
Abstract
Phosphatase and tensin homolog (PTEN), which negatively regulates tumorigenic phosphatidylinositol (3,4,5)-trisphosphate (PIP3) signaling, is a commonly mutated tumor suppressor. The majority of cancer-associated PTEN mutations block its essential PIP3 phosphatase activity. However, there is a group of clinically identified PTEN mutations that maintain enzymatic activity, and it is unknown how these mutations contribute to tumor pathogenesis. Here, we show that these enzymatically competent PTEN mutants fail to translocate to the plasma membrane where PTEN converts PIP3 to PI(4,5)P2. Artificial membrane tethering of the PTEN mutants effectively restores tumor suppressor activity and represses excess PIP3 signaling in cells. Thus, our findings reveal a novel mechanism of tumorigenic PTEN deficiency.
Similar content being viewed by others


Introduction
More than 1500 different mutations in the phosphatase and tensin homolog (PTEN) gene have been identified in different cancers (http://cancer.sanger.ac.uk).1, 2, 3, 4, 5, 6 Approximately 40% of these are substitution missense mutations. These cancer-associated amino acid substitutions are distributed along the entire length of PTEN with the highest frequency in the lipid phosphatase catalytic residues, mutations of which abolish lipid phosphatase activity. However, there is a group of PTEN substitutions that maintain PTEN enzymatic activity,7, 8, 9 and it is currently unknown how these mutations disrupt the tumor suppressor function of PTEN.
The membrane recruitment of PTEN is important for its lipid phosphatase function in phosphatidylinositol (3,4,5)-trisphosphate (PIP3) signaling.10, 11, 12, 13, 14, 15 Membrane association of PTEN is negatively regulated by phosphorylation of the inhibitory C-terminal tail domain. When phosphorylated at four serine/threonine residues, the C-terminal tail binds to the membrane-binding regulatory interface and masks the membrane-binding domain containing the CBR3 loop of the C2 domain.10,11,16 When the phosphorylation sites are mutated to alanine (termed PTENA4), the C-terminal tail dissociates from the membrane-binding regulatory interface and promotes membrane recruitment of PTEN.10,16 Therefore, we hypothesized that the cancer-associated mutations that retain enzymatic activity might affect the membrane recruitment of PTEN. To test our hypothesis and determine whether the enzymatically competent mutants represent a new class of tumorigenic PTEN deficiency, we took advantage of PTENA4. Although functionally important, PTEN membrane association is difficult to analyze because a large number of PTEN molecules are present in the cytosol.10,11 The use of PTENA4 dramatically decreases the cytosolic pool of PTEN and allows us to visualize membrane localization.
Results and Discussion
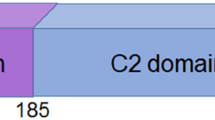
We focused on a group of PTEN missense mutants selected by two criteria. We first chose mutations that do not inhibit phosphatase activity.7, 8, 9 Among these, we further narrowed the choice down to four mutations, S10N, G20E, L42R and F90S, which are located in or near the membrane-binding regulatory interface (Figure 1a and Supplementary Table S1).10 We first biochemically confirmed intact PIP3 phosphatase activity of these mutants using purified PTEN as we described previously.10 Wild-type (WT) and mutant forms of human PTEN fused with green fluorescent protein (GFP) were expressed and immunopurified from Dictyostelium cells using agarose bead-coupled anti-GFP antibodies. PTENS10N, PTENG20E, PTENL42R and PTENF90S showed enzymatic activities indistinguishable from that of WT PTEN (Figure 1b). Activity was completely abolished in the negative control PTENC124S, which carries a substitution in the catalytic residue C124. Fluorescence microscopy of Dictyostelium cells (Figures 1c and d) and the human cell line HEK293T (Figures 1f and g) expressing PTEN mutants fused to GFP showed that PTEN–GFP, PTENS10N–GFP, PTENG20E–GFP, PTENL42R–GFP and PTENF90S–GFP were mainly located in the cytosol and nucleus.

Cancer-associated PTEN mutations inhibit membrane association. (a) The domain structure of PTEN. S10N is located in the lipid-binding domain (LBD), whereas G20E, L42R and F90S are present in the phosphatase domain. The same mutations are indicated in the 3-D structure of PTEN with the phosphatase and C2 domains (R14-V351)28 (http://www.ncbi.nlm.nih.gov/Structure/mmdb/mmdbsrv.cgi?uid=11638). (b) The S10N, G20E, L42R and F90S mutations do not decrease the PIP3 phosphatase activity of purified PTEN in vitro. (c–h) The indicated PTEN–GFP constructs were expressed in Dictyostelium cells (c–e) and HEK293T cells (f–h). Scale bar, 10 μm. Fluorescence intensity in the plasma membrane (blue bars) and the nucleus (red bars) was quantified relative to that in the cytosol (n=8).
To determine whether the S10N, G20E, L42R and F90S mutations affect membrane association, we combined them with the A4 mutations. PTENA4–GFP was located at the plasma membrane in addition to the nucleus in Dictyostelium cells (Figures 1c and e) and HEK293T cells (Figures 1f and h), as previously reported.10,11 In contrast, PTENS10N,A4–GFP, PTENG20E,A4–GFP, PTENL42R,A4–GFP and PTENF90S,A4–GFP were unable to localize at the plasma membrane in both cell types (Figures 1c and e for Dictyostelium, f and g for HEK293T). Their nuclear localization was increased, as expected from our previous study showing that dissociation of PTEN from the plasma membrane leads to accumulation in the nucleus.10 The negative control PTENCBR3(K5A)–GFP, which carries mutations in the CBR3 loop of the membrane binding C2 domain,10 showed cytosolic localization (Figures 1c–h).
Because decreased stability of PTEN contributes to tumorigenesis,17 we tested whether the S10N, G20E, L42R and F90S mutations affect the turnover of PTEN. Immunoblotting revealed similar levels of PTEN–GFP, PTENS10N-GFP, PTENG20E-GFP, PTENL42R-GFP and PTENF90S-GFP in HEK293T cells (Figures 2a). In contrast, PTENA4-GFP, which has decreased stability, showed lower steady-state levels (Figure 2a). Consistent with these results, PTEN–GFP, PTENS10N–GFP, PTENG20E–GFP, PTENL42R–GFP and PTENF90S–GFP had comparable half-lives in the presence of the protein synthesis inhibitor cycloheximide, whereas PTENA4–GFP showed a shorter half-life (Figure 2b). PTEN binds to another tumor suppressor, p53.18,19 We tested whether the four mutations defective in membrane localization affected this protein–protein interaction using co-immunoprecipitation. The results showed similar binding for WT and mutant PTEN to p53 (Supplementary Figure S1).

S10N, G20E, L42R and F90S mutations block interaction of PTEN with membrane lipids. (a) HEK293T cells were transfected with the indicated PTEN–GFP constructs. Whole-cell lysates were analyzed by immunoblotting with antibodies to PTEN and actin. The band intensity was quantified relative to WT PTEN–GFP. Values represent the mean±s.d. (n=3). (b) HEK293T cells were transfected with expression constructs containing GFP fused to the indicated forms of PTEN and were incubated with 100 μg/ml cycloheximide. Whole-cell lysates were analyzed by immunoblotting with anti-GFP antibodies at the indicated time points. Band intensity was quantified relative to a 0-h sample (n=3). (c and d) Liposome-binding assay. Liposomes (PC:PS:PI(4,5)P2:PE-biotin=70:20:5:5) were incubated with whole-cell lysates of Dictyostelium expressing the indicated PTEN–GFP proteins. Liposomes were collected using streptavidin-conjugated magnetic beads. The cell lysate (input) and bound fractions were analyzed by SDS–polyacrylamide gel electrophoresis and immunoblotting using anti-GFP antibodies. Quantification of band intensity is shown in d. Values represent the mean±s.d. (n=3). (e) Association of the C-terminal tail of PTEN with full-length PTEN was examined by an ‘in trans’ interaction assay. Associations between the core and tail domains of the same PTEN molecule (close conformation) are maintained through phosphorylation of the tail (indicated by red asterisks). In contrast, the core region of PTENA4 dissociates from the tail (open conformation) and binds to an exogenously added tail domain. (f) Whole-cell lysates expressing the indicated PTEN–GFP proteins were incubated with PTEN352–403–YFP–FLAG. PTEN352–403–YFP–FLAG was immunoprecipitated with beads coupled to anti-FLAG antibodies. Bound fractions (IP) were analyzed with antibodies to GFP and FLAG. (g) Quantification of band intensity (n=3).
To probe the mechanism by which the S10N, G20E, L42R and F90S mutations inhibited membrane localization of PTEN, we performed an in vitro liposome-binding assay. Liposomes containing PC (70%), PS (20%), PI(4,5)P2 (5%) and PE-biotin (5%) were incubated with lysates of Dictyostelium cells expressing different PTEN–GFP proteins (Figures 2c and d). Consistent with its cytoplasmic localization, WT PTEN–GFP bound only weakly to the liposomes. In contrast, PTENA4, which was enriched at the plasma membrane, showed approximately 10-fold higher association with the liposomes (Figures 2c and d). The PTENA4–liposome interactions were reduced to the WT level by the mutations S10N, G20E, L42R and F90S, thus PTENS10N,A4–GFP, PTENG20E,A4–GFP, PTENL42R,A4–GFP and PTENF90S,A4–GFP only weakly interacted with the liposomes. Therefore, the decreased localization of the PTEN mutants at the plasma membrane likely results from their inability to bind to membrane phospholipids.
The C-terminal tail of PTEN binds to the membrane-binding regulatory interface and closes the protein conformation, physically limiting access of the membrane-binding site to the membrane (Figure 2e).10,16 Mutations in the C-terminal phosphorylation sites in PTENA4 release this inhibitory interaction (Figure 2e).11 To determine whether the S10N, G20E, L42R and F90S mutations restore this interaction in PTENA4, we performed an ‘in trans’ interaction assay and examined the binding of PTEN–GFP to a separate tail domain fused to FLAG after immunoprecipitation with anti-FLAG antibodies. As previously reported,10,11 although PTEN–GFP did not interact with PTEN352–403–FLAG, PTENA4–GFP showed a strong interaction (Figures 2f and g). The S10N, G20E, L42R and F90S mutations did not affect the interaction of PTENA4–GFP with PTEN352–403–FLAG (Figures 2f and g). A negative control, PTENCBR3(K5A),A4–GFP that carries mutations in the tail-binding CBR3 loop,10 did not interact with PTEN352–403–FLAG. Therefore, S10N, G20E, L42R and F90S mutations do not stimulate interaction of the membrane-binding regulatory interface with the C-terminal tail.
To determine the function of PTENS10N, PTENG20E, PTENL42R and PTENF90S, we first investigated cell proliferation and starvation-induced differentiation of Dictyostelium cells. PTEN function is evolutionarily conserved in Dictyostelium and human cells.10,20,21 The loss of PTEN in Dictyostelium causes defects in cell proliferation and differentiation into stress-resistant fruiting bodies under starvation conditions.20 The expression of WT PTEN and PTENA4 restored these phenotypes in Dictyostelium PTEN-null cells (Figures 3a and b). However, PTENS10N, PTENG20E, PTENL42R and PTENF90S were unable to rescue the defects in the presence or absence of the A4 mutation (Figures 3a and b). We then examined the role of PTEN in PIP3 signaling in HEK293T cells, in which PIP3 is overproduced, by measuring phosphorylation of AKT, a major downstream event of PIP3 signaling.22 PTENA4 suppressed AKT phosphorylation by approximately 2.5-fold, whereas addition of the mutations (S10N, G20E, L42R or F90S) blocked the suppressive activity of PTENA4 (Figures 3c and d). Furthermore, we examined the effect of these cancer mutations on PIP3-dependent cell proliferation and migration in the MCF-10A PIK3CA cell line, which carries a constitutively active form of PI3Kα and has increased PIP3 signaling.23,24 When WT PTEN–GFP was expressed in MCF-10A PIK3CA cells, cell proliferation was significantly decreased approximately 2.5-fold compared with expression of the negative controls of GFP alone or the catalytic mutant PTENC124S (Figure 3e). S10N, G20E, L42R and F90S mutations completely blocked the ability of PTEN to decrease proliferation rates. Similarly, the function of PTEN in cell migration was significantly impaired by these four cancer mutations (Figure 3f).

S10N, G20E, L42R and F90S mutations interfere with PTEN function in the regulation of PIP3 signaling. (a) The proliferation of PTEN-null Dictyostelium cells expressing different PTEN–GFP constructs is shown. The numbers of cells were normalized relative to those at day 1. Values represent the mean±s.d. (n=3). (b) PTEN-null Dictyostelium cells expressing different PTEN–GFP constructs were starved to induce differentiation into fruiting bodies. Pictures were taken at 36 h after the onset of starvation. PTEN-null cells expressing WT PTEN or PTENA4 formed fruiting bodies, whereas PTEN-null cells expressing vector alone did not aggregate and remained undifferentiated. PTEN and PTENA4 molecules carrying the cancer mutations did not differentiate into fruiting bodies. Inserts show side views of fruiting bodies. (c) HEK293T cell lysates containing the indicated PTEN–GFP constructs were analyzed by immunoblotting with antibodies to phospho-AKT, AKT, GFP (PTEN–GFP) and actin. (d) Quantification of band intensity (n=3). (e and f) MCF-10A PIK3CA cells were infected with lentiviruses carrying the indicated PTEN constructs and analyzed for cell proliferation by counting the number of cells in individual colonies (e) and for cell migration using transwell plates (f). Values represent the mean±s.d. (n=4).
Finally, we tested whether artificial tethering of PTEN to the plasma membrane restored the function of PTENS10N, PTENG20E, PTENL42R and PTENF90S. For this, we added a myristoylation motif derived from the plasma membrane protein PKBR125 to the N terminus of PTEN. The lipid anchor motif effectively recruited WT and mutant PTEN to the plasma membrane as revealed by fluorescence microscopy (Figure 4a). To assess the effect of membrane recruitment on the amount of PIP3 in the plasma membrane, we co-expressed a biosensor for PIP3, red fluorescent protein fused to the PIP3-binding PH domain from AKT1.26 Bringing PTEN to the plasma membrane significantly improved the ability of PTENS10N,A4, PTENG20E,A4, PTENL42R,A4 and PTENF90S,A4 to remove PIP3 in the plasma membrane. These data further show that the primary defect of the PTEN cancer mutations can be traced to the defects in their membrane association.

Artificial tethering to the plasma membrane neutralizes the effect of S10N, G20E, L42R and F90S on PIP3 signaling and PIP3-regulated cancer cell behavior. (a) HEK293T cells expressing a PIP3 biosensor (RFP–PHAKT) together with different PTEN–GFP constructs were observed by fluorescence microscopy. Myristoylation motif derived from the plasma membrane protein PKBR1 was added to the N terminus of PTEN (+Myr). Scale bar, 10 μm. (b) Intensity of RFP at the plasma membrane was quantified relative to that in the cytosol. Values represent the mean±s.d. (n=8). RFP, red fluorescent protein.
In summary, we identified a new class of cancer-associated PTEN mutations that specifically inhibit the recruitment of PTEN to the plasma membrane as a result of impaired interactions with phospholipids. Our findings reveal the importance of membrane binding of PTEN in tumorigenic PIP3 signaling. As this class of mutations maintains normal PIP3 phosphatase activity and can be rescued by tethering to the plasma membrane, we envision that the pharmacologic promotion of membrane localization may be an effective strategy for anticancer therapy for this class of PTEN deficiency. According to the cBioportal and COSMIC databases, the four PTEN mutations analyzed in our study occur in ~1% (11/1210, COSMIC) of all human cancer sequencing studies. Thus, associations with progression and prognosis cannot currently be assessed given the relatively small number of patients in these studies and the lack of follow-up information for most sequencing studies outside of the Cancer Genome Atlas. However, the fact that these mutations recur, albeit at low frequencies, substantiates the functional relevance. In addition, as stated in our Supplementary Table S1, these mutations appear to occur in a variety of cancer types. The cBioportal database suggests that these mutations are more frequent in breast, lung, brain and uterine cancers. Although the overall frequency is small, 1% of breast cancers is equivalent to ~15 000 cases globally. This demonstrates the impact of our work; low-frequency mutations, which were previously of uncertain significance, now appear to be mutations of functional consequence. Thus, these mutations offer new insights into molecular mechanisms of cancer and possible new avenues for therapeutic intervention.
References
Chalhoub N, Baker SJ . PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol 2009; 4: 127–150.
Ali IU, Schriml LM, Dean M . Mutational spectra of PTEN/MMAC1 gene: a tumor suppressor with lipid phosphatase activity. J Natl Cancer Inst 1999; 91: 1922–1932.
Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C . Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res 2012; 18: 400–407.
Hollander MC, Blumenthal GM, Dennis PA . PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer 2011; 11: 289–301.
Song MS, Salmena L, Pandolfi PP . The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol 2012; 13: 283–296.
Leslie NR, Batty IH, Maccario H, Davidson L, Downes CP . Understanding PTEN regulation: PIP2, polarity and protein stability. Oncogene 2008; 27: 5464–5476.
Denning G, Jean-Joseph B, Prince C, Durden DL, Vogt PK . A short N-terminal sequence of PTEN controls cytoplasmic localization and is required for suppression of cell growth. Oncogene 2007; 26: 3930–3940.
Han SY, Kato H, Kato S, Suzuki T, Shibata H, Ishii S et al. Functional evaluation of PTEN missense mutations using in vitro phosphoinositide phosphatase assay. Cancer Res 2000; 60: 3147–3151.
Rodriguez-Escudero I, Oliver MD, Andres-Pons A, Molina M, Cid VJ, Pulido R . A comprehensive functional analysis of PTEN mutations: implications in tumor- and autism-related syndromes. Hum Mol Genet 2011; 20: 4132–4142.
Nguyen HN, Afkari Y, Senoo H, Sesaki H, Devreotes PN, Iijima M . Mechanism of human PTEN localization revealed by heterologous expression in Dictyostelium. Oncogene (e-pub ahead of print 2 December 2013; doi:10.1038/onc.2013.507).
Rahdar M, Inoue T, Meyer T, Zhang J, Vazquez F, Devreotes PN . A phosphorylation-dependent intramolecular interaction regulates the membrane association and activity of the tumor suppressor PTEN. Proc Natl Acad Sci USA 2009; 106: 480–485.
Gericke A, Leslie NR, Losche M, Ross AH . PtdIns(4,5)P2-mediated cell signaling: emerging principles and PTEN as a paradigm for regulatory mechanism. Adv Exp Med Biol 2013; 991: 85–104.
Das S, Dixon JE, Cho W . Membrane-binding and activation mechanism of PTEN. Proc Natl Acad Sci USA 2003; 100: 7491–7496.
Keniry M, Parsons R . The role of PTEN signaling perturbations in cancer and in targeted therapy. Oncogene 2008; 27: 5477–5485.
Vazquez F, Matsuoka S, Sellers WR, Yanagida T, Ueda M, Devreotes PN . Tumor suppressor PTEN acts through dynamic interaction with the plasma membrane. Proc Natl Acad Sci USA 2006; 103: 3633–3638.
Nguyen HN, Yang JM, Afkari Y, Park BH, Sesaki H, Devreotes PN et al. Engineering ePTEN, an enhanced PTEN with increased tumor suppressor activities. Proc Natl Acad Sci USA 2014; 111: E2684–E2693.
Vazquez F, Ramaswamy S, Nakamura N, Sellers WR . Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol 2000; 20: 5010–5018.
Freeman DJ, Li AG, Wei G, Li HH, Kertesz N, Lesche R et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell 2003; 3: 117–130.
Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y et al. Regulation of PTEN transcription by p53. Mol Cell 2001; 8: 317–325.
Iijima M, Devreotes P . Tumor suppressor PTEN mediates sensing of chemoattractant gradients. Cell 2002; 109: 599–610.
Iijima M, Huang YE, Devreotes P . Temporal and spatial regulation of chemotaxis. Dev Cell 2002; 3: 469–478.
Hawkins PT, Anderson KE, Davidson K, Stephens LR . Signalling through Class I PI3Ks in mammalian cells. Biochem Soc Trans 2006; 34 (Pt 5): 647–662.
Gustin JP, Karakas B, Weiss MB, Abukhdeir AM, Lauring J, Garay JP et al. Knockin of mutant PIK3CA activates multiple oncogenic pathways. Proc Natl Acad Sci USA 2009; 106: 2835–2840.
Wang GM, Wong HY, Konishi H, Blair BG, Abukhdeir AM, Gustin JP et al. Single copies of mutant KRAS and mutant PIK3CA cooperate in immortalized human epithelial cells to induce tumor formation. Cancer Res 2013; 73: 3248–3261.
Kamimura Y, Xiong Y, Iglesias PA, Hoeller O, Bolourani P, Devreotes PN . PIP3-independent activation of TorC2 and PKB at the cell’s leading edge mediates chemotaxis. Curr Biol 2008; 18: 1034–1043.
Chen CL, Wang Y, Sesaki H, Iijima M . Myosin I links PIP3 signaling to remodeling of the actin cytoskeleton in chemotaxis. Sci Signal 2012; 5: ra10.
Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer 2004; 91: 355–358.
Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y et al. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell 1999; 99: 323–334.
Wang Y, Senoo H, Sesaki H, Iijima M . Rho GTPases orient directional sensing in chemotaxis. Proc Natl Acad Sci USA 2013; 110: E4723–E4732.
Zhang P, Wang Y, Sesaki H, Iijima M . Proteomic identification of phosphatidylinositol (3,4,5) triphosphate-binding proteins in Dictyostelium discoideum. Proc Natl Acad Sci USA 2010; 107: 11829–11834.
Kim JS, Xu X, Li H, Solomon D, Lane WS, Jin T et al. Mechanistic analysis of a DNA damage-induced, PTEN-dependent size checkpoint in human cells. Mol Cell Biol 2011; 31: 2756–2771.
Cai H, Huang CH, Devreotes PN, Iijima M . Analysis of chemotaxis in Dictyostelium. Methods Mol Biol 2012; 757: 451–468.
Wang Y, Steimle PA, Ren Y, Ross CA, Robinson DN, Egelhoff TT et al. Dictyostelium huntingtin controls chemotaxis and cytokinesis through the regulation of myosin II phosphorylation. Mol Biol Cell 2011; 22: 2270–2281.
Iijima M, Huang YE, Luo HR, Vazquez F, Devreotes PN . Novel mechanism of PTEN regulation by its phosphatidylinositol 4,5-bisphosphate binding motif is critical for chemotaxis. J Biol Chem 2004; 279: 16606–16613.
Acknowledgements
We thank Dr Todd Waldman (Georgetown University School of Medicine) for lentiviral constructs. The mutation data was obtained from the Sanger Institute Catalogue Of Somatic Mutations In Cancer (COSMIC) web site (http://www.sanger.ac.uk/cosmic).27 This work was supported by NIH grants to MI (GM084015), PND (GM28007 and GM34933), HS (GM089853 and NS084154) and to RP (CA082783 and CA155117).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Rights and permissions
About this article

Cite this article
Nguyen, HN., Yang Jr, JM., Rahdar, M. et al. A new class of cancer-associated PTEN mutations defined by membrane translocation defects. Oncogene 34, 3737–3743 (2015). https://doi.org/10.1038/onc.2014.293
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2014.293
- Springer Nature Limited
This article is cited by
-
Integrating thousands of PTEN variant activity and abundance measurements reveals variant subgroups and new dominant negatives in cancers
Genome Medicine (2021)
-
Multiplex assessment of protein variant abundance by massively parallel sequencing
Nature Genetics (2018)
-
Characterization of PTEN mutations in brain cancer reveals that pten mono-ubiquitination promotes protein stability and nuclear localization
Oncogene (2017)
-
Opening the conformation is a master switch for the dual localization and phosphatase activity of PTEN
Scientific Reports (2015)