
Abstract
In recent years, the increased availability of the DNA sequences has given the possibility to develop and explore the expressed sequence tags (ESTs) derived SSR markers. In the present study, a total of 1956 ESTs of finger millet were used to find the microsatellite type, distribution, frequency and developed a total of 545 primer pairs from the ESTs of finger millet. Thirty-two EST sequences had more than two microsatellites and 1357 sequences did not have any SSR repeats. The most frequent type of repeats was trimeric motif, however the second place was occupied by dimeric motif followed by tetra-, hexa- and penta repeat motifs. The most common dimer repeat motif was GA and in case of trimeric SSRs, it was CGG. The EST sequences of NBS-LRR region of finger millet and rice showed higher synteny and were found on nearly same positions on the rice chromosome map. A total of eight, out of 15 EST based SSR primers were polymorphic among the selected resistant and susceptible finger millet genotypes. The primer FMBLEST5 could able to differentiate them into resistant and susceptible genotypes. The alleles specific to the resistant and susceptible genotypes were sequenced using the ABI 3130XL genetic analyzer and found similarity to NBS–LRR regions of rice and finger millet and contained the characteristic kinase-2 and kinase 3a motifs of plant R-genes belonged to NBS–LRR region. The In-silico and comparative analysis showed that the genes responsible for blast resistance can be identified, mapped and further introgressed through molecular breeding approaches for enhancing the blast resistance in finger millet.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Finger millet (Eleusine coracana (L.) Gaertn.) (2n = 4, x = 36), sub-species coracana, belongs to the family Poaceae and the genus Eleusine in the tribe Eragrostideae. Finger millet commonly known as ragi (India), bulo (Uganda), wimbi (Swahili), and tellebun (Sudan) and is an important cereal crop for subsistence agriculture in dry areas of Eastern Africa, India, and Sri Lanka. It has nutritional qualities superior to that of rice and is on par with that of wheat [1]. It is rich in protein (6–13 %) and calcium (0.3–0.4 %) and serves as an important staple food for rural populations in developing tropical countries where calcium deficiency and anaemia are widespread [2]. Plant diseases are the greatest deterrents to crop production worldwide. Among them, Magnaporthe grisea (anamorph: Pyricularia oryzae), a filamentous ascomycetes fungus, causes blast disease in economically important crops like rice. One of the major biotic constraints in the finger millet crop is the high seed yield loss (50 %) due to blast disease caused by the pathogenic fungus, M. grisea. This demands genetic improvement of finger millet by transferring plant resistant genes against fungus [3]. Fungus affects leaves, fingers, neck and discolors the seeds. Hence, major efforts have been devoted for understanding the molecular mechanisms of genetic resistance and incorporating them into breeding programs to avoid yield loss caused by pathogen.
The analysis of DNA sequence variation is of major importance in genetic studies. In this context, molecular markers are a useful tool for assaying genetic variation, and have greatly enhanced the genetic analysis of crop plants. Microsatellites or simple sequence repeat (SSR) markers have been useful for integrating the genetic, physical and sequence-based physical maps in plant species, and simultaneously have provided molecular breeders with an efficient tool to link phenotypic and genotypic variation. The disadvantages of SSR markers are that they are often tedious and costly cloning and enrichment procedures required for their generation [4–6]. Microsatellites developed from expressed sequence tags (ESTs), popularly known as EST–SSRs or genic microsatellites, represent functional molecular markers as a putative function for a majority of such markers can be deduced by database searches and other in silico approaches. Furthermore, EST–SSR markers are expected to possess high inter specific transferability as they belong to relatively conserved genic regions of the genome. The EST databases have become particularly attractive resources for such In-silico mining, as was demonstrated in, e.g., citrus [7], coffee [8], and particularly in the cereals [9], which can be further effectively being used in diversity studies [10], linkage map construction [11], QTL mapping studies [12] and marker assisted breeding programmes for disease resistance.
Disease resistance is frequently governed by specific recognition between pathogen AVIRULENCE (Avr) genes and corresponding plant disease RESISTANCE (R) genes. This type of gene for gene interaction usually is accompanied by a hypersensitive response leading to the restriction of pathogen growth [13]. Forty-eight R-genes have been cloned from numerous plant species (Arabidopsis, rice, wheat, etc.) using map-based cloning and transposon tagging [14]. The nucleotide binding site—leusine rich repeat (NBS–LRR) disease resistance genes belong to a large and diverse super family of genes, which can be subdivided based on characteristic N-terminal features of their products. Conserved amino acid sequence motifs in the NBS domain have been widely used to isolate and classify NBS–LRR encoding genes [15–18]. The NBS–LRR class resistance genes are proposed to act as receptors in signal transduction pathways that are triggered in response to pathogen attack. The NBS region of characterized R-genes and R-gene analogs (RGAs) contains several highly conserved motifs, in spite of the diversity of pathogens against which they act. Comparative genomics is a powerful tool for genome analysis, annotation with an objective to understand the detailed process of evolution at the gross level and to translate DNA sequence data into proteins of known functions. The rationale here is that DNA sequences encoding important cellular functions are likely to be conserved between species than the non-coding sequences. Thus the present study aimed at (1) Identifying the type, frequency and distribution of EST–SSRs of finger millet; (2) Comparative genomic analysis of NBS–LRR regions of finger millet with rice.
Materials and methods
Plant materials and genomic DNA isolation
For comparative genomic analysis, a set of 16 finger millet genotypes (ten resistant and six susceptible) were taken for the polymorphism analysis of NBS–LRR regions for blast resistance. The details of the genotypes used in the study along with the blast scoring data were given in Table 1. The genomic DNA of different accessions of finger millet was isolated by standard method [19], quantified and analyzed on agarose gel electrophoresis [20].
EST data mining
By July 2012 a total of 1956 EST sequences were available for finger millet in NCBI (GenBank) database. The downloaded sequences were obtained in FASTA format for sequence assembly and SSR analysis. The SSR identification was done using one pipeline tool “Websat” software [21]. Primers designed to flanking sequences using the Websat software which uses the Primer3 software. Five classes of SSRs, that is, di-, tri-, tetra-, penta-, and hexa nucleotide repeats were targeted for identification using this tool. Two types of search criteria were taken for identification of EST–SSRs. The first one was: the minimum number of repeats was 6 for dinucleotide, 4 for trinucleotide, and three for tetra, penta and hexanucleotides. While the second criterion was changed for tetra, penta and hexa with a minimum number of repeats as two was considered. The main parameters for primer designing were: GC content of 40–60 %, annealing temperature (Tm) of 50–60 °C and expected amplified products size of 100–450 bp. All other parameters were set to default values.
SSR amplification and detection
PCR was performed in 20 μL reaction volume containing 2 μL 10× buffer having 15 mM MgCl2, 0.2 μM of each forward and reverse primer, 2 μL of 2 mM dNTPs, 0.2 μL of 0.5 U/μL of Taq DNA polymerase (Invitrogen, USA), and about 50 ng of template DNA. Amplifications were performed in a Thermocycler (MJ Research, USA) programmed for an initial denaturation of 4 min at 95 °C followed by 35 cycles of 30 s at 95 °C, 30 s at (different annealing temperatures for different primers), 1.0 min at 72 °C, and a final extension of 10 min at 72 °C, and hold at 4 °C. The PCR products were fractioned on 3.5 % agarose gel. The electrophoresis was held at 100 volts for 3 h at room temperature. Gels were stained with ethidium bromide and visualized using Bio Imaging System (SynGene, USA) and the primers that amplified and showed detectable length polymorphism were identified. Only those bands that were clear and reproducible were scored for data analysis. Molecular weight of the bands was estimated using 100 bp DNA ladder as standard.
Strategy of comparative analysis of NBS–LRR regions
The EST sequences of finger millet were retrieved from the NCBI website. These sequences were used for BLASTn analysis and identified the Oryza sativa sequences producing high similarity at an E value more than 4e-13. Then identified the positions of these finger millet and rice EST sequences on the rice chromosome maps in the gramene website (www.gramene.org) and compared with the positions of different rice blast genes positions.
Sequencing by ABI 3130XL genetic analyzer
All primer pairs were initially tested via PCR and agarose gel analysis aimed at identifying those pairs producing single amplicons. The primers which produced single amplicons and showed polymorphism between resistant and susceptible genotypes were used for further analysis. The PCR products were purified using a QIAquick® PCR purification kit (Qiagen Inc., Valencia, CA, USA) according to the manufacturer’s protocol. Dideoxy cycle sequencing was performed using the chain-termination method and an ABI Prism Big Dye reaction kit (ver. 3.1) according to the manufacturer’s protocols (Applied Biosystems). The sequencing products were run on an ABI 3130XL genetic analyzer. Sequence editing and assembly of the contigs were performed using Sequencher 4.10. For sequence comparisons among the sequences of genotypes, BioEdit (ver. 7.0.5.3; [22]) with the Clustal W multiple alignment option was used and then adjusted manually by the authors. The PCR products were sequenced from both ends and the resulting termination products were analyzed on an ABI 3130XL genetic analyzer. The two resulting sequence traces derived from opposite ends of each amplicon were analyzed and aligned with standard DNA analysis software Phred and Phrap (http://www.phrap.org/).
Results and discussion
SSR mining and EST-SSRs frequency and distribution in finger millet
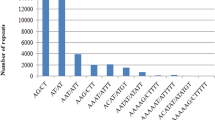
Simple sequence repeats (SSRs) were searched among the 1956 EST sequences of finger millet available in the NCBI website using the software Websat. The search was performed by considering two types of criteria. The first criteria includes repeat motif as: di- (with a repeat count n ≥ 6 repeat units), tri- (n ≥ 4), tetra- (n ≥ 3), penta- (n ≥ 3) and hexa- (n ≥ 3) nucleotides. According to this criteria, a total of 599 (30.6 %) out of 1956 ESTs had SSRs, of which 545 SSR primer pairs were designed. The remaining ESTs had SSRs at very close to the 5′ end or 3′ end of the sequences hence primer pairs could not be designed. Different workers used different criteria for finding the SSRs in the EST sequences. Recently, Reddy et al. [23] found that 324 ESTs had SSRs and developed 132 primer pairs out of the then available ESTs (1927) by using the criteria as n ≥ 5 for di-, tri-, tetra-, penta-, and hexa- repeats. However, we have taken the most common criteria used by several workers in different crops [24]. Thirty-two ESTs had more than two SSRs. As expected, the most frequent type of microsatellites corresponded to trimeric SSRs (320, i.e. represents 53.5 % of the detected SSRs and 16.4 % of total ESTs). The percentage of different SSR repeat motifs present in finger millet has been presented in Fig. 1. Similar findings have been reported by Victoria et al. [24], where they also identified trimer repeat motifs were the most frequent repeats among green algae and mosses; monocots and dicots. In case of medicinal plants also trimer motifs are the most abundant repeats. Zheng et al. [25] found 55 % of the ESTs of Chinese medicinal plant (Epimedium sagittatum (Sieb. Et Zucc.) Maxim) consisted of trimeric repeats. This showed that trimers are the most frequent motifs among crop species. Reddy et al. [23] found dimeric repeats as the most frequent among all the repeats which may be due to that, minimum of repeats was given five for finding the SSRs. In the present study, trimers were followed by the dimeric (102, 17 % of detected SSRs) and tetrameric (75, 12.5 % of detected SSRs) microsatellite repeats. The frequency of pentameric and hexameric SSRs was lower, representing only 23 (3.9 %) and 25 (4.2 %) of the detected microsatellites respectively. However, Reddy et al. [23] found only 11 penta nucleotides and no hexa nucleotide repeats since they used minimum criteria as 5 repeats. So, these results showed that minimum three repeats is sufficient for penta- and hexa- nucleotide repeat SSRs as evidenced in other crops [24, 25]. The second criteria have given different results in comparison to first criteria. According to the second criteria, the repeat motif used as follows: penta- (with a repeat count n ≥ 2 repeat units) and hexa- (n ≥ 2) nucleotides and the remaining di-, tri- and tetra repeats were same as first criteria. According to this criteria, a total of 1248 (64 %) out of 1956 ESTs had microsatellites, and ninety-five ESTs had more than two microsatellites which were higher than the first criteria. The most frequent type of microsatellites observed corresponding to pentameric SSRs (765, i.e. represents 39.1 % of the total ESTs) which was followed by hexameric (630, 32.2 % of the total ESTs).

The details of the repeat motifs (in percentage) present in finger millet
Among the dimeric SSRs, GA was found as the most common motif, followed by AG repeat motif and similar results were found by Reddy et al. [23]. GA was the most abundant motif which was in accordance with the reports in other crops [26–28]. However, Maia et al. [29], found AG/CT and GA/TC were the most frequent dimeric repeat motifs in the crops belonged to the family Poaceae. In case of Algae species, the most frequent dimer motif was AC/GT and CA/TG and in vascular plants like rice AG and GA was the most frequent dimer motif [24]. This showed that among the lower plant species AC and CA; and among higher plant species GA and AG were the frequent dimer motifs. The GA/CT motif represent codons GAG, AGA, UCU, and CUC, in mRNA which translate into the amino acids Arg, Glu, Ala, and Leu, respectively. In proteins, Ala and Leu are present in higher frequencies which results in the abundance of GA/CT motifs in EST sequences. Among trimeric SSRs, the most common motifs were CGG, followed by CTC, CCG and GCA and also supported by Reddy et al. [23]. In rice, CCG/CGG is the predominant trimeric motif and it appears to be high in the members of the grass family [27]. The motifs CCG/CGG, CGC/GCG, and GCC/GGC seem to be less common in other families [29]. For the three other classes, the most common SSR types corresponded to GAGC (for tetrameric SSRs), AAGAG (for pentameric SSRs), and CAGCTC (for hexameric SSRs). The details of the repeat type, frequency distribution have been presented in Table 2. Among the grasses, 0.85 % of all motifs were either CCTC/GAGG, AGGA/TCCT, or CATC/GATG. Other reports have shown ACGT as the most abundant in barley [30] and AAAG/CTTT and AAGG/CCTT in perennial ryegrass [31]. The pentamer repeat AAGAG is the most common repeat in finger millet and also in solanaceous crops [29]. These results showed that most of the higher crops share the common microsatellite repeat motifs.
Comparative genomic analysis of NBS-LRR of finger millet with rice
Comparative genomics has progressed the discovery and understanding of orthologues, and also it has brought to light many fast evolving ‘orphan’ genes of unknown function and evolutionary history. In Eleusine species, comparative analysis provides an opportunity to study rapid genome changes, function of the newly isolated gene sequences and their further utilization in molecular breeding assisted crop improvement programmes for developing high yielding, stress resistant genotypes. Srinivasachary et al. [32] studied the comparative genomic analysis of finger millet and rice genomes through molecular markers and showed that 85 % synteny present between these two crops. In this concern, there is a need to exploit the comparative genomic strategy to identify the molecular markers associated with important agronomic, physiological traits of finger millet. The blast fungus M. grisea causes major economic drain and main pathological threats to rice and finger millet crop over a greater part of the world. In the present study, 45 EST sequences of NBS–LRR region for disease resistance (including blast resistance) in finger millet were retrieved from the NCBI website. The EST sequences were clustered using the MEGA4 [33] software for finding the similar sequences. The MEGA4 analysis could group all the 45 EST sequences into five major clusters viz., A, B, C, D and E (Fig. 2). The major cluster A further divided into two minor clusters A1 and A1. The cluster A contains the 12 ESTs, cluster B 10 ESTs and cluster C, D and E contained 12, 7 and 4 EST sequences respectively. To identify the homology between finger millet and rice chromosomes, BLASTn analysis was performed with these EST sequences in the NCBI website and selected those had E value more than 4e–13. The finger millet ESTs clustered under ‘A’ exhibited homology to the rice EST sequences of AY337926, AY337898 and AF392824 (Table 3). To know the map locations of the finger millet EST sequences on rice chromosome map, these were used for BLASTn search engine of gramene website and obtained their positions on the rice chromosome maps and also same strategy was applied for rice EST sequences. The ESTs of finger millet clustered under C were showed homology with rice PiKh contig (GU258508) and four other sequences (DQ272576, AF220745, Y09812, AF074892) where both the finger millet and rice ESTs were hit on the eleventh chromosome. These results indicated that PiKh gene orthologs may be playing important role for the blast resistance in finger millet also. In a similar way, the finger millet EST sequences (GU301915, EU075236, EU075221) of cluster D showed homology to the rice ESTs (AB430853, DD461353 (both Pi21)) with a putative function related to Pi21 gene influencing the blast resistance and all were hit on the same rice chromosome, i.e. fourth.

Clustering pattern of EST sequences of NBS–LRR regions of finger millet generated by MEGA4
However, the ESTs of finger millet clustered in ‘A’ were hit on the 6th chromosome of rice, while the homologous rice EST sequences were hit on the 11th chromosome. Similarly, the ESTs of finger millet clustered in B were hit on the 2nd chromosome of rice, while the homologous rice EST sequences were hit on the 11th chromosome. Most of the ESTs of rice sequences were hit on the 11th chromosome; however finger millet ESTs were hit on 11th, 6th and 2nd chromosomes. This showed that the finger millet blast resistance may be governed by the orthologous regions of rice blast genes. There was high synteny between rice and finger millet chromosomes as reported by Srinivasachary et al. [32] and the same was authenticated by our results.
Functional marker based characterization and sequencing analysis
A total of 22 SSRs were identified, and 15 SSR loci were designed from sixteen GenBank accessions representing the NBS–LRR region of rice which showed similarity with the finger millet EST sequences. Of the 15, two primer pairs were not amplified among the selected resistant and susceptible genotypes of finger millet. The finger millet genotypes used in the present study were thoroughly characterized for years together for their response to blast resistance (Table 1). Hence, for the present study, 13 SSR primers were used for molecular characterization of ten resistant and six susceptible finger millet genotypes. Out of the thirteen SSR loci, eight were found polymorphic (61 %). The details of the polymorphic primers have been given in Table 4. The SSR loci FMBLEST5 designed from the rice EST contig (DQ272576.1) could able to clearly differentiate the susceptible and resistant genotypes (Fig. 3). The alleles which were unique to the resistant and susceptible genotypes were further used for sequencing and analysis by purifying the PCR product. A total of two sequences were obtained from two different resistant genotypes and one from the susceptible genotype with a Phred score above 40. The Clustal W was performed to know the similarity among the sequences and found high similarity among the resistant genotype sequences. However, sequence from the susceptible genotype did not show any similarity with the other two sequences and also did not hit with the rice resistant EST sequences after BLASTn analysis. Recently, Panwar et al. [34] studied the functional markers based molecular characterization of resistance gene analogs encoding NBS–LRR disease resistance proteins from a large collection of finger millet genotypes for association of NBS sequences with the blast disease resistance and susceptibility caused by blast fungus. The study established genetic relationships among the closely related genotypes and thus help build a more complete picture of the molecular characterization of finger millet genotypes. Such a characterization of genetic variation within natural populations and among breeding lines proves to be crucial for the effective blast management in finger millet or any other target crops. Reddy et al. [35] isolated resistance gene homologues from finger millet (Eleusine coracana L.) using degenerate oligonucleotide primers designed to the conserved regions of the nucleotide binding site of the previously cloned plant disease resistance genes. Of the 107 clones sequenced, 41 showed homology to known R-genes, and are denoted as EcRGHs (Eleusine coracana resistance gene homologues), while 11 showed homology to pollen signalling proteins (PSiPs), and are denoted as EcPSiPs (Eleusine coracana pollen signalling proteins).

Molecular profiling of susceptible and resistant finger millet genotypes based on the primer FMBLEST5 (sequencing was done with VR708 (Susceptible), VL 333 (R), VL 324 (R) genotypes)
The alleles sequenced from the resistant (three alleles) and susceptible genotypes (one allele) were analyzed by performing BLASTn analysis to find the homologous sequences (sequences yet to submit to NCBI). The allelic sequences from resistant genotypes could find high similarity with the rice NBS-LRR regions (some were Pi genes of blast resistance), while the sequence obtained from the susceptible allele did not show any similarity with the any NBS-LRR region. The sequences obtained from the resistant alleles were hit on 11th chromosome of rice and the rice ESTs also hit on the 11th rice chromosome. Along with In-silico analysis, our results also showed the eleventh chromosomal region sequences of rice could be playing important role in the blast disease resistance in finger millet. The nucleotide sequence was further analyzed by conserved domain (CD) search available in the NCBI website and resulted in identification of the NB-ARC (nucleotide binding-APAF-1, R proteins, and CED-4) domain in our sequence. The NB-ARC domain is a novel signalling motif shared by plant resistance gene products and regulators of cell death in animals. This region includes a nucleotide-binding (NB) domain, consisting of kinase 1a (P-loop), 2 and 3a motifs [36], and several other short conserved motifs with unknown function. R gene products are key components in plant defence, which appears macroscopically as rapid localised host cell death at the site of pathogen ingress [37]. This hypersensitive response (HR) is a form of programmed cell death that is thought to impede further infection. The amino acid sequence of finger millet resistant genotype was further compared with the previously cloned plant disease resistance genes and it contained the characteristic NBS motifs of kinase-2 and kinase 3a of plant R-genes (Fig. 4) confirming that the sequences characterized in the present study belong to the NBS-LRR gene super family. Reddy et al. [35] also noticed kinase-2 and kinase-3a motifs in all the EcRGHs isolated from finger millet (Fig. 4). The NBS-LRR genes are usually grouped into two different subfamilies [38]; subfamily I contains the TIR element (Toll-interleukin-1 receptor-like domain) and has been found only in dicots, whereas the subfamily II which lacks the TIR domain (called non-TIR) was found in both monocots and dicots. The sequences converted to amino acid sequences also had a tryptophan residue (W) at the end of the kinase-2 motif, which indicates that these belong to the non-TIR sub-class of R-genes. The last amino acid residue of the kinase-2 domain can be used to predict with 95 % accuracy whether an R-gene belongs to the TIR–NBS–LRR class or the non-TIR–NBS–LRR family; conservation of tryptophan (W) at this location is tightly linked to the non-TIR class of R-genes (RPS2, RPS5 and RPP8) of A. thaliana, whereas conservation of aspartic acid reside (D) or its uncharged derivative asparagines (N) is characteristic of the TIR class of R-genes (N and L6) [39, 40]. Thus the present results showed that comparative genomic analysis of finger millet with rice genome sequences will be very much useful in exploiting the molecular marker studies for identification of the genes responsible for blast resistance, their mapping and for further introgression through marker assisted breeding approaches for enhancing the blast resistance in finger millet genotypes of locally well adopted germplasm.

The kinase2 and kinase3a motifs of NB–ARC domain present in the sequence of finger millet genotype as obtained from CD domain search
References
Latha MA, Venkateswara Rao K, Dashavantha Reddy V (2005) Production of transgenic plants resistant to leaf blast disease in finger millet (Eleusine coracana (L.) Gaertn.). Plant Sci 169:657–667
Chennaveeraiah MS, Hiremath SC (1991) Cytogenetics of minor millets. In: Tsuchiya T, Gupta PK (eds) Chromosome engineering in plants genetics breeding and evolution. Elsevier, Amsterdam, pp 613–627
Ceasar SA, Ignacimuthu S (2008) Efficient somatic embryogenesis and plant regeneration from shoot apex explants of different Indian genotypes of finger millet (Eleusine coracana (L.) Gaertn.). In Vitro Cell Dev Biol Plant 44:427–435
Zane L, Bargelloni L, Patarnello T (2002) Strategies for microsatellite isolation: a review. Mol Ecol 11:1–16
Squirrell J, Hollingsworth PM, Woodhead M, Russell J, Lowe AJ, Gibby M, Powell W (2003) How much effort is required to isolate nuclear microsatellites from plants? Mol Ecol 12:1339–1348
Weising K, Nybom H, Wolff K, Kahl G (2005) Application of DNA fingerprinting in plant sciences. DNA Fingerprinting in plants-principles, methods, and applications. CRC Press, Boca Raton, pp 235–276
Chen C, Zhou P, Choi YA, Huang S, Gmitter FG Jr (2006) Mining and characterizing microsatellites from citrus ESTs. Theor Appl Genet 112:1248–1257
Poncet V, Rondeau M, Tranchant C, Cayrel A, Hamon S, de Kochko A, Hamon P (2006) SSR mining in coffee tree EST databases: potential use of EST-SSRs as markers for the Coffea genus. Mol Gen Genomics 276:436–449
Yu J-K, Dake TM, Singh S, Benscher D, Li W, Gill B, Sorrells ME (2004) Development and mapping of EST-derived simple sequence repeat markers for hexaploid wheat. Genome 47:805–818
Mishra RK, Gangadhar BH, Jae Woong Yu, Doo Hwan Kim, Se Won Park (2011) Development and characterization of EST based SSR markers in Madagascar periwinkle (Catharanthus roseus) and their transferability in other medicinal plants. Plant Omics J 4(3):154–162
Durand J, Catherine B, Emilie C, Frigerio JM et al (2010) A fast and cost effective approach to develop and map EST-SSR markers: oak as a case study. BMC Genom 11:570–583
Senthilvel S, Jayashree B, Mahalakshmi V, Sathish Kumar P, Nakka S, Nepolean T, Hash CT (2008) Development and mapping of Simple Sequence Repeat markers for pearl millet from data mining of Expressed Sequence Tags. BMC Plant Biol 8:119
Carine AT, Wang BB, Majesta SO, Shweta D, Zhu HY, Bruce R, Nevin DY, Steven BC (2008) Identification and characterization of nucleotide-binding site-leucine-rich repeat genes in the model plant Medicago truncatula. Plant Physiol 146:5–21
Okuyama Y, Kanzaki H, Abe A, Yoshida K, Tamiru M, Saitoh H, Fujibe T, Matsumura H, Shenton M, Galam DC, Undan J, Ito A, Sone T, Terauchi R (2011) A multifaceted genomics approach allows the isolation of the rice Pia-blast resistance gene consisting of two adjacent NBS-LRR protein genes. Plant J 66:467–479
Meyers BC, Kozik A, Griego A, Kuang H, Michelmore RW (2003) Genome-wide analysis of NBS–LRR encoding genes in Arabidopsis. Plant Cell 15:809–834
Meyers BC, Dickerman AW, Michelmore RW, Sivaramakrishnan S, Sobral BW, Young ND (1999) Plant disease resistance genes encode members of an ancient and diverse protein family within the nucleotide-binding superfamily. Plant J 20:317–332
He L, Du C, Covaleda L, Xu Z, Robinson AF, Yu JZ, Kohel RJ, Zhang HB (2004) Cloning, characterization, and evolution of the NBSLRR—encoding resistance gene analogue family in polyploidy cotton (Gossypium hirsutum L.). Mol Plant Microbe Interact 17:1234–1241
Cannon SB, Shu H, Baumgarten AM, Spangler R, May G, Cook DR, Young ND (2002) Diversity, distribution, and ancient taxonomic relationships within the TIR and non-TIR NBS-LRR resistance gene subfamilies. J Mol Evol 54:548–562
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8:4321–4326
Maniatis T, Sambrook J, Fritsch EF (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory, New York
Martins WS, Lucas DCS, Neves KFS, Bertioli DJ (2009) WebSat—A Web Software for microsatellite marker development. Bioinformation 3(6):282–283
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser 41:95–98
Reddy BLIN, Lakshmi NM, Sivaramakrishnan S (2012) Identification and characterization of EST–SSRs in finger millet (Eleusine coracana (L.) Gaertn.). J Crop Sci Biotech 15(1):9–16
Victoria FC, da Maia Luciano C, de Oliveira Antonio C (2011) In silico comparative analysis of SSR markers in plants. BMC Plant Biol 11:15–30
Zeng S, Xiao G, Guo J, Fei Z, Xu Y, Bruce A Roe, Wang Y (2010) Development of a EST dataset and characterization of EST-SSRs in a traditional Chinese medicinal plant, Epimedium sagittatum (Sieb. Et Zucc.) maxim. BMC Genom 11:94–105
Jia XP, Shi YS, Song YC (2007) Development of EST–SSR in foxtail millet (Setaria italica) [J]. Genet Resour Crop Evol 54:233–236
Kantety RV, La Rota M, Matthews DE, Sorrells ME (2002) Data mining for simple sequence repeats in expressed sequence tags from barley, maize, rice, sorghum and wheat. Plant Mol Biol 48:501–510
Thiel T, Michalek W, Varshney RK, Graner A (2003) Exploiting EST database for the development and characterization of gen-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet 106:411–422
Maia LC, Souza VQ, Kopp MM, Carvalho FIF, Oliveira AC (2009) Tandem repeat distribution of gene transcripts in three plant families. Genet Mol Biol 32:1–12
Varshney RK, Thiel T, Stein N, Langridge P, Graner A (2002) In silico analysis on frequency and distribution of microsatel lites in ESTs of some cereal species. Cell Mol Biol Lett 7:537–546
Asp T, Frei UK, Didion T, Nielsen KK, Lubberstedt T (2007) Frequency, type, and distribution of EST–SSRs from three genotypes of Lolium perenne, and their conservation across orthologous sequences of Festuca arundinacea, Brachypodium distachyon, and Oryza sativa. BMC Plant Biol 7:36–47
Srinivasachary Dida MM, Gale MD, Devos KM (2007) Comparative analyses reveal high levels of conserved colinearity between the finger millet and rice genomes. Theor Appl Genet 115:489–499
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Panwar P, Jha AK, Pandey PK, Gupta Arun K, Kumar Anil (2011) Functional markers based molecular characterization and cloning of resistance gene analogs encoding NBS-LRR disease resistance proteins in finger millet (Eleusine coracana). Mol Biol Rep 38:3427–3436
Reddy BL, Srinivas Reddy D, Lakshmi Narasu M, Sivaramakrishnan S (2011) Characterization of disease resistance gene homologues isolated from finger millet (Eleusine coracana L. Gaertn). Mol Breed 27(3):315–328
Traut TW (1994) The function and consensus motifs of nine types of peptide segments that form different types of nucleotide binding sites. Eur J Biochem 222:9–19
Morel JB, Dangl JL (1997) The hypersensitive response and the induction of cell death in plants. Cell Death Differ 4:671–683
Pan Q, Wendel J, Fluhr R (2000) Divergent evolution of plant NBS–LRR resistance gene homologues in dicot and cereal genomes. J Mol Evol 50:203–213
Jeong SC, Hayes AJ, Biyashev RM, Saghai Maroof MA (2001) Diversity and evolution of a non-TIR-NBS sequence family that clusters to a chromosomal ‘hotspot’ for disease resistance genes in soybean. Theor Appl Genet 103:406–414
Penuela S, Danesh D, Young ND (2002) Targeted isolation, sequence analysis, and physical mapping of non-TIR NBSLRR genes in soybean. Theor Appl Genet 104:261–272
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Kalyana Babu, B., Pandey, D., Agrawal, P.K. et al. In-silico mining, type and frequency analysis of genic microsatellites of finger millet (Eleusine coracana (L.) Gaertn.): a comparative genomic analysis of NBS–LRR regions of finger millet with rice. Mol Biol Rep 41, 3081–3090 (2014). https://doi.org/10.1007/s11033-014-3168-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-014-3168-8