
Abstract
Marine pelagic fishes are usually characterized by subtle but complex patterns of genetic differentiation, which are influenced by both historical process and contemporary gene flow. Genetic population differentiation of chub mackerel, Scomber japonicus, was examined across most of its range in the Northwestern Pacific by screening variation of eight microsatellite loci. Our genetic analysis detected a weak but significant genetic structure of chub mackerel, which was characterized by areas of gene flow and isolation by distance. Consistent with previous estimates of stock structure, we found genetic discontinuity between Japan and China samples. Local-scale pattern of genetic differentiation was observed between samples from the Bohai Sea and North Yellow Sea and those from the East China Sea, which we ascribed to differences in spawning time and migratory behavior. Furthermore, the observed homogeneity among collections of chub mackerel from the East and South China Seas could be the result of an interaction between biological characteristics and marine currents. The present study underlies the importance of understanding the biological significance of genetic differentiation to establish management strategies for exploited fish populations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Marine pelagic fishes generally exhibit large effective population sizes, high dispersal capability, prolific fecundity as well as long planktonic larval phases that are subject to passive transport by marine currents. The biological peculiarities of these fishes combined with the apparent lack of physical barriers in the marine realm, are thought to facilitate extensive gene flow and low genetic differentiation among populations [1–4]. However, this long held-view was challenged by recent studies that cryptic population structuring has been increasingly detected in highly abundant and widely distributed migratory marine fishes [5–9]. The complicated interaction between marine environment and biological characteristics of marine fishes could sufficiently account for the differentiation processing. Specifically, these complicated factors include historical vicariance events [10, 11], ocean fronts and currents [12], environmental gradients (e.g., salinity and temperature) in water bodies [13, 14], larval retention [15], natural homing as well as local habitat dependence [16, 17]. Hence, to understand the complex population structuring and dynamics of marine migratory fish requires discerning the relative significance of different mechanisms in shaping and maintaining the pattern of population structure.
Considering the complexity of the physical peculiarities of the marine environment and life history traits of marine pelagic fishes, it is difficult to detect distinct populations in marine ecosystems. Lack of detectable differentiation could be attributed to extensive gene flow, and it does not necessarily imply the absence of structuring [18, 19]. Information on population structure is essential for the development of rational and effective management strategies for vulnerable fishery resources [20, 21]. Failure to detect population units could lead to local overexploitation and subsequently to resource declines [22]. An effective way to obtain further insights to the factors that form and maintain genetic divergence is to combine genetic data with information on the physical characteristics of habitats as well as biological peculiarities of marine fishes, to study how these factors impact population structure patterns [7, 13, 16, 23].
Chub mackerel, Scomber japonicus (Houttuyn 1782) is a broadly exploited pelagic fish species and has a cosmopolitan distribution along warm and temperate waters of Indo-Pacific Ocean [24–26]. In the Northwestern Pacific, it is distributed from Japan along the Chinese coast south to the Malay Archipelago [27], and it is commercially exploited in waters of the East China Sea from Japan to northern Taiwan as important fishery resources for China, Korea, and Japan. However, increasing fishing effort has led to a reduction in abundance of S. japonicus, and collapse of chub mackerel fisheries was reported in the East China Sea [28, 29]. The need to establish appropriate management strategy for chub mackerel has led to several genetic studies using a range of molecular markers. A preliminary mitochondrial DNA study conducted by Tzeng et al. [30] suggested that chub mackerel off Taiwan belonged to a single gene pool. Shao and Chen [31] undertook further studies on genetic variations of chub mackerel based on random amplified polymorphic DNA (RAPD) in the Yellow Sea and East China Sea. Their results revealed the existence of two stocks of chub mackerel in the Yellow Sea and East China Sea. Zhang et al. [32] employed amplified fragment length polymorphism (AFLP) analysis on collection of chub mackerel from the Taiwan Strait and its adjacent waters, and found no significant genetic heterogeneity among samples. A more recent microsatellite analysis detected the presence of population structure in the East and South China Seas [33]. These studies have resulted in different conclusions regarding the genetic structure and population identities of chub mackerel at fine geographical scales. There has been no comprehensive study on the Northwestern Pacific-wide population structure of chub mackerel.
Microsatellite DNA has proven to be a useful marker in uncovering population structure that was not apparent using less variable markers [13, 34–36]. We estimated genetic variation and population structure of chub mackerel in the Northwestern Pacific using eight microsatellite loci with the purpose to infer the relative role of biological characteristics and environmental factors involved in shaping contemporary population genetic structure of this species.
Materials and methods
Sample collection
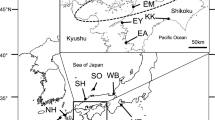
A total of 213 individuals were collected at 10 localities along Chinese and Japanese coastal waters during 2008–2009 (Table 1; Fig. 1). Taxonomic status of the fishes was identified morphologically [37]. A piece of skeletal muscle was excised from each individual and preserved in 95 % ethanol for DNA extraction.

Map showing sample locations of chub mackerel, samples are marked by abbreviations that correspond to Table 1. Ocean currents are shown: 1 Kuroshio Current; 2 Taiwan Warm Current; 3 Tsushima Current; 4 Yellow Sea Warm Current; 5 Oyashio Current; 6 China Coastal Current
Microsatellite genotyping
Genomic DNA was isolated from the muscle tissue by proteinase K digestion followed by the standard phenol/chloroform method [38]. Eight microsatellite loci were PCR amplified using primers Sja3, Sja4, Sja5, Sja6, Sja8, Sja9 developed from chub mackerel [39] and Sa2599, Sa2770 from its congener, spotted mackerel Scomber australasicus [40]. PCR reactions were carried out in 25 μl reaction mixture containing 20 ng template DNA, 1.5 mM MgCl2, 0.2 mM of each dNTPs, 0.5 µm of each primer, 1U Taq polymerase (Takara). All PCRs were performed in an Eppendorf thermal cycler following optimized reaction conditions [39, 40]. PCR products were separated on 8 % non-denaturing vertical polyacrylamide gel electrophoresis and visualized with silver staining as described by Bassam et al. [41]. A sizing standard (100–300 base pairs) was run in the center and at both ends of each gel to calibrate allele size. Furthermore, a reference sample was run on each gel to ensure consistency in genotype scoring across runs.
Statistical analysis
Microsatellite genetic diversity was quantified as number of alleles (A), observed heterozygosity (H O) and expected heterozygosity (H E) for each locus and sample site using POPGENE 1.32 [42]. Polymorphism information content (PIC) was calculated using allele frequencies according to the formula given by Botstein et al. [43]. The allelic richness (R S), which is a standardized index of the mean number of alleles per locus irrespective of sample size, was calculated by Fstat 2.9.3 [44]. Deviations from Hardy–Weinberg equilibrium (HWE) expectation and linkage disequilibrium were tested for each site at each locus using GENEPOP 3.4 [45] with 10,000 burn-in steps, and 500 batches of 5,000 Monte Carlo Markov Chain (MCMC) steps per batch. Critical significance levels for multiple simultaneous tests were adjusted using the sequential Bonferroni correction [46].
To test whether mutation or genetic drift contributed to genetic differentiation of chub mackerel populations, we performed the allele size permutation test to compare allele identity-based statistic F ST (IAM) [47] and allele size-based statistic R ST (SMM) [48]. The procedure was implemented in the program SPAGeDi 1.3 [49]. R ST were calculated for each locus and compared with the distribution of ρR ST values generated by 1000 permutations. A significant one-side test establishes the alternative hypothesis that genetic differentiation is caused mainly by SMM-like mutation (R ST > ρR ST), otherwise no contribution of SMM to genetic differentiation. In this study, a non-significant result showed that stepwise mutations were not informative (P = 0.0866). Therefore, F ST rather than R ST estimates were used for the subsequent genetic analysis, as estimated by the program Fstat 2.9.3 [44] and significance assessed with permutation tests (1,000 replicates). To test for isolation by distance in chub mackerel, correlation between genetic differentiation and geographical distance between sampling sites was assessed using a Mantel procedure. The logarithm of geographical distance in kilometers was regressed against F ST/(1 − F ST) as computed in IBDWS (http://ibdws.sdsu.edu/~ibdws/) [50, 51]. Significance of the regression slopes was tested with 1,000 permutations.
Three dimensional factorial correspondence analysis (3D-FCA) was performed in GENETIX 4.05 [52] to explore population divisions and relationships of chub mackerel, independent from a prior knowledge of their relationships. A model-based Bayesian clustering algorithm was undertaken using STRUCTURE 2.1 [53] to determine the number of genetic discrete populations (K) with the highest posterior probability. The simulated K values ranged from 1 to 10 (total sites). Ten independent runs were implemented for each specific K-value in order to verify the consistency of the results. The simulations were conducted assuming an admixture model with correlated allele frequencies, which is considered as the superior model for detecting structure among closely related populations [54]. MCMC consisted of 100,000 burn-in iterations followed by 1,000,000 iterations. Finally, we conducted a hierarchical analysis of molecular variance (AMOVA) in ARLEQUIN 2.000 [55] to test population structure across the entire geographic sample range and among putative regional grouping of samples. For all calculations, significance was assessed by 1,000 random permutations and P values from multiple comparisons were Bonferroni adjusted [46].
Results
Genetic variability
Genotyping of 213 individuals of chub mackerel from 10 locations enabled scoring of 113 alleles at eight microsatellite loci. The amount of genetic variability in terms of average number of alleles, observed and expected heterozygosities was similar among sampling sites for the same microsatellite locus (Table 1). However, large difference was detected among microsatellite loci. The average number of alleles per locus (A) over samples ranged from 4.1 alleles for Sja9 to 12.1 for Sja5 and the average number of alleles per population ranged from 5.9 (DD) to 11.1 (RC). The allelic richness varied between 4.2 (DD) and 5.7 (RC). The observed and expected heterozygosities ranged from 0.369 to 0.556 and 0.596 to 0.784 per population, respectively. The population with the highest polymorphism information content (0.749) was the population NB, whereas that with the lowest value (0.562) was the population RC.
Thirty of 80 locus-by-population tests exhibited significant departure from HWE with heterozygote excess, but none of which remained significant after correction for multiple tests. Significant linkage disequilibrium was detected between Sja3 and Sja9 over all populations. In addition, such case was also found between Sja3 and Sa2770 in the population RC and NB. Thus, locus Sja3 was excluded from subsequent analyses.
Population genetic differentiation
The allele size permutation test did not detect any R ST value significantly >ρR ST (Table 2), suggesting that F ST should be preferred over R ST for the measurement of genetic differentiation between chub mackerel populations. The global test for genetic differentiation (F ST = 0.049, P < 0.05, 95 % C.I. 0.029–0.068) over all loci suggested population structuring of the analyzed samples. Pairwise F ST values between 10 populations ranged from 0.0109 to 0.161, and most of them were significant after sequential Bonferroni correction except 7 comparisons (Table 3). F ST estimates between population pairs that included RS samples were significant, whereas many non-significant outcomes were observed among samples collected from the East and South China Seas. The MNP population was significantly differentiated from other samples with pairwise F ST values ranging from 0.0457 to 0.161, except from the geographically closest HA samples.
According to the Mantel test, a significant correlation (r = 0.549, R2 = 0.301, P < 0.05) was observed between genetic distance determined as F ST/(1 − F ST) and geographical distance based on all loci, indicating isolation by distance among chub mackerel populations (Fig. 2). The linear regression explained 30.1 % of the variation in genetic differentiation of chub mackerel populations. Three dimensional factorial correspondence analysis (3D-FCA) explaining 47.25 % of the overall variation, separated the three Bohai Sea and North Yellow Sea populations (DD, RC and RS) from the five populations (YC, NB, WZ, XM and SW), with the remaining two Japan populations (MNP and HA) in the top (Fig. 3). The Bayesian algorithm implemented in the program STRUCTURE indicated that all individuals could be assigned to four main genetic clusters (Fig. 4a). The four clusters showed continuous distributions, with neighboring populations assigning to the same cluster. As shown in Fig. 4b, most individuals from four populations (NB, WZ, XM and SW) were assigned into the first cluster regardless of geographic origin, whereas most individuals from three populations (DD, RC and RS) collected from the Bohai Sea and North Yellow Sea were assigned to the second cluster. Most individuals from the remaining two populations (HA and MNP) were assigned to a single cluster, respectively. The hypothetical grouping of populations was further explored by an analysis of molecular variance (AMOVA) tests (Table 4). Most of the total genetic variance was found within populations. Potential geographic structuring (organized as China versus Japan samples) was supported by the AMOVA (P = 0.029). The hierarchical AMOVA conducted with populations clustered according to the 3D-FCA analysis revealed that 1.37 % of genetic variance was distributed among groups (P = 0.012), whereas 4.48 % was partitioned among populations within group. An alternative AMOVA model, with populations clustered according to the Bayesian clustering analysis, showed comparatively more suitable variance partitioning with higher percentage of variance (1.89 %) among groups (P = 0.038). When the AMOVA was performed without considering population disjunction, 5.43 % of the genetic variability was apportioned among populations, and 94.57 % was detected among individuals within the population (P = 0.000).

Genetic isolation by distance in chub mackerel populations inferred from pairwise estimates of F ST/(1 − F ST) based on seven microsatellite loci and geographic distance (r = 0.549, R2 = 0.301, P = 0.020)

Three dimensional factorial correspondence analysis (3D-FCA) showing relationships among chub mackerel populations based on seven microsatellite loci. Refer to Table 1 for abbreviations of sampling sites

a Number of chub mackerel populations with the highest posterior probability expressed as the mean likelihood Ln P(D) for each assumed population (K); b Graphical results of the STRUCTURE analysis of seven microsatellite loci in chub mackerel populations. Vertical lines are proportional to the probability of individual membership in simulated cluster. Refer to Table 1 for abbreviations of sampling sites. (Color figure online)
Discussion
The eight microsatellite loci used in this study showed relatively moderate level of polymorphism in chub mackerel compared to other marine pelagic fishes [36, 56, 57]. No geographical trend appeared in the levels of microsatellite diversity. F ST and R ST are two commonly used estimators to assess the amount of genetic differentiation among populations. Comparisons of F ST with R ST values on microsatellite data have been suggested for checking the relative contribution of mutation versus migration rates to population differentiation [58, 59]. The allele size permutation test did not reveal any significant contribution of stepwise mutations to genetic differentiation of chub mackerel populations, indicating that migration rates and drift become important relative to mutation in contributing to population differentiation of chub mackerel.
Overall F ST detected weak but significant genetic differentiation of chub mackerel populations. Further evidence for population structuring was obtained by plotting pairwise estimates of F ST/(1 − F ST) against geographic distances. The Mantel test demonstrated that chub mackerel exhibited a clear isolation-by-distance pattern, which implied restricted gene flow among chub mackerel from different sampled locations. It should be taken into account that when long-distance samplings have been made, a significant IBD model can be obtained even if geographic distance is not the main factor causing the genetic differences [60, 61]. Thus, the present geographical pattern of genetic differentiation was further tested using 3D-FCA and Bayesian clustering analysis. Both tests suggested a grouping of samples according to geographical positions, with the Bohai Sea and North Yellow Sea populations (DD, RC and RS) forming one group, and the East and South China Seas populations (NB, WZ, XM and SW) forming another group, with the remaining two Japan populations (HA and MNP) being distinct from these collected from China coastal waters. This pattern was further confirmed by the hierarchical AMOVA analysis. What was unexpected was the assignment of individuals from the South Yellow Sea population (YC), indicating a complicated genetic composition of this population. However, inclusion of mitochondrial DNA analysis and more comprehensive sampling in the South Yellow Sea and East China Sea is essential to investigate this problem further in the future.
Marine pelagic fishes generally exhibit low levels of genetic subdivision, eventually leading to panmictic populations [62]. Nevertheless, numerically small but significant genetic differentiation has been detected in many marine pelagic fishes despite high dispersal capabilities and large effective population sizes [5, 6, 9, 63]. In this study, multiple analyses showed that a weak but significant genetic structure existed in chub mackerel populations. The pertinent features of species biology and marine environment, coupled with their interactions, need be considered to explain the genetic patterns observed in chub mackerel. According to differences in the patterns of seasonal migration and spawning grounds, chub mackerel in Japan is managed as two separate stocks: the Pacific stock and the Tsushima Current stock [64]. The Tsushima Current stock of chub mackerel is distributed from the southern East China Sea to the northern Japan Sea, the Yellow Sea and Bohai Gulf. Multiple analyses showed genetic discontinuity between Japan and China samples of S. japonicus in our study, adding additional evidence for supporting this management policy. In the case of chub mackerel off Japan’s Pacific coast, genetic differentiation between the two Japan populations (MNP and HA) was detected from the Bayesian clustering analysis (Fig. 4b). Ocean currents are expected to be the predominant environmental factor influencing contemporary levels of gene flow between populations, especially in species with pelagic eggs or larvae, or in species with highly migratory adults [12, 65, 66]. There are two main ocean currents off the eastern coast of Japan, the Kuroshio-extension current and the Oyashio-extension current (Fig. 1). The former originates in the tropics further south and warm (25–27 °C at the surface layer in summer), while the latter originates in the boreal seas to the north and cold (6–10 °C in summer). Such currents might have the potential to facilitate larval retention relatively close to spawning ground [67]. Accordingly, genetic differentiation may arise, at least partly, by the combined adaptation of eggs and early larval stages to local environmental conditions on the spawning and nursery grounds due to hydrological features. However, a more refined insight into this assumption requires additional evidence.
For chub mackerel off China coast, all tests based on microsatellites, including pairwise F ST, 3D-FCA and Bayesian clustering analysis, illustrated that samples from the Bohai Sea and North Yellow Sea (DD, RC and RS) were divergent from those from the East China Sea, a pattern consistent with previous estimates of population structure derived from RAPD analysis [31]. Chub mackerel in Bohai Sea and Yellow Sea are generally from the wintering grounds off the waters of western Kyushu. There is a geographic variation in the spawning season, with chub mackerel in the southern East China Sea spawning approximately 4 months earlier than those in the western Japan Sea [68]. This difference in spawning time and natal homing to different spawning grounds might be responsible for the observed genetic differentiation between theses areas.
Previous analysis of tagging data indicated that chub mackerel in Fujian offshore waters should be divided into two stocks, the East China Sea stock and South Fujian-East Guangdong stock [69]. However, our genetic analysis was inconsistent with this view. Samples collected from the East and South China Seas appeared to constitute a genetically homogenous group. This agreed with the absence of genetic structure reported for chub mackerel sampled around Taiwan waters inferred from AFLP analysis [32]. The similar genetic pattern was also assessed by Zeng et al. [33]. They observed that S. japonicus individuals were strongly admixed between the populations from the East China Sea extending to the eastern coast of Hainan Island in the South China Sea. However, the population off the southern coast of Hainan Island in the central South China Sea was genetically different from the other populations. Further sampling in the central South China Sea will be necessary to clarify the population structure of chub mackerel in these areas.
Like many marine pelagic fishes, chub mackerel exhibits highly migratory behavior, large population size and long high potential for dispersal during early life stage (about 29 days-period passive larval stage). Major currents along the China coast are the Kuroshio and the coastal currents (Fig. 1). The warm and highly saline Kuroshio current, originating from east of Philippine Islands, flows northward along the east coast of Taiwan and reaches the steep east–west continental shelf break of the East China Sea. Then, it splits into two branches, the major one flowing northeast towards the southern coast of Kyushu, and the minor one flowing northward as a shallow surface current [70]. The path of the Kuroshio northeast of Taiwan shows large seasonal variations. It moves seaward in spring and summers and shoreward in fall and winter [71]. The cold and low-saline China Coastal Current flows southwards along the Chinese coast from the Bohai Sea to the Taiwan Strait [72]. At the mercy of these currents, individuals of chub mackerel from the East China Sea may migrate southward along the Taiwan coast to the northern South China Sea. Furthermore, pelagic larvae from the South China Sea could follow the northwards current to the East China Sea for recruitment into the local population [32]. The importance of ocean currents in shaping the genetic structure of spotted mackerel S. australasicus populations has recently been demonstrated [65]. It is likely that the same hydrographic features have shaped the genetic patterns in these two closely related species, which have similar biological characteristics. Consequently, the interaction between biological characteristics and marine currents likely facilitates genetic homogeneity of chub mackerel populations of the East and South China Seas.
In conclusion, microsatellite marker is proven to be an effective method to delineate genetic diversity and structure of highly migratory fish species. The results of the present study provided evidence that the biological characteristics of chub mackerel and the oceanographic properties of the Northwestern Pacific could be responsible for the observed population differentiation of chub mackerel. It is noteworthy that the investigation of temporal genetic differentiation in the analyzed locations is needed to test whether the observed population genetic patterns in chub mackerel are stable on a temporal scale. Although sample sizes for microsatellites are small, these sample sizes were still large enough and the microsatellite marker polymorphic enough to provide power to detect population structure of chub mackerel in the studied areas. However, the combined use of nuclear and mitochondrial markers, more comprehensive sampling integrated with local ecological investigations could help to shed more light on the exact ecological process in shaping genetic population structure of chub mackerel.
References
Palumbi SR (1994) Genetic divergence, reproductive isolation, and marine speciation. Annu Rev Ecol Syst 25:547–572
Arnason E, Pálsson S (1996) Mitochondrial cytochrome b DNA sequence variation of Atlantic cod Gadhus morhua, from Norway. Mol Ecol 5:715–724
Vis ML, Carr SM, Bowering WR, Davidson WS (1997) Greenland halibut (Reinhartius hippoglossides) in the North Atlantic are genetically homogeneous. Can J Fish Aquat Sci 54:1813–1821
Smedbol RK, McPerson A, Hansen MM, Kenchington E (2002) Myths and moderation in marine ‘metapopulations’? Fish Fish 3:20–35
Nielsen EE, Nielsen PH, Meldrup D, Hansen MM (2004) Genetic population structure of turbot (Scophthalmus maximus L.) supports the presence of multiple hybrid zones for marine fishes in the transition zone between the Baltic Sea and the North Sea. Mol Ecol 13:585–595
Bekkevold D, André C, Dahlgren TG, Clausen LA, Torstensen E, Mosegaard H, Carvalho GR, Christensen TB, Norlinder E, Ruzzante DE (2005) Environmental correlates of population differentiation in Atlantic herring. Evolution 59:2656–2668
Was A, Gosling E, McCrann K, Mork J (2008) Evidence for population structuring of blue whiting (Micromesistius poutassou) in the Northeast Atlantic. ICES J Mar Sci 65:216–225
Cunningham KM, Canino MF, Spies IB, Hauser L (2009) Genetic isolation by distance and localized fjord population structure in Pacific cod (Gadus macrocephalus): limited effective dispersal in the northeastern Pacific Ocean. Can J Fish Aquat Sci 66:153–166
André C, Larsson LC, Laikre L, Bekkevold D, Brigham J, Carvalho GR, Dahlgren TG, Hutchinson WF, Mariani S, Mudde K, Ruzzante DE, Ryman N (2011) Detecting population structure in a high gene-flow species, Atlantic herring (Clupea harengus): direct, simultaneous evaluation of neutral vs putatively selected loci. Heredity 106:270–280
Viñas J, Alvarado Bremer J, Pla C (2004) Phylogeography of the Atlantic bonito (Sarda sarda) in the northern Mediterranean: the combined effects of historical vicariance, population expansion, secondary invasion, and isolation by distance. Mol Phylogenet Evol 33:32–42
Liu JX, Tatarenkov A, Beacham TD, Gorbachev V, Wildes S, Avise JC (2011) Effects of Pleistocene climatic fluctuations on the phylogeographic and demographic histories of Pacific herring (Clupea pallasii). Mol Ecol 20:3879–3893
Magoulas A, Castilho R, Caetano S, Marcato S, Patarnello T (2006) Mitochondrial DNA reveals a mosaic pattern of phylogeographical structure in Atlantic and Mediterranean populations of anchovy (Engraulis encrasicolus). Mol Phylogenet Evol 39:734–746
Jørgensen HB, Hansen MM, Bekkevold D, Ruzzante DE, Loeschcke V (2005) Marine landscapes and population genetic structure of herring (Clupea harengus L.) in the Baltic Sea. Mol Ecol 14:3219–3234
Han Z, Yanagimoto T, Zhang Y, Gao T (2012) Phylogeography study of Ammodytes personatus in Northwestern Pacific: pleistocene isolation, temperature and current conducted secondary contact. PLoS ONE 7(5):e37425. doi:10.1371/journal.pone.0037425
Zardoya R, Castilho R, Grande C, Favre-Krey L, Caetano S, Marcato S, Krey G, Patarnello T (2004) Differential population structuring of two closely related fish species, the mackerel (Scomber scombrus) and the chub mackerel (Scomber japonicus), in the Mediterranean Sea. Mol Ecol 13:1785–1798
Nesbø CL, Rueness EK, Iversen SA, Skagen DW, Jakobsen KS (2000) Phylogeography and population history of Atlantic mackerel (Scomber scombrus L.): a genealogical approach reveals genetic structuring among eastern Atlantic stocks. Proc R Soc Lond B 267:281–292
Natoli A, Birkun A, Aguilar A, Lopez A, Hoelzel AR (2005) Habitat structure and the dispersal of male and female bottlenose dolphins (Tursiops truncatus). Proc R Soc Lond B 272:1217–1226
Waples RS (1998) Separating the wheat from the chaff: patterns of genetic differentiation in high gene flow species. J Hered 89:438–450
Avise JC (2000) Phylogeography: The history and formation of species. Harvard University Press, Cambridge
Carvalho GR, Hauser L (1994) Molecular genetics and the stock concept in fisheries. Rev Fish Biol Fish 4:326–350
Ward RD, Grewe PM (1994) Appraisal of molecular genetic techniques in fisheries. Rev Fish Biol Fish 4:300–325
Hutchings JA (2000) Collapse and recovery of marine fishes. Nature 406:882–885
Liu M, Lin LS, Gao TX, Yanagimoto T, Sakurai Y, Grant WS (2012) What maintains the central North Pacific genetic discontinuity in Pacific herring? PLoS ONE 7:e50340. doi:10.1371/journal.pone.0050340
Scoles DR, Collette BB, Graves JE (1998) Global phylogeography of mackerels of the genus Scomber. Fish Bull 96:823–842
Infante C, Blanco E, Zuasti E, Crespo A, Manchado M (2007) Phylogenetic differentiation between Atlantic Scomber colias and Pacific Scomber japonicus based on nuclear DNA sequences. Genetica 130:1–8
Catanese G, Manchado M, Infante C (2010) Evolutionary relatedness of mackerels of the genus Scomber based on complete mitochondrial genomes: Strong support to the recognition of Atlantic Scomber colias and Pacific Scomber japonicus as distinct species. Gene 452:35–43
Collette BB, Nauen CE (1983) Scombrids of the world: An annotated and illustrated catalogue of tunas, mackerels, bonitos and related species known to date. FAO Fish Synop 125:1–137
Li CS, Li YZ, Li GF, Chen WZ, Chen GB, Chen CH, Jin XS, Yu LF, Lin LY (2006) Pelagic fishes. In: Tang QS (ed) Marin biology resources and habitat environments in the EEZ of China. Science Press, Beijing, pp 599–608
Yukami R, Ohshimo S, Yoda M, Hiyama Y (2009) Estimation of the spawning grounds of chub mackerel Scomber japonicus and spotted mackerel Scomber australasicus in the East China Sea based on catch statistics and biometric data. Fish Sci 75:167–174
Tzeng TD, Haung HL, Wang D, Yeh SY (2007) Genetic diversity and population expansion of the common mackerel (Scomber japonicus) off Taiwan. J Fish Soc Taiwan 34:237–245
Shao F, Chen XJ (2008) Genetic variations of Scomber japonicus and S. australasicus based on random amplified polymorphic DNA in the Yellow Sea and the East China Sea. J Guangdong Ocean Univ 28:83–87 (in Chinese with English abstract)
Zhang LY, Su YQ, Wang HJ, Wang J (2011) Population genetic structure of Pneumatophorus japonicus in the Taiwan Strait. Acta Ecol Sin 31:7097–7103 (in Chinese with English abstract)
Zeng L, Cheng Q, Chen X (2012) Microsatellite analysis reveals the population structure and migration patterns of Scomber japonicus (Scombridae) with continuous distribution in the East and South China Seas. Biochem Syst Ecol 42:83–93
Ruzzante DE, Taggart CT, Cook D (1999) A review of the evidence for genetic structure of cod (Gadus morhua) populations in the NW Atlantic and population affinities of larval cod off Newfoundland and the Gulf of St Lawrence. Fish Res 43:79–97
Nielsen EE, Hansen MM, Schmidt C, Meldrup D, Grønkjaer P (2001) Population of origin of Atlantic cod. Nature 413:272
Carlsson J, McDowell JR, Diaz-Jaimes P, Carlsson JE, Bole SB, Gold JR, Graves J (2004) Microsatellite and mitochondrial DNA analyses of Atlantic bluefin tuna (Thunnus thynnus thynnus) population structure in the Mediterranean Sea. Mol Ecol 13:3345–3356
Matsui T (1967) Review of the mackerel genera Scomber and Rastrelliger with description of a new species of Rastrelliger. Copeia 1:71–83
Sambrook J, Russell DW (2001) Molecular cloning: A laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, New York
Yagishita N, Kobayashi T (2008) Isolation and characterization of nine microsatellite loci from the chub mackerel, Scomber japonicus (Perciformes, Scombridae). Mol Ecol Resour 8:302–304
Tang CY, Tzeng CH, Chen CS, Chiu TS (2009) Microsatellite DNA markers for population genetic studies of blue mackerel (Scomber australasicus) and cross-specific amplification in S. japonicus. Mol Ecol Resour 9:824–827
Bassam BJ, Caetano-Anolles G, Gresshoff PM (1991) Fast and sensitive silver staining of DNA in polyacrylamide gels. Anal Biochem 19:680–683
Yeh FC, Yang R, Boyle TJ, Ye Z, Xiyan JM (2000) POPGENE 32, Microsoft Windows-based freeware for population genetic analysis. Molecular Biology and Biotechnology Centre, University of Alberta, Edmonton
Botstein D, White RL, Skolnick M, Davis RW (1980) Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet 32:314–331
Goudet J (2001) FSTAT V.2.9.3, a program to estimate and test gene diversities and fixation indices (Updated from Goudet 1995). University of Lausanne, Lausanne
Raymond M, Rousset F (1995) GENEPOP Version 1.2: population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Rice WR (1989) Analysing tables of statistical tests. Evolution 43:223–225
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Slatkin M (1995) A measure of population subdivision based on microsatellite allele frequencies. Genetics 130:457–462
Hardy OJ, Vekemans X (2002) SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620
Bohonak AJ (2002) IBD (isolation by distance): a program for analyses of isolation by distance. J Hered 93:153–154
Jensen JL, Bohonak AJ, Kelley ST (2005) Isolation by distance, web service. BMC Genet 6:13
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F (2004) GENETIX 4.05, logiciel sous Windows TM pour la génétique des populations. Laboratoire Génome, Populations, Interactions, Université de Montpellier II, Montpellier
Pritchard JK, tephens MS, onnelly PD (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Schneider S, Roessli D, Excoffier L (2000) ARLEQUIN, Version 2.0: a software for population genetic data analysis. University of Geneva, Geneva
Cárdenas L, Silva AX, Magoulas A, Cabezas J, Poulin E, Ojeda FP (2009) Genetic population structure in the Chilean jack mackerel, Trachurus murphyi (Nichols) across the South-eastern Pacific Ocean. Fish Res 100:109–115
Fauvelot C, Borsa P (2011) Patterns of genetic isolation in a widely distributed pelagic fish, the narrow-barred Spanish mackerel (Scomberomorus commerson). Biol J Linn Soc 104:886–902
Ross KG, Krieger MJ, Shoemaker DD, Vargo EL, Keller L (1997) Hierarchical analysis of genetic structure in native fire ant populations: results from three classes of molecular markers. Genetics 147:643–655
Estoup A, Angers B (1998) Microsatellites and minisatellites for molecular ecology: theoretical and empirical considerations. In: Carvalho GR (ed) Advances in molecular ecology. IOS Press, Amsterdam, pp 55–86
Templeton A, Routman E, Phillips C (1995) Separating population structure from population history: a cladistic analysis of the geographical distribution of mitochondrial DNA haplotypes in the tiger salamander, Ambystoma tigrinum. Genetics 140:767–782
Templeton A (2001) Using phylogeographic analyses of gene trees to test species status and processes. Mol Ecol 10:779–791
McQuinn IH (1997) Metapopulations and the Atlantic herring. Rev Fish Biol Fish 7:297–329
Knutsen H, Jorde PE, André C, Stenseth NC (2003) Fine-scaled geographical population structuring in a highly mobile marine species: the Atlantic cod. Mol Ecol 12:385–394
Shiraishi T, Okamoto K, Yoneda M, Sakai T, Ohshimo S, Onoe S, Yamaguchi A, Matsuyama M (2008) Age validation, growth and annual reproductive cycle of chub mackerel Scomber japonicus off the waters of northern Kyushu and in the East China Sea. Fish Sci 74:947–954
Tzeng TD (2007) Population structure and historical demography of the spotted mackerel (Scomber australasicus) off Taiwan inferred from mitochondrial control region sequencing. Zool Stud 46:656–663
Zhan A, Hu J, Hu X, Zhou Z, Hui M, Wang S, Peng W, Wang M, Bao Z (2009) Fine-scale population genetic structure of Zhikong scallop (Chlamys farreri): do local marine currents drive geographical differentiation? Mar Biotechnol 11:223–235
Liu M, Lu ZC, Gao TX, Yanagimoto T, Sakurai Y (2010) Remarkably low mtDNA control-region diversity and shallow population structure in Pacific cod Gadus macrocephalus. J Fish Biol 77:1071–1082
Ouchi A, Hamasaki S (1979) Population analysis of the common mackerel, Scomber japonicus, based on the catch statistics and biological information in the western Japan Sea and the East China Sea. Bull Seikai Natl Fish Res Inst 53:125–152
Yan YM (1997) Biology of Pneumatophorus japonicus in Fujian offshore waters. Mar Fish 19:69–73 (in Chinese with English abstract)
Hsueh Y, Wang J, Chern CS (1992) The intrusion of the Kuroshio across the continental shelf northeast of Taiwan. J Geophys Res 97:14320–14323
Sun X (1987) Analysis of the surface path of the Kuroshio in the East China Sea. In: Sun X (ed) Essays on the investigation of Kuroshio. Ocean Press, Beijing, pp 1–14
Wu B (1982) Some problems on circulation study in Taiwan Strait. Taiwan Str 1:1–7
Acknowledgments
The present study could not have been carried out without the willing help of those listed below in collecting specimens: Mr. Xi Han, Mr. Dian-Rong Sun, Dr. Xiao Chen, Dr. Long-Shan Lin, Dr. Bo-Nian Shui. This study was supported by the International Cooperation and Exchange of the National Natural Science Foundation of China (No. 31061160187) and the Fundamental Research Funds for the Central Universities (No. 201262022).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Cheng, J., Yanagimoto, T., Song, N. et al. Population genetic structure of chub mackerel Scomber japonicus in the Northwestern Pacific inferred from microsatellite analysis. Mol Biol Rep 42, 373–382 (2015). https://doi.org/10.1007/s11033-014-3777-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-014-3777-2