic stability and cell cycle arrest [61, 86]. A previous study showed that despite the loss of NEDD4-1 in NEDD4-1 knockout cells, PTEN protein levels and localization do not change, suggesting that other E3 ligases may contribute to PTEN ubiquitylation [87, 88]. Other E3 ligases, WWP2 (WW domain containing protein 2) [89], X-linked inhibitor of apoptosis (XIAP) [90], CHIP (Carboxyl terminus of Hsc70 interacting protein) [91], SPOP [92] have been identified which mediate ubiquitination and degradation of PTEN. E3 ligase RFP (Ret finger protein also called TRIM27) can also ubiquitylate PTEN at various lysine sites and inhibit its phosphatase activity with no change in PTEN location and stability [93, 94]. Lys 13 and 289 have been identified as monoubiquitylation sites of PTEN that are important for cytoplasmicnuclear shuttling [86, 95]. Studies have shown that context-dependent regulation of PTEN by various E3 ligases to achieve specific functions is possible [87]. A recent study identified Lys 66 as a new site of PTEN ubiquitylation. This site has a major role in the stability and polyubiquitylation of PTEN in comparison with other previously recognized sites (Lys 13 and 289) in many cell types. Mutation of Lys 66 leads to significant enhanced PTEN stability while combined mutation of Lys 13 and 289 affect slightly [96]. Regulation of PTEN protein stability has been widely studied, but detailed information about the mechanism of controlling is in infancy. As discussed above, although it is clear that the phosphorylation state of PTEN and PTEN-interacting proteins plays a critical role in PTEN protein stability [26, 97] but ubiquitin-mediated proteasomal degradation of PTEN has a dominant role. PTEN contains two PEST sequences, a landmark of short half-life proteins degraded through ubiquitination [98]. Disorders in controlling PTEN protein stability may lead to decreasing PTEN protein levels. Inhibition of proteasomes function as a therapeutic way can improve protein level and stability in many cell types. HAUSP (herpesvirus associated ubiquitin specific protease also known as USP7) as a deubiquitylase can reverse monoubiquitylation of PTEN and prevent the nucleus transportation of PTEN [99, 100]. In Acute promyelocytic leukemia (APL) and prostate cancer, inhibition of HAUSP by promyelocytic leukemia results in the absence of nuclear PTEN and promotes aggressive tumors [100]. Other ubiquitin proteases, such as OTUD3 [101] and USP13 [102] have been identified to be able to deubiquitinate PTEN.
Sumoylation of PTEN
SUMOs (Small ubiquitin like modifiers), or SUMOylation, are able to regulate PTEN activity through covalent attachment of related proteins to C2 domain of PTEN at Lys254 and 266 sites. Covalent modification of PTEN at Ly266 leads to an increase in membrane affinity. Consequently, PTEN binds to PIP3 resulting in downregulation PI3K/AKT pathway and suppressing cell proliferation and tumor progression [62]. SUMOlyation at Lys254 regulates the nuclear localization of PTEN and contributes to DNA repairing mechanism. Therefore, the existence of PTEN in the nucleus is important to decrease sensitivity to DNA damage in cells [68].
PTEN regulation by protein–protein interactions
PTEN from gene to protein at all levels, including transcriptional, translational and post-translational is regulated. PTEN activity also regulated through interaction with other proteins [18]. Some researchers have shown that PTEN protein levels and activities can be regulated by several PTEN-interacting proteins through binding to PTEN. These interactions can affect the tumor suppressor functions of PTEN through alteration in conformation, location and stability of PTEN. MC1R is one of the PTEN-interacting proteins, which increases PTEN stability through binding to PTEN and preventing PTEN ubiquitylation and degradation by the E3 ligase WWP2 in melanocytes [103]. In the same way, PTEN ubiquitylation by NEDD4-1 can be inhibited by FRK, a tyrosine kinase that phosphorylates PTEN, probably through preventing the binding of NEDD4-1 to PTEN [72]. Deletion of NEDD4-1 and FRK has been revealed in various cancers [104,105,106]. PTEN-interacting proteins can also influence PTEN function and activity through the regulation of PTEN localization. Scaffolding proteins such as β-arrestins and membrane associated guanylate kinase inverted 2 (MAGI2), which are stimulated by ROCK, increase PTEN membrane localization resulting in activating PTEN phosphatase activity by binding to it [98, 104, 107]. Adaptor protein NHERF (Na+/H+ exchanger regulatory factor also called SLC9A3R1) recruits PTEN to platelet-PDGFR through interaction between PTEN PDZ-domain and NHERF at the membrane and forming a ternary complex with the PDGFR. Stimulation of PDGFR as a part of a ternary complex including NHERF, PDGFR and PTEN can limit activation of the PI3K–AKT pathway [108]. Interestingly, NHERF1 interacts with PHLPP1 (PH domain leucine-rich repeat protein phosphatase 1) and reduces AKT activity by a phosphatase function in a PTEN independent manner [109]. The motor protein myosin V binds to PTEN directly and regulates its movement to the membrane resulting in enhancing PTEN activity by converting PIP3 to PIP2 [110]. The interaction between mammalian DLG1 (disks large homologue 1) and PTEN exhibited increased PTEN tumor suppressor function and axonal stimulation of myelination in Schwann cells. DLG1–PTEN interactions probably inhibit PTEN degradation and enhance its stability [111]. PI3K/AKT pathway can be downregulated through p85, the regulatory subunit of PI3K associated with PTEN. The interaction between p85 and PTEN is stimulated by EGF resulting in activation of PTEN lipid phosphatase function [112, 113]. Previous studies reported that mutation in p85 gene reveals disruption in p85 binding to PTEN and increases PIP3 levels and AKT phosphorylation [114]. Microtubule-associated Ser/Thr kinase 2 (MAST2) also binds to the PDZ binding motif of PTEN and negatively regulates neuronal survival pathways through increasing PTEN phosphorylation and changing its intracellular transference [115, 116]. Other PTEN regulators that influence its lipid phosphatase activity and function through forming a complex with PTEN including PREX2a (PIP3 dependent RAC exchanger factor 2a), [117] SIPL1 (Shank-interacting protein-like 1 also known as SHARPIN), which is a part of the NF-κB-stimulating linear ubiquitin chain gathering complex, [118] and MAN2C1 (α-mannosidase 2C1) [119].
PTEN localization
Firstly, researches revealed that PTEN localizes between the cytosol and the plasma membrane and negatively controls intracellular levels of PIP3 and downregulates PI3K-AKT pathway via converting PIP3 to PIP2 [120, 121]. But recent studies showed that PTEN can be found in cytoplasmic organelles, for example, the nucleus, nucleolus, the mitochondria, the Mitochondria-associated membranes (MAMs) and the Endoplasmic reticulum (ER) [86]. Recently, several studies discovered that PTEN is also able to exit the cell and present in the extracellular matrix and uptake by receiver cells [16, 17].
Plasma membrane PTEN
PTEN binds to the plasma membrane and inhibits the PI3K/AKT pathway signaling through phosphatase activity and converting PIP3 to PIP2. Different elements regulate PTEN association with the plasma membrane and PIP3 access [122, 123]. Main residues of the C2 domain are necessary for connecting PTEN and plasma membrane [20]. Moreover, the N-terminal PIP2-binding motif of PTEN can recruit PTEN to the plasma membrane in response to PIP2 and PIP3 gradients [122, 123]. The PDZ-binding motif of PTEN can also drive PTEN to the plasma membrane while various membrane-anchored PDZ proteins are bound to it [124, 125]. Additionally, the C-terminal tail plays an important role in the recruitment of PTEN to the plasma membrane as a flexible part in the opened conformation of PTEN [26, 126].
Nuclear PTEN
It has been demonstrated that the nucleus is another location of PTEN. PTEN presence in the nucleus has a critical role as a tumor suppressor independent of its phosphatase activity and loss of PTEN associated with tumorigenesis. PTEN entrance to the nucleus is controlled by NEDD4-mediated monoubiquitylation while PTEN is deubiquitinated and eliminated by HAUSP in the nucleus. HAUSP-mediated PTEN deubiquitination causes nuclear elimination [86, 100]. Recently, it has been reported that sumoylation also maintains nuclear PTEN subpopulation [68]. PTEN conserves genomic stability through interacting with the CENP-C (Centromere-specific binding protein C). This interaction is necessary for the stability of centromere and stimulates the expression of RAD51, which has a critical role in DNA repair in double strand break (DSB) [127]. Additionally, the cell cycle progression is regulated by nuclear PTEN through interaction with Anaphase-promoting complex/cyclosome (APC/C), an E3 ubiquitin ligase, which increases APC/C activity and affinity to its activator CDH1 (also known as FZR1). PTEN helps the formation of APC/C–CDH1 complex [128, 129] and induces G0–G1 arrest by decreasing cyclin D1 levels through APC/C–CDH1-mediated protein degradation [15]. These functions explain that PTEN-mediated tumor suppression can be independent on its phosphatase activity [130]. In early studies, it is observed that nuclear PTEN is mostly found in primary, differentiated and quiescent cells and decreased in the nucleus of cancer cells by rapidly dividing and cycling, [131, 132]. This point indicates that PTEN location may be dependent on the cell cycle. Therefore, PTEN expression level in the nucleus in G0–G1 phase is higher than S phase [133]. This finding implies that nuclear PTEN deficiency can be involved in aggressive cancers [132, 134, 135]. Recently, researchers have reported that major vault protein (MVP) as a carrier molecule is involved in PTEN entrance to the nucleus [136]. PTEN interaction with MVPs in a calcium-dependent manner resulted in PTEN entrance to the nucleus [137], whereas other data propose that PTEN nuclear import occurs through passive diffusion [138]. Additionally, the entry of PTEN in to the nucleus needs importins and Ran-GTPase-dependent pathway activity [139]. The first 32 amino acid residues of PTEN, Which are essential for its membrane targeting due to including PIP2-binding domain, also include a functional nuclear localization signal [139, 140]. PTEN, despite the existence of pools of PIP3 and activated PI3K in the nucleus, PTEN is not involved in PI3K/AKT pathway, suggesting PTEN functions mainly beyond its lipid phosphatase activity [134, 141].
Cytoplasmic organelles PTEN
In recent years, an alternate translation of PTEN was discovered, named PTEN-α or PTEN-Long or PTEN-L, which is translated from different initiation sites at a CUG site in the 5′ untranslated region (5′ UTR) of PTEN mRNA. This alternative CUG start codon adds 173 additional amino-terminal amino acids and generates an N-terminally extended form of PTEN, which is membrane-permeable and is able to associate with intracellular membrane-containing organelles. PTEN-L localizes to the cytoplasm and the mitochondria [142]. Mitochondrial localization of PTEN is important to the conservation of mitochondrial structure and cooperates with canonical PTEN to regulate mitochondrial bioenergetic functions [95]. Additionally, the accumulation of PTEN in the mitochondria can contribute to apoptosis through sustaining ROS production. Increased PTEN level in mitochondria is detected in primary rat hippocampal neurons in response to using staurosporine which is an apoptosis inducer [95, 143]. Also, studies revealed that ER and MAMs are other PTEN localized cytoplasmic organelles [95]. Calcium (Ca2+) released from the ER is controlled by PTEN [144,145,146]. Ca2+ entrance to the intracellular space occurs through two ways: Ca2+ influx from the extracellular space and Ca2+ release from the ER. Ca2+ concentration in intracellular space has a critical role in metabolism, proliferation, differentiation, and apoptosis [95, 145, 146]. Accumulation of PTEN at the ER increases the release of Ca2+ and subsequently leads to enhance mitochondrial Ca2+ overload and (Ca2+)-dependent apoptosis induction. It is shown that PTEN can control the Ca2+ release from ER by interaction with Inositol-1,4,5-trisphosphate receptors (IP3Rs) and dephosphorylates it in a protein phosphatase-dependent manner [95]. Indeed, PTEN counteracts Akt-mediated phosphorylation of IP3R3 [147,148,149,150]. ER Ca2+ release is impaired in PTEN silencing that leads to decrease in Ca2+ accumulation in the cytosol and mitochondria, thus Ca2+-mediated apoptotic induction is lessened [95].
Nucleolus PTEN
More recently, researchers identified another isoform of PTEN, which confers an additional 146 N-terminal amino acids to the canonical PTEN and is named PTEN-b. This translational variant of PTEN initiates to translate from an AUU codon, 438 bp upstream of the AUG starting site of canonical PTEN translation. This different translation type of PTEN (PTEN-b) is predominantly found in the nucleolus and prevents pre-rRNA production and affects cellular proliferation by nucleolin dephosphorylation. Taken together, it seems likely that other PTEN isoforms may exist and more studies are needed to completely understand the function of them [151].
Extracellular PTEN and paracrine roles of it in tumor microenvironment
Recently, researchers have reported that in addition to intracellular localization of PTEN-L, it is able to exit the cell. This longer form of PTEN is secreted in exosomes and microvesicles of endosomal origin and exists outside the cell [17]. NEDD4-1, an adaptor protein for the ubiquitin ligases, controls PTEN secretion in the exosomes. In addition, alternately translated region (ATR) of PTEN-L due to having a signal sequence with an accepted cleavage site permits secretion of PTEN-L [16] which is detected in human serum and plasma [152]. On the other hand, tumors change their own environment with production oncogenic growth factors which increase cancer cells growth. Adjacent cell to tumor initiating cell may also trigger a response to prevent the aberrant proliferation of tumor cells. PTEN secretion is a way that neighboring cells can respond to the aberrant proliferation of tumor cells through it. Enhancement of PTEN-L protein in histiocytes adjacent to tumor cells supports this hypothesis [17]. The secretion of intercellular factors by adjacent cells may be one of the first defense responses against tumor growth and could be helpful as diagnostic biomarkers of tumor initiation, although some tumors might be resistant to this type of response [153]. PTEN-L is membrane permeable and can be transferred to adjacent cells [17]. Consistent with this, the presence of a polyarginine motif in the PTEN-L ATR, which is similar to polybasic residues in permeable peptides, helps to PTEN-L entrance in to the cell [16].
It was shown that uptake of this PTEN variant in neighboring cells as a therapeutic agent helps tumor regression which could decrease AKT phosphorylation through antagonizing PI3K/AKT signaling pathway and reduce cell proliferation and induce apoptosis in tumor cells in in vitro and in vivo [154, 155]. Therefore, this type of PTEN may be generated in the cell for the purpose of exiting and activation in another cell as a modulating exogenous agent. Functional PTEN in the tumor microenvironment may effect on active PTEN level in tumor cells and may have a tumor-suppressive role which is observed in macrophage-like cells in the tumor microenvironment [156]. It has also been shown that PTEN reduction in the tumor microenvironment promotes tumor development in tumor cells with wild type PTEN [157, 158]. Further investigations are needed for a more precise understanding of PTEN-L activity and function comparing with canonical PTEN to improve novel therapeutic applications through manipulation of tumor microenvironments and intercellular regulation to tumor-suppressive status.
References
Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DH, Tavtigian SV (1997) Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet 15(4):356–362. https://doi.org/10.1038/ng0497-356
Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R (1997) PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275(5308):1943–1947
Myers MP, Stolarov JP, Eng C, Li J, Wang SI, Wigler MH, Parsons R, Tonks NK (1997) P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc Natl Acad Sci 94(17):9052–9057
Yin Y, Shen W (2008) PTEN: a new guardian of the genome. Oncogene 27(41):5443–5453
Maehama T, Dixon JE (1998) The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem 273(22):13375–13378
Myers MP, Pass I, Batty IH, Van der Kaay J, Stolarov JP, Hemmings BA, Wigler MH, Downes CP, Tonks NK (1998) The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc Natl Acad Sci 95(23):13513–13518
Stambolic V, Suzuki A, De La Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW (1998) Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 95(1):29–39
Leslie NR, Downes CP (2002) PTEN: The down side of PI 3-kinase signalling. Cellular Signal 14(4):285–295
Tamura M, Gu J, Matsumoto K, Aota S-i, Parsons R, Yamada KM (1998) Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science 280(5369):1614–1617
Gu J, Tamura M, Pankov R, Danen EH, Takino T, Matsumoto K, Yamada KM (1999) Shc and FAK differentially regulate cell motility and directionality modulated by PTEN. J Cell Biol 146(2):389–404
Peyrou M, Clément S, Maier C, Bourgoin L, Branche E, Conzelmann S, Kaddai V, Foti M, Negro F (2013) PTEN protein phosphatase activity regulates hepatitis C virus secretion through modulation of cholesterol metabolism. J Hepatol 59(3):420–426
Zhang XC, Piccini A, Myers MP, Van Aelst L, Tonks NK (2012) Functional analysis of the protein phosphatase activity of PTEN. Biochem J 444(3):457–464. https://doi.org/10.1042/bj20120098
Tibarewal P, Zilidis G, Spinelli L (2012) PTEN protein phosphatase activity correlates with control of gene expression and invasion, a tumor-suppressing phenotype, but not with AKT activity. Sci Signal 5:ra18
Bononi A, Pinton P (2015) Study of PTEN subcellular localization. Methods 77:92–103
Planchon SM, Waite KA, Eng C (2008) The nuclear affairs of PTEN. J Cell Sci 121(3):249–253
Putz U, Howitt J, Doan A, Goh CP, Low LH, Silke J, Tan SS (2012) The tumor suppressor PTEN is exported in exosomes and has phosphatase activity in recipient cells. Sci Signal 5(243):ra70. https://doi.org/10.1126/scisignal.2003084
Hopkins BD, Fine B, Steinbach N, Dendy M, Rapp Z, Shaw J, Pappas K, Yu JS, Hodakoski C, Mense S, Klein J, Pegno S, Sulis ML, Goldstein H, Amendolara B, Lei L, Maurer M, Bruce J, Canoll P, Hibshoosh H, Parsons R (2013) A secreted PTEN phosphatase that enters cells to alter signaling and survival. Science 341(6144):399–402. https://doi.org/10.1126/science.1234907
Salmena L, Carracedo A, Pandolfi PP (2008) Tenets of PTEN tumor suppression. Cell 133(3):403–414
Gericke A, Munson M, Ross AH (2006) Regulation of the PTEN phosphatase. Gene 374:1–9
Lee J-O, Yang H, Georgescu M-M, Di Cristofano A, Maehama T, Shi Y, Dixon JE, Pandolfi P, Pavletich NP (1999) Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell 99(3):323–334
Walker SM, Leslie NR, Perera NM, Batty IH, Downes CP (2004) The tumour-suppressor function of PTEN requires an N-terminal lipid-binding motif. Biochem J 379(2):301–307
Campbell RB, Liu F, Ross AH (2003) Allosteric activation of PTEN phosphatase by phosphatidylinositol 4, 5-bisphosphate. J Biol Chem 278(36):33617–33620
Waite KA, Eng C (2002) Protean PTEN: form and function. Am J Hum Genet 70(4):829–844
Zhao X, Greener T, Al-Hasani H, Cushman SW, Eisenberg E, Greene LE (2001) Expression of auxilin or AP180 inhibits endocytosis by mislocalizing clathrin: evidence for formation of nascent pits containing AP1 or AP2 but not clathrin. J Cell Sci 114(2):353–365
Lo SH, Weisberg E, Chen LB (1994) Tensin: a potential link between the cytoskeleton and signal transduction. Bioessays 16(11):817–823
Vazquez F, Ramaswamy S, Nakamura N, Sellers WR (2000) Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol 20(14):5010–5018
Klinghoffer RA, Duckworth B, Valius M, Cantley L, Kazlauskas A (1996) Platelet-derived growth factor-dependent activation of phosphatidylinositol 3-kinase is regulated by receptor binding of SH2-domain-containing proteins which influence Ras activity. Mol Cell Biol 16(10):5905–5914
Frech M, Andjelkovic M, Ingley E, Reddy KK, Falck JR, Hemmings BA (1997) High affinity binding of inositol phosphates and phosphoinositides to the pleckstrin homology domain of RAC/Protein Kinase B and their influence on kinase activity. J Biol Chem 272(13):8474–8481. https://doi.org/10.1074/jbc.272.13.8474
Klarlund JK, Guilherme A, Holik JJ, Virbasius JV, Chawla A, Czech MP (1997) Signaling by phosphoinositide-3, 4, 5-trisphosphate through proteins containing pleckstrin and Sec7 homology domains. Science 275(5308):1927–1930
Keniry M, Parsons R (2008) The role of PTEN signaling perturbations in cancer and in targeted therapy. Oncogene 27(41):5477–5485
Vivanco I, Palaskas N, Tran C, Finn SP, Getz G, Kennedy NJ, Jiao J, Rose J, Xie W, Loda M (2007) Identification of the JNK signaling pathway as a functional target of the tumor suppressor PTEN. Cancer cell 11(6):555–569
Malek M, Kielkowska A, Chessa T, Anderson KE, Barneda D, Pir P, Nakanishi H, Eguchi S, Koizumi A, Sasaki J, Juvin V, Kiselev VY, Niewczas I, Gray A, Valayer A, Spensberger D, Imbert M, Felisbino S, Habuchi T, Beinke S, Cosulich S, Le Novere N, Sasaki T, Clark J, Hawkins PT, Stephens LR (2017) PTEN regulates PI(3,4)P2 signaling downstream of class I PI3K. Mol Cell 68(3):566–580.e510. https://doi.org/10.1016/j.molcel.2017.09.024
Reed DE, Shokat KM (2017) INPP4B and PTEN Loss Leads to PI-3,4-P2 Accumulation and Inhibition of PI3K in TNBC. Mol Cancer Res 15(6):765–775. https://doi.org/10.1158/1541-7786.mcr-16-0183
Hawkins PT, Stephens LR (2016) Emerging evidence of signalling roles for PI(3,4)P2 in Class I and II PI3K-regulated pathways. Biochem Soc Trans 44(1):307–314. https://doi.org/10.1042/bst20150248
Li H, Marshall AJ (2015) Phosphatidylinositol (3,4) bisphosphate-specific phosphatases and effector proteins: a distinct branch of PI3K signaling. Cell Signal 27(9):1789–1798. https://doi.org/10.1016/j.cellsig.2015.05.013
Garcia JM, Silva J, Pena C, Garcia V, Rodriguez R, Cruz MA, Cantos B, Provencio M, Espana P, Bonilla F (2004) Promoter methylation of the PTEN gene is a common molecular change in breast cancer. Genes Chromosom Cancer 41(2):117–124. https://doi.org/10.1002/gcc.20062
Goel A, Arnold CN, Niedzwiecki D, Carethers JM, Dowell JM, Wasserman L, Compton C, Mayer RJ, Bertagnolli MM, Boland CR (2004) Frequent inactivation of PTEN by promoter hypermethylation in microsatellite instability-high sporadic colorectal cancers. Cancer Res 64(9):3014–3021
Kang YH, Lee HS, Kim WH (2002) Promoter methylation and silencing of PTEN in gastric carcinoma. Lab Investig 82(3):285–291
Virolle T, Adamson ED, Baron V, Birle D, Mercola D, Mustelin T, de Belle I (2001) The Egr-1 transcription factor directly activates PTEN during irradiation-induced signalling. Nat Cell Biol 3(12):1124–1128
Patel L, Pass I, Coxon P, Downes CP, Smith SA, Macphee CH (2001) Tumor suppressor and anti-inflammatory actions of PPARγ agonists are mediated via upregulation of PTEN. Curr Biol 11(10):764–768
Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, Benchimol S, Mak T (2001) Regulation of PTEN transcription by p53. Mol Cell 8(2):317–325
Shen YH, Zhang L, Gan Y, Wang X, Wang J, LeMaire SA, Coselli JS, Wang XL (2006) Up-regulation of PTEN (phosphatase and tensin homolog deleted on chromosome ten) mediates p38 MAPK stress signal-induced inhibition of insulin signaling A cross-talk between stress signaling and insulin signaling in resistin-treated human endothelial cells. J Biol Chem 281(12):7727–7736
Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, Caparros E, Buteau J, Brown K, Perkins SL (2007) Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med 13(10):1203–1210
Xia D, Srinivas H, Ahn Y-h, Sethi G, Sheng X, Yung WA, Xia Q, Chiao PJ, Kim H, Brown PH (2007) Mitogen-activated protein kinase kinase-4 promotes cell survival by decreasing PTEN expression through an NFκB-dependent pathway. J Biol Chem 282(6):3507–3519
Hettinger K, Vikhanskaya F, Poh M, Lee M, De Belle I, Zhang J-T, Reddy S, Sabapathy K (2007) c-Jun promotes cellular survival by suppression of PTEN. Cell Death Differ 14(2):218–229
Chow JY, Dong H, Quach KT, Van Nguyen PN, Chen K, Carethers JM (2008) TGF-β mediates PTEN suppression and cell motility through calcium-dependent PKC-α activation in pancreatic cancer cells. Am J Physiol 294(4):G899–G905
Moorehead RA, Hojilla CV, De Belle I, Wood GA, Fata JE, Adamson ED, Watson KL, Edwards DR, Khokha R (2003) Insulin-like growth factor-II regulates PTEN expression in the mammary gland. J Biol Chem 278(50):50422–50427
Yu J, Zhang SS, Saito K, Williams S, Arimura Y, Ma Y, Ke Y, Baron V, Mercola D, Feng GS, Adamson E, Mustelin T (2009) PTEN regulation by Akt-EGR1-ARF-PTEN axis. EMBO J 28(1):21–33. https://doi.org/10.1038/emboj.2008.238
Ma J, Sawai H, Matsuo Y, Ochi N, Yasuda A, Takahashi H, Wakasugi T, Funahashi H, Sato M, Takeyama H (2010) IGF-1 mediates PTEN suppression and enhances cell invasion and proliferation via activation of the IGF-1/PI3K/Akt signaling pathway in pancreatic cancer cells. J Surg Res 160(1):90–101
Freeman DJ, Li AG, Wei G, Li H-H, Kertesz N, Lesche R, Whale AD, Martinez-Diaz H, Rozengurt N, Cardiff RD (2003) PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and-independent mechanisms. Cancer Cell 3(2):117–130
Yoshimi A, Goyama S, Watanabe-Okochi N, Yoshiki Y, Nannya Y, Nitta E, Arai S, Sato T, Shimabe M, Nakagawa M (2011) Evi1 represses PTEN expression and activates PI3K/AKT/mTOR via interactions with polycomb proteins. Blood 117(13):3617–3628
Whelan JT, Forbes SL, Bertrand FE (2007) CBF-1 (RBP-Jk) binds to the PTEN promoter and regulates PTEN gene expression. Cell Cycle 6(1):80–84
Chappell WH, Green TD, Spengeman JD, McCubrey JA, Akula SM, Bertrand FE (2005) Increased protein expression of the PTEN tumor suppressor in the presence of constitutively active Notch-1. Cell Cycle 4(10):1389–1395
Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136(2):215–233. https://doi.org/10.1016/j.cell.2009.01.002
Meng F, Henson R, Lang M, Wehbe H, Maheshwari S, Mendell JT, Jiang J, Schmittgen TD, Patel T (2006) Involvement of human micro-RNA in growth and response to chemotherapy in human cholangiocarcinoma cell lines. Gastroenterology 130(7):2113–2129. https://doi.org/10.1053/j.gastro.2006.02.057
Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T (2007) MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 133(2):647–658. https://doi.org/10.1053/j.gastro.2007.05.022
Hong L, Lai M, Chen M, Xie C, Liao R, Kang YJ, Xiao C, Hu WY, Han J, Sun P (2010) The miR-17-92 cluster of microRNAs confers tumorigenicity by inhibiting oncogene-induced senescence. Cancer Res 70(21):8547–8557. https://doi.org/10.1158/0008-5472.can-10-1938
Jin HY, Oda H, Lai M, Skalsky RL, Bethel K, Shepherd J, Kang SG, Liu WH, Sabouri-Ghomi M, Cullen BR, Rajewsky K, Xiao C (2013) MicroRNA-17 ~ 92 plays a causative role in lymphomagenesis by coordinating multiple oncogenic pathways. EMBO J 32(17):2377–2391. https://doi.org/10.1038/emboj.2013.178
Lee S-R, Yang K-S, Kwon J, Lee C, Jeong W, Rhee SG (2002) Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem 277(23):20336–20342
Okumura K, Mendoza M, Bachoo RM, DePinho RA, Cavenee WK, Furnari FB (2006) PCAF modulates PTEN activity. J Biol Chem 281(36):26562–26568
Wang X, Trotman LC, Koppie T, Alimonti A, Chen Z, Gao Z, Wang J, Erdjument-Bromage H, Tempst P, Cordon-Cardo C (2007) NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell 128(1):129–139
Huang J, Yan J, Zhang J, Zhu S, Wang Y, Shi T, Zhu C, Chen C, Liu X, Cheng J, Mustelin T, Feng G-S, Chen G, Yu J (2012) SUMO1 modification of PTEN regulates tumorigenesis by controlling its association with the plasma membrane. Nat Commun 3:911. https://doi.org/10.1038/ncomms1919
Miller SJ, Lou DY, Seldin DC, Lane WS, Neel BG (2002) Direct identification of PTEN phosphorylation sites. FEBS Lett 528(1–3):145–153
Odriozola L, Singh G, Hoang T, Chan AM (2007) Regulation of PTEN activity by its carboxyl-terminal autoinhibitory domain. J Biol Chem 282(32):23306–23315. https://doi.org/10.1074/jbc.M611240200
Maccario H, Perera NM, Davidson L, Downes CP, Leslie NR (2007) PTEN is destabilized by phosphorylation on Thr366. Biochem J 405(3):439–444. https://doi.org/10.1042/bj20061837
Jang HD, Noh JY, Shin JH, Lin JJ, Lee SY (2013) PTEN regulation by the Akt/GSK-3β axis during RANKL signaling. Bone 55(1):126–131
Al-Khouri AM, Ma Y, Togo SH, Williams S, Mustelin T (2005) Cooperative phosphorylation of the tumor suppressor phosphatase and tensin homologue (PTEN) by casein kinases and glycogen synthase kinase 3beta. J Biol Chem 280(42):35195–35202. https://doi.org/10.1074/jbc.M503045200
Bassi C, Ho J, Srikumar T, Dowling R, Gorrini C, Miller S, Mak T, Neel B, Raught B, Stambolic V (2013) Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science 341(6144):395–399
Mehenni H, Lin-Marq N, Buchet-Poyau K, Reymond A, Collart MA, Picard D, Antonarakis SE (2005) LKB1 interacts with and phosphorylates PTEN: a functional link between two proteins involved in cancer predisposing syndromes. Hum Mol Genet 14(15):2209–2219
Li Z, Dong X, Wang Z, Liu W, Deng N, Ding Y, Tang L, Hla T, Zeng R, Li L (2005) Regulation of PTEN by Rho small GTPases. Nat Cell Biol 7(4):399–404
Papakonstanti EA, Ridley AJ, Vanhaesebroeck B (2007) The p110δ isoform of PI 3-kinase negatively controls RhoA and PTEN. Embo J 26(13):3050–3061
Yim E-K, Peng G, Dai H, Hu R, Li K, Lu Y, Mills GB, Meric-Bernstam F, Hennessy BT, Craven RJ (2009) Rak functions as a tumor suppressor by regulating PTEN protein stability and function. Cancer Cell 15(4):304–314
Okahara F, Itoh K, Nakagawara A, Murakami M, Kanaho Y, Maehama T (2006) Critical role of PICT-1, a tumor suppressor candidate, in phosphatidylinositol 3, 4, 5-trisphosphate signals and tumorigenic transformation. Mol Biol Cell 17(11):4888–4895
Yim JH, Kim YJ, Ko JH, Cho YE, Kim SM, Kim JY, Lee S, Park JH (2007) The putative tumor suppressor gene GLTSCR2 induces PTEN-modulated cell death. Cell Death Differ 14(11):1872–1879. https://doi.org/10.1038/sj.cdd.4402204
Ross SH, Lindsay Y, Safrany ST, Lorenzo O, Villa F, Toth R, Clague MJ, Downes CP, Leslie NR (2007) Differential redox regulation within the PTP superfamily. Cell Signal 19(7):1521–1530
Tonks NK (2005) Redox redux: revisiting PTPs and the control of cell signaling. Cell 121(5):667–670
Cho S-H, Lee C-H, Ahn Y, Kim H, Kim H, Ahn C-Y, Yang K-S, Lee S-R (2004) Redox regulation of PTEN and protein tyrosine phosphatases in H2O2-mediated cell signaling. FEBS Lett 560(1–3):7–13
Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG (2002) Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem 277(23):20336–20342. https://doi.org/10.1074/jbc.M111899200
Cao J, Schulte J, Knight A, Leslie NR, Zagozdzon A, Bronson R, Manevich Y, Beeson C, Neumann CA (2009) Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. EMBO J 28(10):1505–1517. https://doi.org/10.1038/emboj.2009.101
Hui STY, Andres AM, Miller AK, Spann NJ, Potter DW, Post NM, Chen AZ, Sachithanantham S, Jung DY, Kim JK, Davis RA (2008) Txnip balances metabolic and growth signaling via PTEN disulfide reduction. Proc Natl Acad Sci USA 105(10):3921–3926. https://doi.org/10.1073/pnas.0800293105
Kim YC, Kitaura H, Taira T, Iguchi-Ariga SM, Ariga H (2009) Oxidation of DJ-1-dependent cell transformation through direct binding of DJ-1 to PTEN. Int J Oncol 35(6):1331–1341
Ding L, Chen S, Liu P, Pan Y, Zhong J, Regan KM, Wang L, Yu C, Rizzardi A, Cheng L (2014) CBP loss cooperates with PTEN haploinsufficiency to drive prostate cancer: implications for epigenetic therapy. Cancer Res 74(7):2050–2061
Ikenoue T, Inoki K, Zhao B, Guan K-L (2008) PTEN acetylation modulates its interaction with PDZ domain. Cancer Res 68(17):6908–6912
Qu Y, Zhang J, Wu S, Li B, Liu S, Cheng J (2012) SIRT1 promotes proliferation and inhibits apoptosis of human malignant glioma cell lines. Neurosci Lett 525(2):168–172
Chae H-D, Broxmeyer HE (2010) SIRT1 deficiency downregulates PTEN/JNK/FOXO1 pathway to block reactive oxygen species-induced apoptosis in mouse embryonic stem cells. Stem Cells Dev 20(7):1277–1285
Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H, Pavletich NP, Carver BS, Cordon-Cardo C, Erdjument-Bromage H, Tempst P, Chi SG, Kim HJ, Misteli T, Jiang X, Pandolfi PP (2007) Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 128(1):141–156. https://doi.org/10.1016/j.cell.2006.11.040
Drinjakovic J, Jung H, Campbell DS, Strochlic L, Dwivedy A, Holt CE (2010) E3 ligase Nedd4 promotes axon branching by downregulating PTEN. Neuron 65(3):341–357
Fouladkou F, Landry T, Kawabe H, Neeb A, Lu C, Brose N, Stambolic V, Rotin D (2008) The ubiquitin ligase Nedd4-1 is dispensable for the regulation of PTEN stability and localization. Proc Natl Acad Sci 105(25):8585–8590
Maddika S, Kavela S, Rani N, Palicharla VR, Pokorny JL, Sarkaria JN, Chen J (2011) WWP2 is an E3 ubiquitin ligase for PTEN. Nat Cell Biol 13(6):728–733
Van Themsche C, Leblanc V, Parent S, Asselin E (2009) X-linked inhibitor of apoptosis protein (XIAP) regulates PTEN ubiquitination, content, and compartmentalization. J Biol Chem 284(31):20462–20466
Ahmed SF, Deb S, Paul I, Chatterjee A, Mandal T, Chatterjee U, Ghosh MK (2012) The chaperone-assisted E3 ligase C terminus of Hsc70-interacting protein (CHIP) targets PTEN for proteasomal degradation. J Biol Chem 287(19):15996–16006. https://doi.org/10.1074/jbc.M111.321083
Li G, Ci W, Karmakar S, Chen K, Dhar R, Fan Z, Guo Z, Zhang J, Ke Y, Wang L, Zhuang M, Hu S, Li X, Zhou L, Li X, Calabrese MF, Watson ER, Prasad SM, Rinker-Schaeffer C, Eggener SE, Stricker T, Tian Y, Schulman BA, Liu J, White KP (2014) SPOP promotes tumorigenesis by acting as a key regulatory hub in kidney cancer. Cancer Cell 25(4):455–468. https://doi.org/10.1016/j.ccr.2014.02.007
Lee JT, Shan J, Zhong J, Li M, Zhou B, Zhou A, Parsons R, Gu W (2013) RFP-mediated ubiquitination of PTEN modulates its effect on AKT activation. Cell Res 23(4):552–564
Maccario H, Perera NM, Gray A, Downes CP, Leslie NR (2010) Ubiquitination of PTEN (phosphatase and tensin homolog) inhibits phosphatase activity and is enhanced by membrane targeting and hyperosmotic stress. J Biol Chem 285(17):12620–12628
Bononi A, Bonora M, Marchi S, Missiroli S, Poletti F, Giorgi C, Pandolfi PP, Pinton P (2013) Identification of PTEN at the ER and MAMs and its regulation of Ca(2+) signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ 20(12):1631–1643. https://doi.org/10.1038/cdd.2013.77
Gupta A, Leslie NR (2016) Controlling PTEN (Phosphatase and Tensin Homolog) stability: a dominant role for lysine 66. J Biol Chem 291(35):18465–18473. https://doi.org/10.1074/jbc.M116.727750
Torres J, Pulido R (2001) The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J Biol Chem 276(2):993–998. https://doi.org/10.1074/jbc.M009134200
Tolkacheva T, Boddapati M, Sanfiz A, Tsuchida K, Kimmelman AC, Chan AM (2001) Regulation of PTEN binding to MAGI-2 by two putative phosphorylation sites at threonine 382 and 383. Cancer Res 61(13):4985–4989
Gunaratne J, Goh MX, Swa HLF, Lee FY, Sanford E, Wong LM, Hogue KA, Blackstock WP, Okumura K (2011) Protein interactions of phosphatase and tensin homologue (PTEN) and its cancer-associated G20E mutant compared by using stable isotope labeling by amino acids in cell culture-based parallel affinity purification. J Biol Chem 286(20):18093–18103
Song MS, Salmena L, Carracedo A, Egia A, Lo-Coco F, Teruya-Feldstein J, Pandolfi PP (2008) The deubiquitinylation and localization of PTEN are regulated by a HAUSP–PML network. Nature 455(7214):813–817
Yuan L, Lv Y, Li H, Gao H, Song S, Zhang Y, Xing G, Kong X, Wang L, Li Y, Zhou T, Gao D, Xiao ZX, Yin Y, Wei W, He F, Zhang L (2015) Deubiquitylase OTUD3 regulates PTEN stability and suppresses tumorigenesis. Nat Cell Biol 17(9):1169–1181. https://doi.org/10.1038/ncb3218
Zhang J, Zhang P, Wei Y, Piao HL, Wang W, Maddika S, Wang M, Chen D, Sun Y, Hung MC, Chen J, Ma L (2013) Deubiquitylation and stabilization of PTEN by USP13. Nat Cell Biol 15(12):1486–1494. https://doi.org/10.1038/ncb2874
Cao J, Wan L, Hacker E, Dai X, Lenna S, Jimenez-Cervantes C, Wang Y, Leslie NR, Xu GX, Widlund HR (2013) MC1R is a potent regulator of PTEN after UV exposure in melanocytes. Mol Cell 51(4):409–422
Lima-Fernandes E, Enslen H, Camand E, Kotelevets L, Boularan C, Achour L, Benmerah A, Gibson LC, Baillie GS, Pitcher JA (2011) Distinct functional outputs of PTEN signalling are controlled by dynamic association with β-arrestins. EMBO J 30(13):2557–2568
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6(269):pl1
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. AACR 2(5):401–404
Wu X, Hepner K, Castelino-Prabhu S, Do D, Kaye MB, Yuan X-J, Wood J, Ross C, Sawyers CL, Whang YE (2000) Evidence for regulation of the PTEN tumor suppressor by a membrane-localized multi-PDZ domain containing scaffold protein MAGI-2. Proc Natl Acad Sci 97(8):4233–4238
Takahashi Y, Morales FC, Kreimann EL, Georgescu MM (2006) PTEN tumor suppressor associates with NHERF proteins to attenuate PDGF receptor signaling. EMBO J 25(4):910–920
Molina JR, Agarwal NK, Morales FC, Hayashi Y, Aldape KD, Cote G, Georgescu M-M (2012) PTEN, NHERF1 and PHLPP form a tumor suppressor network that is disabled in glioblastoma. Oncogene 31(10):1264–1274
Van Diepen MT, Parsons M, Downes CP, Leslie NR, Hindges R, Eickholt BJ (2009) MyosinV controls PTEN function and neuronal cell size. Nat Cell Biol 11(10):1191–1196
Cotter L, Özçelik M, Jacob C, Pereira JA, Locher V, Baumann R, Relvas JB, Suter U, Tricaud N (2010) Dlg1-PTEN interaction regulates myelin thickness to prevent damaging peripheral nerve overmyelination. Science 328(5984):1415–1418
Chagpar RB, Links PH, Pastor MC, Furber LA, Hawrysh AD, Chamberlain MD, Anderson DH (2010) Direct positive regulation of PTEN by the p85 subunit of phosphatidylinositol 3-kinase. Proc Natl Acad Sci 107(12):5471–5476
Rabinovsky R, Pochanard P, McNear C, Brachmann SM, Duke-Cohan JS, Garraway LA, Sellers WR (2009) p85 Associates with unphosphorylated PTEN and the PTEN-associated complex. Mol Cell Biol 29(19):5377–5388
Taniguchi CM, Winnay J, Kondo T, Bronson RT, Guimaraes AR, Alemán JO, Luo J, Stephanopoulos G, Weissleder R, Cantley LC (2010) The phosphoinositide 3-kinase regulatory subunit p85α can exert tumor suppressor properties through negative regulation of growth factor signaling. Cancer Res 70(13):5305–5315
Terrien E, Chaffotte A, Lafage M, Khan Z, Préhaud C, Cordier F, Simenel C, Delepierre M, Buc H, Lafon M (2012) Interference with the PTEN-MAST2 interaction by a viral protein leads to cellular relocalization of PTEN. Sci Signal 5(237):ra58–ra58
Valiente M, Andrés-Pons A, Gomar B, Torres J, Gil A, Tapparel C, Antonarakis SE, Pulido R (2005) Binding of PTEN to specific PDZ domains contributes to PTEN protein stability and phosphorylation by microtubule-associated serine/threonine kinases. J Biol Chem 280(32):28936–28943
Fine B, Hodakoski C, Koujak S, Su T, Saal LH, Maurer M, Hopkins B, Keniry M, Sulis ML, Mense S (2009) Activation of the PI3K pathway in cancer through inhibition of PTEN by exchange factor P-REX2a. Science 325(5945):1261–1265
He L, Ingram A, Rybak AP, Tang D (2010) Shank-interacting protein-like 1 promotes tumorigenesis via PTEN inhibition in human tumor cells. J Clin Investig 120(6):2094–2108
He L, Fan C, Kapoor A, Ingram AJ, Rybak AP, Austin RC, Dickhout J, Cutz J-C, Scholey J, Tang D (2011) α-Mannosidase 2C1 attenuates PTEN function in prostate cancer cells. Natu Commun 2:307. https://doi.org/10.1038/ncomms1309
Vazquez F, Matsuoka S, Sellers WR, Yanagida T, Ueda M, Devreotes PN (2006) Tumor suppressor PTEN acts through dynamic interaction with the plasma membrane. Proc Natl Acad Sci USA 103(10):3633–3638. https://doi.org/10.1073/pnas.0510570103
Iijima M, Huang YE, Luo HR, Vazquez F, Devreotes PN (2004) Novel mechanism of PTEN regulation by its phosphatidylinositol 4,5-bisphosphate binding motif is critical for chemotaxis. J Biol Chem 279(16):16606–16613. https://doi.org/10.1074/jbc.M312098200
Redfern RE, Redfern D, Furgason ML, Munson M, Ross AH, Gericke A (2008) PTEN phosphatase selectively binds phosphoinositides and undergoes structural changes. Biochemistry 47(7):2162–2171
Walker SM, Leslie NR, Perera NM, Batty IH, Downes CP (2004) The tumour-suppressor function of PTEN requires an N-terminal lipid-binding motif. Biochem J 379(Pt 2):301–307. https://doi.org/10.1042/bj20031839
Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, Chaudhuri S, Guan Y, Janakiraman V, Jaiswal BS (2012) Recurrent R-spondin fusions in colon cancer. Nature 488(7413):660–664
Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B (2010) Integrative genomic profiling of human prostate cancer. Cancer Cell 18(1):11–22
Vazquez F, Grossman SR, Takahashi Y, Rokas MV, Nakamura N, Sellers WR (2001) Phosphorylation of the PTEN tail acts as an inhibitory switch by preventing its recruitment into a protein complex. J Biol Chem 276(52):48627–48630. https://doi.org/10.1074/jbc.C100556200
Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, Yin Y (2007) Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 128(1):157–170
Manchado E, Eguren M, Malumbres M (2010) The anaphase-promoting complex/cyclosome (APC/C): cell-cycle-dependent and-independent functions. Biochem Soc Trans 38(1):65–71
Wäsch R, Robbins JA, Cross FR (2010) The emerging role of APC/CCdh1 in controlling differentiation, genomic stability and tumor suppression. Oncogene 29(1):1–10
Song MS, Carracedo A, Salmena L, Song SJ, Egia A, Malumbres M, Pandolfi PP (2011) Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell 144(2):187–199
Perren A, Komminoth P, Saremaslani P, Matter C, Feurer S, Lees JA, Heitz PU, Eng C (2000) Mutation and expression analyses reveal differential subcellular compartmentalization of PTEN in endocrine pancreatic tumors compared to normal islet cells. Am J Pathol 157(4):1097–1103
Gimm O, Perren A, Weng L-P, Marsh DJ, Yeh JJ, Ziebold U, Gil E, Hinze R, Delbridge L, Lees JA (2000) Differential nuclear and cytoplasmic expression of PTEN in normal thyroid tissue, and benign and malignant epithelial thyroid tumors. Am J Pathol 156(5):1693–1700
Ginn-Pease ME, Eng C (2003) Increased nuclear phosphatase and tensin homologue deleted on chromosome 10 is associated with G0-G1 in MCF-7 cells. Cancer Res 63(2):282–286
Lindsay Y, McCoull D, Davidson L, Leslie NR, Fairservice A, Gray A, Lucocq J, Downes CP (2006) Localization of agonist-sensitive PtdIns (3, 4, 5) P3 reveals a nuclear pool that is insensitive to PTEN expression. J Cell Sci 119(24):5160–5168
Baker SJ (2007) PTEN enters the nuclear age. Cell 128(1):25–28. https://doi.org/10.1016/j.cell.2006.12.023
Chung J-H, Ginn-Pease ME, Eng C (2005) Phosphatase and tensin homologue deleted on chromosome 10 (PTEN) has nuclear localization signal–like sequences for nuclear import mediated by major vault protein. Cancer Res 65(10):4108–4116
Minaguchi T, Waite KA, Eng C (2006) Nuclear Localization of PTEN is regulated by Ca2+ through a tyrosil phosphorylation–independent conformational modification in major vault protein. Cancer Res 66(24):11677–11682
Liu F, Wagner S, Campbell RB, Nickerson JA, Schiffer CA, Ross AH (2005) PTEN enters the nucleus by diffusion. J Cell Biochem 96(2):221–234
Gil A, Andrés-Pons A, Fernández E, Valiente M, Torres J, Cervera J, Pulido R (2006) Nuclear localization of PTEN by a Ran-dependent mechanism enhances apoptosis: involvement of an N-terminal nuclear localization domain and multiple nuclear exclusion motifs. Mol Biol Cell 17(9):4002–4013
Denning G, Jean-Joseph B, Prince C, Durden D, Vogt P (2007) A short N-terminal sequence of PTEN controls cytoplasmic localization and is required for suppression of cell growth. Oncogene 26(27):3930–3940
Tanaka K, Horiguchi K, Yoshida T, Takeda M, Fujisawa H, Takeuchi K, Umeda M, Kato S, Ihara S, Nagata S (1999) Evidence that a phosphatidylinositol 3, 4, 5-trisphosphate-binding protein can function in nucleus. J Biol Chem 274(7):3919–3922
Liang H, He S, Yang J, Jia X, Wang P, Chen X, Zhang Z, Zou X, McNutt MA, Shen WH (2014) PTENα, a PTEN isoform translated through alternative initiation, regulates mitochondrial function and energy metabolism. Cell Metabol 19(5):836–848
Zhu Y, Hoell P, Ahlemeyer B, Krieglstein J (2006) PTEN: a crucial mediator of mitochondria-dependent apoptosis. Apoptosis 11(2):197–207
Marchi S, Patergnani S, Pinton P (2014) The endoplasmic reticulum–mitochondria connection: one touch, multiple functions. Biochim Biophys Acta (BBA) 1837(4):461–469
Patergnani S, Suski JM, Agnoletto C, Bononi A, Bonora M, De Marchi E, Giorgi C, Marchi S, Missiroli S, Poletti F (2011) Calcium signaling around mitochondria associated membranes (MAMs). Cell Commun Signal 9(1):19
Giorgi C, Baldassari F, Bononi A, Bonora M, De Marchi E, Marchi S, Missiroli S, Patergnani S, Rimessi A, Suski JM (2012) Mitochondrial Ca2+ and apoptosis. Cell Calcium 52(1):36–43
Giorgi C, Ito K, Lin H-K, Santangelo C, Wieckowski MR, Lebiedzinska M, Bononi A, Bonora M, Duszynski J, Bernardi R (2010) PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science 330(6008):1247–1251
Marchi S, Rimessi A, Giorgi C, Baldini C, Ferroni L, Rizzuto R, Pinton P (2008) Akt kinase reducing endoplasmic reticulum Ca2+ release protects cells from Ca2+-dependent apoptotic stimuli. Biochem Biophys Res Commun 375(4):501–505
Marchi S, Marinello M, Bononi A, Bonora M, Giorgi C, Rimessi A, Pinton P (2012) Selective modulation of subtype III IP3R by Akt regulates ER Ca2+ release and apoptosis. Cell Death Dis 3(5):e304
Szado T, Vanderheyden V, Parys JB, De Smedt H, Rietdorf K, Kotelevets L, Chastre E, Khan F, Landegren U, Söderberg O (2008) Phosphorylation of inositol 1, 4, 5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc Natl Acad Sci 105(7):2427–2432
Liang H, Chen X, Yin Q, Ruan D, Zhao X, Zhang C, McNutt MA, Yin Y (2017) PTENbeta is an alternatively translated isoform of PTEN that regulates rDNA transcription. Nat Commun 8:14771. https://doi.org/10.1038/ncomms14771
Malaney P, Uversky VN, Davé V (2013) The PTEN Long N-tail is intrinsically disordered: increased viability for PTEN therapy. Mol BioSyst 9(11):2877–2888
Hopkins BD, Parsons RE (2014) Molecular pathways: intercellular PTEN and the potential of PTEN restoration therapy. Clin Cancer Res 20(21):5379–5383. https://doi.org/10.1158/1078-0432.ccr-13-2661
Bolduc D, Rahdar M, Tu-Sekine B, Sivakumaren SC, Raben D, Amzel LM, Devreotes P, Gabelli SB, Cole P (2013) Phosphorylation-mediated PTEN conformational closure and deactivation revealed with protein semisynthesis. Elife 2:e00691
Howitt J, Lackovic J, Low L-H, Naguib A, Macintyre A, Goh C-P, Callaway JK, Hammond V, Thomas T, Dixon M (2012) Ndfip1 regulates nuclear Pten import in vivo to promote neuronal survival following cerebral ischemia. J Cell Biol 196(1):29–36
Wen S, Stolarov J, Myers MP, Su JD, Wigler MH, Tonks NK, Durden DL (2001) PTEN controls tumor-induced angiogenesis. Proc Natl Acad Sci USA 98(8):4622–4627. https://doi.org/10.1073/pnas.081063798
Alimonti A, Carracedo A, Clohessy JG, Trotman LC, Nardella C, Egia A, Salmena L, Sampieri K, Haveman WJ, Brogi E, Richardson AL, Zhang J, Pandolfi PP (2010) Subtle variations in Pten dose determine cancer susceptibility. Nat Genet 42(5):454–458. https://doi.org/10.1038/ng.556
Berger AH, Knudson AG, Pandolfi PP (2011) A continuum model for tumour suppression. Nature 476(7359):163–169. https://doi.org/10.1038/nature10275
Acknowledgements
This work funded by Tabriz University of Medical Sciences (Grant Code: 5/104/996). We thank the Stem Cell Research Center for technical support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Naderali, E., Khaki, A.A., Rad, J.S. et al. Regulation and modulation of PTEN activity. Mol Biol Rep 45, 2869–2881 (2018). https://doi.org/10.1007/s11033-018-4321-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-018-4321-6