
Abstract
The new transcriptomes provided comprehensive sequence profiling data of transcriptomic variation during vernalization in Lily Asiatic Hybrids ‘Tiny ghost’. A number of 52,277,184 sequencing raw reads totaling 5.11 Gbp of the chilling treatment (4 °C) sample and 39,466,176 sequencing raw reads totaling 3.85 Gbp of room temperature control (25 °C) sample were assembled de novo into 68,718 unigenes with a mean length of 984 bp, and a total of 33,208 (45.6 %) unigenes were annotated by using public protein databases with a cut-off E value about 10−5. There are 6,153 unigenes of which were assigned to specific metabolic pathways by the Kyoto encyclopedia of genes and genomes. Gene Ontology analysis of the annotated unigenes revealed that the majority of sequenced genes were associated with signal transduction mechanisms, posttranslational modification, protein turnover and chaperones. In addition, the genes expression levels were compared just after vernalization completion between the cold treatment and room temperature control. There are 68,116 unigenes were differentially expressed, and hierarchical clustering analysis arranged 7,301 significantly differentially-expressed unigenes into 56 groups. Six genes related to the vernalization were selected to confirm their expression levels by using quantitative real-time polymerase chain reaction. Furthermore, typical vernalization unigenes VRN1 and VRN2 were identified, and also some vernalization-associated unigenes, such as CBF, SOC, TaAGL, AP2, LEA, LIM et al. were also annotated in the present study. As for VRN1 and VRN2, their expressions were consistent with some previous related studies. Also, this was the first time the vernalization genes VRN1 and VRN2 were founded in lily. According to the results of the present studies, we predicted that they would play an important role during vernalization in Lily Asiatic Hybrids; these data provided the foundation for the future studies of metabolism during vernalization of Asiatic lily.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Vernalization is a critical developmental process that allows plants to flower by exposing to cold temperatures. Lily is one of the most important flowering crops and is second to the rose in the cut flower market worldwide [1]. Among the species in Lilium, Lily Asiatic Hybrids is loved by people for its richest variations in colors and beautiful shapes. But the molecular mechanisms regulating vernalization of these species remain unclear. As a powerful modern genetic research tool, transcriptome sequencing has been used to analyze plenty of species, e.g. Gossypium hirsutum [2], Pyrus (Pyrus pyrifolia ear group) [3], Chrysanthemum lavandulifolium [4], Prunus mume [5], Cymbidium sinense [6], but transcriptome and genomic information for lily is currently unavailable. Transcriptome and expression profiling data for this species would be needed as an important resource to identify genes and to better understand the biological mechanisms of vernalization in lily. Vernalization is the acquisition of a plant’s ability to flower by exposing to the prolonged cold of winter. That is to say, plants grown in temperate climates which require vernalization must experience a period of low winter temperature to initiate or accelerate the flowering process [7]. After vernalization, plants have acquired the ability to flower, but they may require additional seasonal cues or weeks of growth before they would actually flower. Vernalization temperatures are usually between 5 and 10 °C. So far, much progress has been made on vernalization.
Genetic analysis of winter wheat found that VRN1, VRN2, VRN3, VRN4 and VRN5 were the major genes that control its vernalization of winter wheat [8–10]. They controlled wheat vernalization habits through combinations and mutual effect of different recessiveness and dominance. VRN1 gene is a flowering promoting factor induced by low temperature in the hexaploid genome wheat, VRN1 gene had three copies, the VRNA1, VRNB1 and VRND1, located on chromosome 5A, 5B and 5D individually [11]. Further study showed that any one of three genes VRNA1, VRNB1 and VRND1 is dominant, it’s growth characteristics of spring, they do not need cold-induced for vernalization, or shorter jarovization could induce blooming. If all three genes are recessive its growth characteristics of winter. In the process of growth, they would take a long time of low temperature jarovization to blossom.
VRN2 is a flowering inhibiting factor, and dominant for winter habit [12]. Any functional copy of VRN2 is sufficient to inhibit flowering. And the product of VRN2 is the repressor of VRN1. VRN2 was cloned by Yang et al. [13] in Arabidopsis, and they found that VRN2 was a single copy gene in Arabidopsis genome. Furthermore, scientist also found that the flowering inhibitor gene FLC in Arabidopsis and rice has a similar function to VRN2, both of whom inhibit flowering and are repressed by vernalization at the same time [9, 14, 15]. Wheat VRT2 gene is a MADS-box transcription factor, with a similar vernalization response mode to VRN2. Danyluk being studied the molecular interaction relationship between VRN1, VRN2 and VRT2 genes and their products by transient expression analysis in Nicotiana benthamiana and found that VRT2 was a transcription repressor of VRN1, VRT2 bonding with CArG domain in promoter region of VRN1 to inhibit the expression of VRN1. Meanwhile, VRN2 neither united with VRN1 promoter region nor affected its activity directly. But it enhance the inhibitory effect of VRT2, indicating that there was a regulation complex joint affecting the expression of VRN1 together [16]. In winter wheat, Timing expression showed that VRT2, VRN2 and VRN1 accumulate in apical meristems and tender leaves. And it also proved that VRT2 and VRN2 inhibit flowering, and VRN1 promotes flowering. While in the short-day condition, the unvernalized spring-habit wheat accumulate VRT2 and postpone flowering, which indicated that VRT2 was adjusted and controlled by light and low temperature separately. In a word, VRT2 and VRN2 inhibit the expression of VRN1 together.
VRN3 gene shows homologous with FT in Arabidopsis and HvFT in barley [10, 11, 17]. VRN3 promotes flowering through the regulation of vernalization pathway and long-day pathway, and VRN3 a dominant allele related to spring habit. Sequence analysis showed that VRN3 in wheat is not mutated in the coding area, but the insertion of a reversal base to the promoter region can distinguish the recessiveness and show dominant allele genes. VRN3 gene links VRN1 and VRN2 in the vernalization pathway and genes in the photoperiod pathway to control the flowering time together.
In 1972, the fourth vernalization gene VRN4 was found by Pugsley in wheat (Triticum aestivum ‘Gabo’) [8], and the existence of VRN4 was proved by Gonch [18]. VRN5 was first proposed by Law and Wolfe [17], and was unified named VRNB4 in 1998.
The vernalization gene FaVRN1 was cloned from Festuca arundinacea by Wang et al. [19], and the vernalization gene VRN2 was cloned and constructed expression vector in Arabidopsis by Zhang et al. [20]. Other vernalization genes, such as FLOWERING LOCUS C (FLC), Arabidopsis vernalization genes FLC belongs to MADS-box gene [21], which encodes a protein transcription factor’s inhibitory effect on flowering, promoting some late flowering in Arabidopsis ecotypes and late flowering mutants flowering through negative regulation of transcription and protein expression levels.
When plants were exposed to low temperature, accompanied with vernalization, they will experience a serious of changes, such as feeling the low temperature, making a series of physiological and biochemical reaction changes, and adjusting genes’ expression, which is the process that transcription factor CBF1 binds to the CRT/DRE motif to induce COR gene expression and then improve the cold resistance for the plants.
To elucidate these mechanisms, advances in the next generation high-throughput sequencing technologies are now being exploited not only to the analysis of static genomes, but also dynamic transcriptomes in an approach termed RNA-seq [22–26]. Because of its higher sensitivity, base-pair resolution and the larger range of expression values can be detected [25–27]. In addition, RNA-Seq requires less prior knowledge of gene sequences. Through these powerful and rapidly evolving technologies, scientists have already made substantial contributions to understanding of genome expression and regulation in a number of plant species. The model species whose RNA-Seq analysis has been applied include Arabidopsis [28, 29], soybean [30, 31], rice [32], maize [33] and Medicago truncatula [34]. There are also some non-model plant species which lack a reference genome using RNA-Seq, such as Eucalyptus grandis [35], grape (Vitis vinifera L.) [36], California poppy (Eschschlozia califonica) [29], avocado (Persea americana) [29], Pachycladon enysii [37], and Artemisia annua [29].
Materials and methods
Plant materials and RNA isolation
Plants of Asiatic lily ‘Tiny ghost’ were collected from Kunming Academy of Agricultural Sciences. The underground bulbs were harvested when the aboveground parts withered, and they were placed in planting boxes (50 × 60 cm), covered with 30 cm planting soil, 20 bulbs per box, a total of 20 boxes. Treatments as follows: 10 boxes were placed in cold storage at 4 °C (treatment), while the other 10 boxes were placed in the room temperature at 25 °C (control). The intermediate scales were sampled every 5 days and the content of soluble sugars were determined by anthrone method. When the soluble sugar content of the cold treatment group reaches peak, 40 stem tips of each group were sampled, and stored in the refrigerator at −80 °C for the isolation of RNA. Total RNA was extracted using the EASYspin Plus plant RNA extraction kit (Aidlab, China). The quantity and quality of the isolated total RNA was examined using spectrophotometry (BIO-RAD SmartSpec Plus, USA), and loaded on a denaturing agarose gel to check the concentration and integrity of the RNA (BIO-RAD, USA).
Library preparation for transcriptome analysis and sequencing
Poly-A-containing mRNAs were purified from the total RNA samples by using the OligoTex mRNA mini kit (Qiagen, China). Then the mRNA was fragmented into small pieces by using RNA fragmentation kit (Ambion, USA). With these short fragments as the templates, the first cDNA strand was synthesized by using random hexamer primers and reverse transcriptase (Invitrogen, China), and the second-strand cDNA was synthesized by using DNA polymerase I and RNase H. The cDNA fragments were purified using the QiaQuick polymerase chain reaction (PCR) extraction kit (Qiagen, China) and resolved with EB buffer for end reparation and poly (A) addition. The short fragments were then connected with sequencing adapters, and the products were subsequently purified and amplified via PCR to create the final cDNA libraries. The cDNA library was sequenced by using Illumina HiSeq™ 2000, and the sequencing-derived raw image data were transformed by base calling into sequence data by using Illumina Pipeline Software v1.6. The raw reads were cleaned by the trimming of adaptor sequences, empty reads and ambiguous nucleotides (‘N’ in the end of the reads). The reads obtained were then assembled by using the SOAPdenovo program [38]. SOAPdenovo first combined those reads of a certain length of overlap to form longer fragments without N, which were called contigs. The resulting contigs were joined into scaffolds by using the paired-end reads. The paired-end reads were used again for the gap filling of scaffolds to obtain sequences with the least Ns that could not be extended at either end. Such sequences were defined as unigenes. In the final step, Basic local alignment search tool X (BLASTX) alignments (E value 0.00001) between unigenes and protein databases, including UniProt, Kyoto encyclopedia of genes and genome (KEGG) and clusters of orthologous groups (COGs), were performed, and the best alignment results were used to decide the sequence direction of the unigenes. When a unigene happened to be unaligned with none of the above databases, ESTScan software was used to predict its coding regions and to decide its sequence direction [39].
De novo assembly
Raw reads were assembled using SOAPdenovo (http://soap.genomics.org.cn/soapdenovo.html). The raw sequence data in fast a format were filtered to remove the following reads: (1) eliminate the low quality reads, they are less than 50 % proportion of the bases which their qualities are less than 20, and Q = −10logerror_ratio, (2) eliminate the reads which containing adapter or the length is less than 20 bp, (3) eliminate the reads with fuzzy N-containing bases, (4) eliminate ribosome RNA, using the Lilium’s rRNA for the comparison template, and the match ones maybe the ribosome RNA reads, and (5) eliminate the 3′ bases which their qualities are less than 20, and the mistake rate of bases are less than 0.01. Only clean reads were used in the following analysis. The contigs are the longest assembled sequences containing no Ns. We mapped the reads back to contigs and combined paired-end information, linked the contigs into scaffolds. Scaffold length was estimated on the basis of the average segment length of each pair of reads. By filling the gaps in scaffolds using paired-end reads, unknown bases were filled with Ns and unigene sequences were created. Unigene sequences were alignment to @blastdb using BLASTX (E value 0.00001). Sequence orientations were determined depending on the best hit in the database. If results from different databases clashed with each other, then the final choice was in line with @blastdb. The orientations and CDSs of sequences that had no hit in BLASTX were predicted using ESTScan [39]. The original transcript sequences (5′–3′) were supplied if their orientations could be determined. Other sequences were provided as assembler outputs functional annotation.
Unigene functional annotation provides information by BLASTX alignment (E value <10−5) between unigenes and protein databases, such as nr, Swiss-Prot, KEGG and Gene Ontology (GO) The unigenes were retrieved from these databases along with their protein functional annotations as long as they showed high sequence similarities to proteins that found in this study. Using this process, protein function could be deduced by comparison with other similar genes. The possible functions of a fasciata unigenes could be predicted and classified by compared to the COG database.
Together with nr annotation, the BLAST2GO program was used to obtain GO annotations for a fasciata unigenes [40], and also was used for GO functional enrichment analysis of certain genes by performing Fisher’s exact test with a robust false discovery rate (FDR) correction to obtain an adjusted P value between certain test gene groups and the whole annotation. After obtaining GO annotations for every fasciata unigene, GO functional classification for all the unigenes were undertaken by WEGO software, and also the distribution of gene function in the species at the macro level was investigated.
Analysis and mapping of DGE reads and differential gene expression
For gene expression analysis, in order to eliminates the influence of different gene lengths and sequencing discrepancies on the calculation of gene expression, the expression level of the unigenes was calculated and then normalized to RPKM (reads per kilobase of exon region per million) [22]. In this way, the calculated gene expressions can be directly compared among samples. The cut-off value for determining gene transcriptional activity was determined according to a 95 % confidence interval for all the RPKM values of each gene. Different expression genes were selected between treatment and control on condition of FDR <0.05, and fold-change ≥2.
Clustering of gene expression profiles
Hierarchical cluster analysis of the 75 gene expression patterns was performed with cluster [41] and Java Treeview [42] software, and the hierarchical clustering were analyzed by the log2Ratio for each gene.
qRT-PCR validation and expression analysis
Total RNAs were isolated from stem tip of the treatment and controlled as described above. First-strand cDNA synthesis was performed using Superscript II reverse transcriptase (Invitrogen) according to the manufacturer’s instructions, using 1 μg of total RNA and oligo (dT) primers. Real-time quantitative reverse transcription PCR (qRT-PCR) was performed using a Rotor-Gene 3000 real-time PCR detection system (Qiagen) using SYBR® qPCR Mix (Toyobo, Tokyo, Japan) according to the manufacturer’s protocol. The primers used in this study were designed by use of Beacon Designer (Premier, Palo Alto, CA, USA) and are listed in Table 1. Real time PCR reaction was carried out using the prepared cDNA (60 μg) with each set of primer and probe and iQTM SYBR® Green Supermix (Cat. number 170-8882, BIO-RAD, Hercules, CA, USA). PCR cycling was at 95 °C (10 s), 56 °C (30 s), and 72 °C (30 s). Three independent replicates were performed for each sample. The comparative CT method was used to determine the relative amount of LaLEA3, LaAGL20, LaAP2, LaCBF, LaLIM1, LaSOC1 and LaVRN2 in Asiatic lily ‘Tiny ghost’, with the expression of Actin used as an internal control.
Results
Annotation of predicted proteins
The output of sequenced data was from two experimental samples of cold treatment and room temperature control. 52,277,184 sequencing raw reads a total number of 5.11 Gbp from cold treatment sample and 39,466,176 sequencing raw reads totaling 3.85 Gbp of room temperature control sample. Total 91,743,360 sequence data had a mean length of 96 bp. These clean reads were assembled and produced to contigs, scaffolds and unigene sequences (Table 2). Finally, unigene data from the cold treatment and room temperature control were summarized into an all-unigene consisting of 68,718 sequences with a mean size of 984 bp, which included all non-redundant unigene sequences from both the cold treatment and room temperature control plant samples.
CDS prediction
All of the unigenes were compared with the sequences in public databases, including the National Center for Biotechnology Information (NCBI) COGs database, the UniProt protein database, and the KEGG database, using the BLASTX algorithm with an E value threshold of 10−5. A total of 33,208 unigenes had significant hits (E value 10−5) to the sequences in the UniProt protein database. The results also showed that 66.78 % of the unigenes over 500 bp in length had BLAST matches, whereas only 21.87 % of the unigenes were shorter than 300 bp. This absence of homology could be caused by several factors. There is limited information about the genomes or transcriptomes of Asiatic lily and its related species. Some unigenes were too short to allow statistically meaningful matches.
GO is an international standardized gene functional classification system that provides a standardized vocabulary that is used to assign function to uncharacterized sequences. BLAST2GO program [40]. A tool that associates GO terms with sequences based on several pieces of annotation evidence, was used to classify gene function in our dataset. Putative functions were assigned to 28,954 unique sequences involved in the categories of biological process (11,931), cellular component (4, 751) and molecular function (12,272; Fig. 1). As for the molecular function, the most two common types of genes were localized to the binding and catalytic activity, and the functions of the identified genes cover various electron carrier activity, transporter activity and structural molecule activity. The sequences encoded a broad set of transcripts represented within the biological process category. Among these, the single-organism process, response to stimulus, biological regulation, regulation of biological process and localization were better represented.

Gene Ontology classification of assembled unigenes. Note the unigenes were assigned to a biological process, b cellular component and c molecular function
Unigenes are aligned to the COG database, a database where orthologous gene products are classified, to predict and classify possible functions. A total of 14,511 sequences were assigned to the COG classifications (Fig. 2). Among the 25 COG categories, the cluster for signal transduction mechanisms (5,838; 15.27 %) represented the largest group, followed by posttranslational modification, protein turnover, chaperones (4,813; 12.59 %), general function prediction only (4,141; 10.83 %), RNA processing and modification (2,416; 6.31 %), intracellular trafficking, secretion, and vesicular transport (2,177; 5.60 %), transcription (1,689; 4.79 %) and secondary metabolites biosynthesis, transport and catabolism (1,598; 4.64 %).

COG functional classification of all unigene sequences. Note out of UniProt hits, 14,511 sequences were assigned to 25 COG classifications
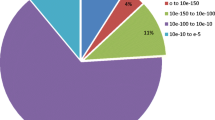
The assembled unigenes were assigned to the biochemical pathways described in KEGG based on their EC numbers. The KEGG database contains a systematic analysis of inner-cell metabolic pathways and functions of gene products. Pathway-based analyses help to understand the biological function of genes further. A total of 19,865 unigenes demonstrated sequence similarities to the genes in the KEGG database, and 3,825 unigenes were assigned to five KEGG biochemical pathways, including metabolic pathways (2,191 unigenes), biosynthesis of secondary metabolites (1,210), microbial metabolism in diverse environments (513), ribosome (324), and pyrimidine metabolism (311; Table 3). These results highlight the immense capacity of Illumina sequencing to discover genes in metabolic pathways.
Difference in genes related to vernalization of lily
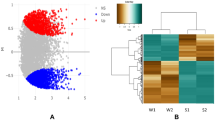
The difference in levels of differentially expressed genes between cold treatment and room temperature control was calculated according to RPKM value of the obtained unigenes. A total of 68,116 differentially expressed genes with FDR ≤0.001 and |log2Ratio| ≥1 were found (Fig. 3), of which, 7,301 differentially expressed genes with FDR ≤0.05 and |log2Ratio| ≥2 were screened, of which 4,401 genes were up-regulated and 2,900 were down-regulated (Fig. 3). These results demonstrated the overall difference in transcriptional expression level of differentially expressed genes between cold treatment and room-temperature control.

Differential expression level comparison of treatment with control
According to the Fig. 4, there is no significant difference in morphology between the cold treatment and room temperature control, but the internal physiological and biochemical has undergone tremendous changes. These specific traits are controlled by vernalization related genes. According to RPKM expression, GO annotation, and KEGG pathway annotation of the differentially expressed genes, literatures, and sequences of other species in GenBank, 59 unigenes defined as genes related to the formation of lily flower architecture were analyzed by cluster analysis (Fig. 5). The acquisition of these candidate genes provided theoretical basis for further understanding the formation mechanism of lily vernalization.

Lily growing point of cold treatment. Note a Before vernalization (cold storage for 0 days). b Just completed vernalization (cold storage for 27 days). (1) Shoot tips, and (2) inner scales

Cluster image of genes related to the vernalization of lily. Note data using RPKM value and taking its logarithm with 0.001 instead of 0 value. Each column represents a test sample, each row represents a change of gene expression of different multiples in different colors, red indicates up regulation, green down-regulation. (Color figure online)
From cold treatment and room temperature control, 4,401 genes were up-regulated and 2,900 were down-regulated. The results revealed that the overall transcription differences of cold treatment and room temperature control during vernalization. GO functional annotation and pathway analysis in KEGG database also revealed similar results as well. Some of the pathway-enriched unigenes encode the key enzymes regulating gibberellin, indole-3-acetic acid, and brassinosteroid biosynthesis, indicating that the architecture formation of lily may be related to hormone metabolic regulation. By comparison, the cold treatment and room temperature control showed significant differences in vernalization. Classical genetic studies showed that the vernalization of lily was a quality trait controlled by the combined effect of some vernalization genes. These differences are controlled by genes. In differentially expressed genes, we found the vernalization gene VRN1 (Contig63927) and VRN2 (Contig27291), VRN1 and VRN2 gene sequence showing homology with Oryza sativa subsp. and Malus domestica, respectively.
In GO functional annotation, VRN1 gets 29 clusters, among which GO:0008152 attributed to metabolic process of biological processes, another 28 clusters were attributable to molecular function (Table 4).
In COG annotation, the function attributed to posttranslational modification, protein turnover, chaperones. The predicted functions is AAA ATP enzyme. The best five results (cut off 10−5 E values) are shown in Table 5.
To further verify the expression profiles of genes in our Illumina sequencing analyses, six DEGs were selected randomly by qRT-PCR using the same samples originally used for RNA-Seq, including late embryogenesis abundant gene (LaLEA3), CBF-like transcription factor (LaCBF1), MADS-box transcription factor AGL6a (LaTaAGL), vernalization gene (VRN2), transcription factors regulate the expression of target gene (LaLIM1) and ethylene-responsive element binding protein (LaAP2). The results presented in Fig. 6 show that the expression levels of four genes were higher in treatment than those in control, including late embryogenesis abundant gene (LaLEA3), MADS-box transcription factor AGL (LaTaAGL), vernalization gene (LaVRN2) and transcription factors regulating the expression of target gene (LaLIM1), CBF-like transcription factor (LaCBF1) and ethylene-responsive element binding proteins (LaAP2) expressed much higher in control than that in treatment.

Expression levels of highly abundant LaAP2, LaCBF1, LaLEA3, LaLIM1, LaTaAGL and LaVRN2 genes in treatment and control of Asiatic lily ‘Tiny ghost’. Note total of RNA was using the same samples originally used for RNA-Seq. Real-time quantitative RT-PCR was performed to assay the gene expression of LaAP2, LaCBF1, LaLEA3, LaLIM1, LaTaAGL and LaVRN2 as indicated. The resulting mean values are relative to the expression of Actin. Legend 25 °C on behalf of the control group, and Legend 4 °C on behalf of the treatment group
Discussion
The advantages of high throughput sequencing technology in the study of lily vernalization
Compared with the chip technology, RNA-seq does not need reference genome information, but also has a higher accuracy. In addition, with the rapid development of NGS technology, the cost of sequencing is rapidly declining, and the data analysis technology has been constantly improved. These characteristics are especially attractive for the study of non-model species transcriptomics [43, 44]. Illumina/Solexa is the most extensive sequencing platform for NGS application, and has been widely used in the plant transcriptome research. In this research, a total number of 91.74 million 90 bp paired-end clean reads were achieved, 68,718 unigenes were generated and compared with the sequences in UniProt databases. 625,149,167 and 14,511 unigenes had significant similarity to existing sequences in GO, KEGG and COG databases, respectively. A bit of cold-induced up-regulated and down-regulated genes were found, which revealed the flower complicated regulation network during lily vernalization at transcriptome levels. According to Fig. 7, the results suggest that this library is a good source for studying the useful characters in the lily breeding.

Schematic diagram of floral induction pathways on Arabidopsis. Genes in red type were found by Ιllumina sequencing. (Color figure online)
Basal metabolism plays an important role in the lily vernalization process
In early twentieth century, scientists proved that the nutritional status of plants could influence plant flowering through a lot of experiments. The results of this study showed that the gene expression in nitrogen metabolism, starch and carbohydrate metabolism were significantly up-regulated during vernalization.
As shown in Table 6, Compared to the treatment sample, VRN1 express higher in control, on the contrary, VRN2 expresses higher in treatment group. According to the results and the previous studies on VRN1 and VRN2, they probably play an important role in vernalization of lily. However, considering the complexity and quantity of lily genome, it is not clear how these genes affect the architecture formation of lily vernalization.
VRN2 is a homeodomain protein of PRC2 complex subunit, is needed to sustain FLC gene silencing and the expression after vernalization in epigenomics [45, 46]. There are four pathways controlling lily flowering, floral repression pathway, autonomous promotion pathway, photoperiodic promotion pathway and vernalization promotion pathway. SUPPRESSOR OF OVEREXPRESSION OF CONSTANS1 (SOC1) can integrate these blossom signaling pathways to regulate flowering time [47, 48], which is presents in both monocotyledonous and dicotyledonous plants, and they all could encode the MADS-box transcription factor [49–54]. At present, some SOC1 homologous genes have been cloned, such as GMGAL1 of soybean (Glycine max) [55], CsCL1 and CsCL2 of orange (Citrus sinensis) [56], UNS of petunia (Petunia hybrid) [57], NAC1, widely found in higher plants. They were characterized by their abilities to regulate development of plants and response to abiotic-stresses, respectively. These genes had been implicated in a plant defense mechanism against environmental stresses such as high temperature and low temperature or drought stress and various pathogens. NAC, found in strawberry and peach, they were induced by abscisic acid or salicylic acid, which mediates plant responses to abiotic stresses [58, 59]. However, their biological function in plants is still unclear.
Genetic regulatory pathway during lily vernalization
Lily is a flower of summer dormancy. It can not be planted annually in natural conditions, and would not germination or rendered blind flower without being broken the dormancy. The dormancy of lily is not compulsive structural dormancy, but endogenous physiological dormancy caused by inhibitor, It needs the stimulation of low temperature, to complete flower bud differentiation to blossom and yield fruit, which limits the ornamental and market application to a certain extent. In this research, genes relate to four pathway controlling the timing of flowering, including floral repression pathway, autonomous promotion pathway, photoperiodic promotion pathway and vernalization promotion pathway [59] were found, such as VRN1, VRN2, LFY, SEP, FT, AP2, AG, SOC1.
According to the results of this research and other related reports, a gene regulation network of lily vernalization to flowering could be preliminary as shown in Fig. 7. There are four main pathways controlling lily flowering: floral repression pathway, autonomous promotion pathway, photoperiodic promotion pathway and vernalization promotion pathway. The first two pathways regulated the internal states; the other two pathways transmitted signals from external environment. In this four pathways, vernalization promotion pathway makes the largest difference on lily. Vernalization process is regulated by a series of genes. FLC was found as one of the key genes in the process of flowing, FLC interacts with CArG boxes of FT, SOC1 and LFY to inhibit their activity, and inhibits flowering induction [60]. FLC is mainly expressed in root and stem tip. Another vernalization related gene is FRI, which plays an important role in Arabidopsis thaliana vernalization through the up-regulation of the expression of FLC gene. The activation of FRI and FRI-like gene on FLC is more significant, which can overcome the plant itself inhibitory effect on FLC [61]. Besides, two genes VRN1 and VRN2, related to vernalization could keep the inhibitory action of FLC. Methylation in general was closely related to vernalization, which might influence flowering time by regulating the expression of FWA. In our study, LaVRN1, LaVRN2, LaSOC1, LaLFY, LaAP1, LaAPX, LaTaAGL genes related to vernalization were found. The interactions and the function in vernalizations pathway are shown in Fig. 7.
Conclusion
In this study, genomics and bio-informatics approaches were used to identify vernalization-related genes that are expressed specifically in reproductive organs of Asiatic lily. The RNA-Seq library revealed a great number of vernalization related genes in the constructed ESTs (Table 3). This is the first time that the vernalization gene VRN1 and VRN2 has been found in lily. According to the results of previous studies, we predict that they play an important role during vernalization in Lily Asiatic Hybrids, and the expression analysis proves that VRN1 gene is a flowering promoting factor, and VRN2 is flowering inhibiting factor. The results are consistent with previous studies. In Fig. 8, the FLC gene was not detected in this study, we speculated that there are two reasons, one is that no significant differences in expression, and the other is that maybe lily does not pass through this pathway, that is to say, there is another pathway to flower of lily which different from the model plant Arabidopsis. More research must be conducted. Whatever, these data provided a basis for future studies of metabolism in the process of vernalization. Furthermore, the highly expressed vernalization genes LoVRN2 was further identified by qRT-PCR and the vernalization regulatory network in Arabidopsis was outlined, thus it provided a scientific basis for further investigation of the molecular mechanisms for the underlying vernalization development of Asiatic lily.
Abbreviations
- BLAST:
-
Basic local alignment search tool
- COG:
-
Cluster of orthologous group Cluster
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- NCBI:
-
National Center for Biotechnology Information
- qRT-PCR:
-
Real-time quantitative reverse transcription polymerase chain reaction
References
Lee IL, Kyong CP, Ye SS, Jae HS, Soon JK, Jong KN, Jong HK, Nam SK (2011) Development of expressed sequence tag derived-simple sequence repeats in the genus Lilium. Genes Genomics 33(6):727–733
Wang G (2011) Transcriptional analysis of young cotton (Gossypium hirsutum) seedlings under salt stress via Solexa sequencing. PhD Dissertation, Shandong Agricultural University
Liu GQ, Li WS, Zheng PH, Xu T, Chen LJ, Liu DF, Hussain S, Teng YW (2012) Transcriptomic analysis of ‘Suli’ pear (Pyrus pyrifolia ear group) buds during the dormancy by NA-Seq. BMC Genomics 13:700–718
Huang H (2012) The molecular mechanism of salt response in Chrysanthemum lavandulifolium (Fisch. ex Trautv.) Makino. PhD Dissertation, Beijing Forestry University
Shi WF (2012) Research on the transcriptome of Prunus mume through RNA-Seq. PhD Dissertation, Beijing Forestry University
Zhang JX, Wu KL, Zeng SJ, Jaime ATDdS, Zhao XL, Tian CE, Xia HQ, Duan J (2013) Transcriptome analysis of Cymbidium sinense and its application to the identification of genes associated with floral development. BMC Genomics 14:279–292
Sung S, He YH, Tifani WE, Tamada Y, Johnson L, Nakahigashi K, Goto K, Steve EJ, Richard MA (2006) Epigenetic maintenance of the vernalized state in Arabidopsis thaliana requires like heterochromatin protein 1. Nat Genet (Nat) 38(6):706–710
Yan L, Loukoianov A, Tranquilili G et al (2003) Positional cloning of the wheat vernalization gene VRN1. Proc Natl Acad Sci USA 100:6263–6268
Yan L, Loukoianov A, Blechl A et al (2004) The wheat VRN2 gene is a flowering repressor down-regulated by vernalization. Science 303:1640–1644
Yan L, Fu D, Li C (2006) From the cover: the wheat and barley vernalization gene VRN3 is an orthologue of FT. Proc Natl Acad Sci USA 103:19581–19586
Mcintosha RA, Hart GE, Devos KM et al (1998) Catalogue of gene symbols for wheat. In: Wheat genetics symposium. University of Saskatchewan, Saskatoon, pp 1–236
Dubcovsky J, Lijavetzky D, Appendino L et al (1998) Comparative RFLP mapping of Triticum monococcum genes controlling vernalization requirement. Theor Appl Genet 97:968–975
Yang Y, Xu BY, Jin ZQ (2003) Cloning and analysis of vernalization-related gene VRN2 in Arabidopsis thaliana. Chin J Trop Crops 24:46–50
Michaelsd A (1999) Flowering locus encodes a novel MADS domain protein that acts as a repressor of flowering. Plant Cell 11:949–956
Sheldon C, Burn E, Perez P et al (1999) The FLC MADS box gene are pressor of flowering in Arabidopsis regulated by vernalization and methylation. Plant Cell 11:445–458
Danyluk J, Kane NA, Breton G, Limin AE, Fowler DB, Sarhan F (2003) TaVRT-1, a putative transcription factor associated with vegetative to reproductive transition in Cereals. Plant Physiol 132(4):1849–1860
Law CN, Wolfe MS (1966) Location of genetic factors for mildew resistance and ear emergence time on chromosome 7B of wheat. Can J Genet Cytol 18:462–470
Gonch AP (2003) Genetics of growth habit (spring vs winter) in common wheat: confirmation of the existence of dominant gene Vrn4. Theor Appl Genet 107:768–772
Wang XL, Chen W, Li WC, Wu JH, Liu XX, Yang YC (2009) Cloning and characterization of vernalization gene FaVRN1 from Tall Fescue. J Nucl Agric Sci 23(5):778–784
Zhang WH, Hong LP, Zeng BY (2011) Cloning vernalization-related gene VRN2 and construction of its plant expression vector in Arabidopsis thaliana subtropical. Plant Sci 40(1):1–4
Liu LN, Liu W, Ye QS (2003) Advances on research of vernalization-related gene FLC. Acta Bot Boreal Occident Sin 23(12):2229–2234
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5(7):621–628
Lister R, Gregory BD, Ecker JR (2009) Next is now: new technologies for sequencing of genomes, trancriptomes, and beyond. Curr Opin Plant Biol 12:107–118
Marguerat S, Bähler J (2010) RNA-seq: from technology to biology. Cell Mol Life Sci 67:569–579
Wilhelm BT, Landry JR (2009) RNA-Seq-quantitative measurement of expression through massively parallel RNA-Sequencing. Methods 48:249–257
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63
Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y (2008) RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res 18:1509–1517
Weber APM, Weber KL, Carr K, Wilkerson C, Ohlrogge JB (2007) Sampling the Arabidopsis transcriptome with massively parallel pyrosequencing. Plant Physiol 144(1):32–42
Wall PK, Leebens MJ, Chanderbali AS, Barakat A, Wolcott E, Liang H, Landherr L, Tomsho LP, Hu Y, Carlson JE, Ma H, Schuster SC, Soltis DE, Soltis PS, Altman N, de Pamphilis CW (2009) Comparison of next generation sequencing technologies for transcriptome characterization. BMC Genomics 10:347
Libault M, Farmer A, Joshi T, Takahashi K, Langley RJ, Franklin LD, He J, Xu D, May G, Stacey G (2010) An integrated transcriptome atlas of the crop model Glycine max, and its use in comparative analyses in plants. Plant J 63:86–99
Severin AJ, Woody JL, Bolon YT, Joseph B, Diers BW, Farmer AD, Muehlbauer GJ, Nelson RT, Grant D, Specht JE, Graham MA, Cannon SB, May GD, Vance CP, Shoemaker RC (2010) RNA-Seq atlas of Glycine max: a guide to the soybean transcriptome. BMC Plant Biol 10:160
Lu T, Lu G, Fan D, Zhu C, Li W, Zhao Q, Feng Q, Zhao Y, Guo Y, Li W, Huang X, Han B (2010) Functional annotation of the rice transcriptome at single-nucleotide resolution by RNA-seq. Genome Res 20:1238–1249
Barbazuk WB, Emrich SJ, Chen HD, Li L, Schnable PS (2007) SNP discovery via 454 transcriptome sequencing. Plant J 51(5):910–918
Cheung F, Haas BJ, Goldberg SMD, May GD, Xiao Y, Town CD (2006) Sequencing Medicago truncatula expressed sequenced tags using 454 Life Sciences technology. BMC Genomics 7:272
Novaes E, Drost DR, Farmerie WG, Pappas GJ Jr, Grattapaglia D, Sederoff RR, Kirst M (2008) High-throughput gene and SNP discovery in Eucalyptus grandis, an uncharacterized genome. BMC Genomics 9:312
Bellin D, Ferrarini A, Chimento A, Kaiser O, Levenkova N, Bouffard P, Delledonne M (2009) Combining next-generation pyrosequencing with microarray for large scale expression analysis in non-model species. BMC Genomics 10:555
Collins LJ, Biggs PJ, Voelckel C, Joly S (2008) An approach to transcriptome analysis of non-model organisms using short-read sequences. Genome Inform 21:3–14
Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, Li Y, Li S, Shan G, Kristiansen K, Li S, Yang H, Wang J (2010) De novo assembly of human genomes with massively parallel short read sequencing. Genome Res 20:265–272
Iseli C, Jongeneel C, Bucher P (1999) ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc Int Conf Intell Syst Mol Biol 1:134–148
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M (2005) BLAST2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676
de Hoon MJL, Imoto S, Nolan J, Miyano S (2004) Open source clustering software. Bioinformatics 20(9):1453–1454
Saldanha AJ (2004) Java Treeview—extensible visualization of microarray data. Bioinformatics 20(17):3246–3248
Okoniewski M, Miller C (2006) Hybridization interactions between probesets in short oligo microarrays lead to spurious correlations. BMC Bioinform 7(1):276
Royce TE, Rozowsky JS, Gerstein MB (2007) Toward a universal microarray: prediction of gene expression through nearest-neighbor probe sequence identification. Nucleic Acids Res 35(15):99
Gendall AR, Levy YY, Wilson A, Dean C (2001) The VERNALIZATION 2 gene mediates the epigenetic regulation of vernalization in Arabidopsis. Cell 107:525–535
Bastow R, Mylne JS, Lister C, Lippman Z, Martienssen RA, Dean C (2004) Vernalization requires epigenetic silencing of FLC by histone methylation. Lett Nat 427:164–167
Simpson GG, Dean C (2002) Arabidopsis, the Rosetta stone of flowering time. Science 296:285–289
Parcy F (2005) Flowering: a time for integration. Int J Dev Biol 49:585–593
Lee H, Suh SS, Park E, Cho E, Ahn JH, Kim SG, Lee JS, Kwon YM, Lee I (2000) The AGAMOUS-LIKE 20 MADS domain protein integrates floral inductive pathways in Arabidopsis. Genes Dev 14:2366–2376
Lee S, Kim J, Han JJ, Han MJ, An G (2004) Functional analyses of the flowering time gene OsMADS50, the putative SUPPRESSOR OF OVEREXPRESSION OF CO 1/AGAMOUS-LIKE 20(SOC1/AGL20)ortholog in rice. Plant J 38:754–764
Lee J, Oh M, Park H, Lee I (2008) SOC1 translocated to the nucleus by interaction with AGL24 directly regulates LEAFY. Plant J 55:832–843
Cseke LJ, Zheng J, Podila GK (2003) Characterization of PTM5 in aspen trees: a MADS-box gene expressed during woody vascular development. Gene 318:55–67
Ferrario S, Busscher J, Franken J, Gerats T, Vandenbussche M, Angenent GC, Immink RGH (2004) Ectopic expression of the petunia MADS-box gene UNSHAVEN accelerates flowering and confers leaf-like characteristics to floral organs in a dominant negative manner. Plant Cell 16:1490–1505
Nakamura T, Song IJ, Fukuda T, Yokoyama J, Maki M, Ochiai T, Kameya T, Kanno A (2005) Characterization of TrcMADS1 gene of Trillium camtschatcense (Trilliaceae) reveals functional evolution of the SOC1/TM3-like gene family. J Plant Res 118:229–234
Zhong XF, Dai X, Xv J, Wu HY, Liu B, Li HY (2012) Cloning and expression analysis of GmGAL1, SOC1 homolog gene in soybean. Mol Biol Rep 9(6):6967–6974
Tan FC, Swain SM (2007) Functional characterization of AP3, SOC1 and WUS homologues from citrus (Citrus sinensis). Physiol Plant 131:481–495
Zhang CH, Liu H, Yu ML, Ge AJ, Dong QH (2011) Bioinformatics analysis of the NAC gene family in strawberry. Jiyinzuxue Yu Yingyong Shengwuxue (Genomics Appl Biol) 30(41):1261–1271
Zhang CH, Shangguan LF, Yu ML, Zhang YP, Ma RJ (2012) Bioinformatics analysis of NAC gene family in peach. Jiangsu J Agric Sci 28(2):406–414
Yaron YL, Caroline D (1998) The transition to flowering. Plant Cell 10:1973–1990
Srikanth A, Schmid M (2011) Regulation of flowering time: all roads lead to Rome. Cell Mol Life Sci 68:2013–2037
Jung C, Muller AE (2009) Flowering time control and applications in plant breeding. Trends Plants Sci 14(10):563–573
Acknowledgments
This work was supported by the China National Natural Science Foundation (Grant numbers 31071815 and 31272204), ‘863’ research program (Grant number 2011AA10020804), and the D. Programs Foundation of the Ministry of Education of China (Grant number 20110014110006). Xiaohua Liu and Huang Jie contributed equally to this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Huang, J., Liu, X., Wang, J. et al. Transcriptomic analysis of Asiatic lily in the process of vernalization via RNA-seq. Mol Biol Rep 41, 3839–3852 (2014). https://doi.org/10.1007/s11033-014-3250-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-014-3250-2