
Abstract
Resistance gene homologues were isolated from finger millet (Eleusine coracana L.) using degenerate oligonucleotide primers designed to the conserved regions of the nucleotide binding site (NBS) of previously cloned plant disease resistance genes (R-genes) using polymerase chain reaction (PCR). Of the eleven primer combinations tested, only five showed amplification of resistance gene homologues in finger millet. BLAST search of cloned finger millet DNA fragments showed strong homology to NBS-LRR-type R-genes of other crop species. Of the 107 clones sequenced, 41 showed homology to known R-genes, and are denoted as EcRGHs (Eleusine coracana resistance gene homologues), while 11 showed homology to pollen signalling proteins (PSiPs), and are denoted as EcPSiPs (Eleusine coracana pollen signalling proteins). The cloned EcRGH sequences were classified into four clusters, and EcPSiPs formed two separate clusters based on sequence homology at the amino acid level. The amino acid sequences of the cloned EcRGHs showed characteristic features of non-TIR-type R-genes, which have been identified in all the monocot species studied so far. Six EcRGHs-specific primers were designed based on the sequences obtained in finger millet; reverse transcription PCR was performed on the cDNA and revealed the expression of EcRGHs in finger millet. The ratio of non-synonymous to synonymous nucleotide substitution (dN/dS) in the NBS domains of finger millet RGHs varied from 0.3 to 0.7 for the different classes, which suggests a purifying selection, though the LRR region also needs to be considered to make predictions. This is the first report on RGHs in finger millet, which will serve as a valuable resource for finger millet improvement using molecular tools.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Finger millet (Eleusine coracana (L.) Gaertn), commonly known as Ragi (India), Bulo (Uganda), Wimbi (Swahili) and Tellebun (Sudan), is an important cereal crop for subsistence agriculture in dry areas of Eastern Africa, India and Sri Lanka. World finger millet production is about 4.5 million tonnes, of which 2.5 million tonnes are produced in Africa alone (ICRISAT and FAO 1996). India is one of the major cereal producing countries in the world and finger millet occupies a total area of about 1.5 million hectares with a production of 1.2 million tonnes (Anonymous 1995, 1996). Finger millet has an excellent nutritional value as its seeds contain 7–14% protein and are particularly rich in methionine, iron and calcium (Barbeau and Hilu 1993; Vadivoo et al. 1998). Even though finger millet is known to be one of the hardiest crops, it is affected by a number of diseases like blast, foot rot, smut, streak and mottling virus (Govindu et al. 1970). Blast of finger millet is a major disease caused by the fungus Pyricularia grisea (Cooke) Sacc. (formerly Pyricularia oryzae Cavara.), an anamorph of Magnaporthe grisea (Hulbert et al. 2001) Barr (Rossman et al. 1990) that causes blast disease in rice. It is a heterothallic, filamentous fungus pathogenic to almost 40 plant species in 30 genera of Poaceae including Eleusine. Blast caused by the fungus Pyricularia grisea is the most devastating disease affecting different aerial parts of the plant at all stages of its growth starting from seedling to grain formation. Yield loss due to blast may be around 28% (Vishwanath et al. 1986). In spite of a great deal of research on the pathogen and the disease, blast still remains a serious constraint to finger millet production in areas with conducive environments where the high-yielding cultivars become susceptible. Incorporation of genes for resistance in breeding populations, varieties and hybrids is the most effective means to manage diseases such as blast.
Plant disease resistance genes (R-genes) are important components of the genetic resistance mechanism in plants (Flor 1956) and have been studied in detail in many plants like Arabidopsis, tobacco, tomato, flax, rice and maize (Grant et al. 1995; Gassmann et al. 1999; Parker et al. 1997; Jones et al. 1994; Dixon et al. 1998; Whitham et al. 1994; Lawrence et al. 1995; Wang et al. 1999: Johal and Briggs 1992). Resistance genes are present as gene clusters of different specificities in the plant genome and are either allelic in nature and/or closely linked. Novel techniques for deploying resistance to various biotic stresses, such as genetic transformation, molecular marker-aided selection and gene transfer from alien sources, have yet to make an impact on finger millet research.
Many biotechnological tools are being increasingly used to study the interaction between plants and potential pathogens at all stages of infection (Michelmore 1995) and considerable progress has been made in the identification and cloning of resistance genes using map-based cloning and transposon tagging (Foote et al. 1997; Seah et al. 1998). The availability of cloned disease resistance genes permits studies of resistance gene structure and function. The majority of the plant disease resistance genes cloned so far show conserved DNA sequences and amino acid domains irrespective of whether they confer resistance to viral, bacterial or fungal diseases. Sequence comparison of known R-genes shows remarkable similarities in general protein structure and conservation of specific domains that participate in protein–protein interaction and signal transduction (Sivaramakrishnan and Seetharama 2001; Baker et al. 1997; Gassmann et al. 1999). The similarity in the resistance gene sequences among the plant species, both at the nucleotide and amino acid levels, has made it possible to isolate such resistance gene homologues (RGHs) from any plant species of interest using polymerase chain reaction (PCR) with oligonucleotide primers designed to the conserved domains (Michelmore 1996). To isolate homologues of the known R-genes, the nucleotide binding site (NBS) of the NBS-LRR family of proteins is normally chosen based on the conserved motifs present (Bai et al. 2002; Cannon et al. 2008; Michelmore and Meyers 1998; Young 2000). This approach, known as the ‘candidate gene approach’, has been successfully used in a variety of plant species, including potato, soybean and lettuce, to isolate disease resistance gene homologues (Yu et al. 1996; Kanazin et al. 1996; Leister et al. 1996; Shen et al. 1998). The availability of rice genome sequence enabled the global characterization of NBS-leucine-rich repeat (NBS-LRR) genes, the largest class of plant disease resistance genes (Goff et al. 2002).
As a first step in the identification and isolation of disease resistance genes from finger millet, the degenerate oligonucleotide primers designed to the conserved motifs in the NBS region were used to amplify DNA fragments. These DNA fragments were cloned, sequenced and screened for sequence homology with other known R-genes in the genome databases. We report here the identification and isolation of such RGHs identified in cultivated finger millet. Identification, cloning and mapping of resistance genes corresponding to major pathogens would contribute significantly to improvement of finger millet and related crops.
Materials and methods
Amplification of NBS-like sequences using degenerate primers
A local popular high-yielding cultivar of finger millet developed by ANGRAU, UR762, was selected for the isolation of RGHs from DNA. Genomic DNA was extracted from 5 g of leaf tissue from 2-week-old finger millet seedlings using a modified cetyltrimethylammonium bromide (CTAB) method (Porebski et al. 1997). The degenerate primers used were designed based on the results of previous studies (Shen et al. 1998) so as to amplify DNA sequences spanning the most conserved domains of the NBS. In each pair, the forward primers were designed to anneal to the sequence encoding the kinase-1a or P-loop domain (Shen et al. 1998) and the reverse primers were designed in the antisense orientation corresponding to the coding sequences of an amino acid domain known as GLPLAL (Lawrence et al. 1995).
A total of 18 individual primers including forward and reverse primers were synthesized (Table 1) and tested in 11 possible combinations (Table 2) for the amplification of RGHs from genomic DNA of finger millet cultivar UR762. PCR was performed with the different primer combinations in a 30 μl reaction volume containing 10 mM of Tris HCl (pH 8.3), 50 mM of KCl, 1.5 mM of MgCl2, 0.2 μM of each forward and reverse primer, 100 μM of dNTPs, 1 U of Taq DNA polymerase (Invitrogen, USA) and about 50 ng of template DNA. PCRs were carried out in a PTC-100 thermal cycler (MJ Research, USA). Thermocycler parameters were: initial denaturation step at 95°C for 5 min, followed by 30 thermal cycles of 95°C for 1 min, 45/51°C for 1 min depending on the primers and 72°C for 1 min, with a final extension step of 10 min.
Cloning of PCR products
Amplified PCR products were separated by electrophoresis on 1.5% TBE agarose gels and stained with ethidium bromide for visualization (Fig. 1). The expected 500-bp size fragments corresponding to RGHs and other major fragments were excised from the gel and purified using Qiagen gel extraction kit (Qiagen, USA). Ligation reaction was set up in 10 μl reaction volume with 50 ng of pGEM®-T Easy vector (Promega, USA) according to the manufacturer’s protocol. Ligation was carried out at 14°C for 16 h.

Agarose gel electrophoresis pattern of PCR amplified finger millet DNA. Lanes 1, 2 S1/AS1, 3 RO1/RO4, 4 S1/AS3, 5 100-bp ladder, 6, 7 S2/AS3, 8, 9 LM637/638. PCR was carried out using 11 degenerate primer combinations on finger millet DNA as the template. The amplified products were run on 1.5% agarose gel in TAE, stained with ethidium bromide and visualized on a UV-transilluminator
Competent cells of E. coli DH10B strain were prepared using 10% glycerol (Sambrook et al. 1989) and bacterial transformation was effected by electroporation. Colony PCR was carried out for the confirmation of DNA inserts in the selected putative recombinant clones. PCR was performed in a total reaction volume of 30 μl with M13 universal forward and reverse primers. The PCR was carried out in a PTC-100™ thermal cycler (MJ research®, USA) and the parameters were: initial denaturation step of 95°C for 5 min followed by 30 thermal cycles of 95°C for 1 min, 55°C for 1 min and 72°C for 1 min, with a final extension step of 10 min. PCR products were verified for cloned inserts by resolving the DNA fragments on a 1.5% TAE agarose gel.
Sequencing and analysis
Initially, plasmids were isolated from five recombinant clones from each ligation event by alkaline lysis using the PEG precipitation method (Sambrook and Russell 2001) and were sequenced. BLAST searches (Altschul et al. 1997) were performed via the National Centre for Biotechnology Information (NCBI) web site.
Dot blot hybridization of putative RGH clones was carried out to select more clones for sequencing according to the method of Sambrook et al. (1989). Ninety-six clones were taken for dot blot analysis from each ligation event and EcRGH clones of the same ligation event were used as probes for hybridization in the dot blots. Additional clones were identified based on the intensity of hybridization in the dot blot and were sequenced. Homology search was done using BLAST in the GenBank database. Eleusine coracana resistance gene homologues (EcRGHs) sequences were aligned by ClustalW (Thompson et al. 1994) method using the MgAlign tool of DNAstar software (Lasergene, USA). The reliability of phylogenetic tree topologies was tested by bootstrapping using the neighbour joining method of CLC Workbench3.1 software (www.clcbio.com). Amino acid sequences of NBS domains of three TIR-class R-genes (L6 of flax, N gene of tobacco, RPS4 of Arabidopsis thaliana) and three non-TIR R-genes (RPS2, RPS5 and RPP8 of A. thaliana) were included in the analysis. Seven rice R-genes and one pollen signalling protein (PSiP) gene were also included in the phylogenetic analysis for comparison.
Finger millet RGH-specific primers
Six RGH-specific primers were designed based on the RGHs isolated from finger millet following multiple alignments of RGHs using Primer3 software. Annealing temperatures were standardized using gradient PCR.
RT-PCR analysis
Total RNA was isolated from leaves of finger millet cultivar UR-762 using the the RNeasy Plant Mini Kit (Qiagen, USA) method. Total RNA was treated with RNAse-free DNase I (Promega, USA) to remove genomic DNA contamination in RNA preparations. For cDNA synthesis, 2 μg of DNA-free RNA was primed with oligo dT 23NV (New England Biolabs, USA) and the first strand cDNA was synthesized using 200 units of MMLV reverse transcriptase (New England Biolabs, USA). The second strand was synthesized using DNA Pol I, and the cDNA was purified and used for amplification with six finger millet RGH-specific primers. Control reactions were performed with RNA and cDNA using primers to check the genomic DNA contamination. PCR amplified products were cloned into pGEM-T Easy vector system (Promega, USA) and sequence analysis was carried out as mentioned earlier.
GenBank submission
Cloned RGH sequences of finger millet were deposited in GenBank with the following accession numbers: EF408611–EF408639 and EU075219–EU075263. The expressed finger millet RGHs identified using six EcRGH-specific primers were also deposited in GenBank EST database (GH612831–GH612847).
Results
Of the 11 primer combinations used to amplify the finger millet template DNA, seven amplified a major band in the expected size range of ~500 bp or more, as reported in the literature for other plant species. Two primer combinations, RO1/RO4 and PLoop/GLPLAL4, did not give any amplification. A representative amplification pattern with different primer combinations is shown in Fig. 1. The recombinant clones were tested for the presence of DNA inserts by colony PCR, which showed size differences in the DNA amplicons. Five recombinant clones from each event were sequenced initially and homology search was carried out to find similarity with R-genes from other plant species. Further, dot blot hybridization was carried out on the recombinant clones using as probes the EcRGHs identified earlier by sequencing. A total of 107 recombinant clones were identified for sequencing based on the results of colony PCR and dot blot hybridization.
Of the seven primer combinations tested with finger millet genomic DNA, only five combinations, S1/AS1, S2/AS1, S2/AS3, LM638/LM637 and Ploop/GLPLAL, yielded amplification of RGHs. The BLAST search indicated that the isolated EcRGHs showed strong homology to NBS-LRR disease resistance protein homologues from Oryza sativa, Saccharum officinarum, Pennisetum glaucum, Zizania latifolia, Ipomoea batatas, Zingiber cernuum, Lycopersicon hirsutum, Musa acuminate and Helianthus annuus (Table 3). The other primer combinations showed DNA fragments of larger size on PCR amplification.
A total of 41 clones, excluding the two short sequences, were aligned by ClustalW into six clusters. Nucleotide sequences of greater than 65% similarity were considered to belong to the same cluster. The construction of a phylogenetic tree based on the nucleotide sequences also revealed six major branches that were considered to be separate clusters of EcRGHs as supported by high bootstrap values. Clones generated from the amplified products of S2/AS1 combination were very divergent and formed five clusters. A major cluster (E) was formed with the cloned RGHs amplified with S2/AS3, S2/AS1 and LM638/LM637 primer combinations that were about 500 bp in size, whereas the clones generated from the 600 bp fragment amplified with S2/AS3 formed a separate cluster (D). Two clusters B and F were formed with single clones. Clones generated from the amplified product with PLoop/GLPLAL primer combinations were included in cluster E (Table 2).
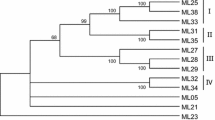
The nucleotide sequences of EcRGHs were translated and the amino acid sequences were aligned by the neighbour joining method (Fig. 2). The six clusters seen at the nucleotide level came down to four major clusters at the amino acid level, after eliminating the clones that did not show any open reading frame (ORF). Two clones from class C and one clone representing class B were eliminated as they did not show ORFs (cluster B and C were eliminated in Fig. 2 that were present in cluster analysis at nucleotide level). Of a total of 41 putative RGHs of finger millet, 31 RGHs showed uninterrupted ORFs after verification of translated amino acid sequences. The remaining RGHs had potential stop codons that resulted in distorted amino acid sequences upon translation; consequently these were unable to align with the other EcRGHs at the amino acid level.

Phylogenetic analysis of EcRGHs at amino acid level. The tree was created by using CLC Workbench software based on the homology at amino acid level RGHs. Six classes at the nucleotide level reduced to four classes at the amino acid level
Eleven clones from the Ploop/GLPLAL combination other than the RGH clones mentioned above showed homology to resistance genes from Oryza sativa and Musa acuminate in BLAST search but they did not have a non-TIR-NBS-LRR group characteristic or TIR characteristic consensus motifs. These 11 clones were aligned by ClustalW into two classes at the nucleotide level and upon translation only five out of the 11 sequences showed uninterrupted ORFs, which were divided into two classes based on 70% homology. These clones separated as different clusters when compared with EcRGHs and other RGHs from rice (Fig. 3).

Phylogenetic analyses of EcRGHs with EcPSiP and known rice R-genes at amino acid level. The tree was created by using the tree construction tool of CLC Workbench software. The algorithm used was UPGMA, and values indicated on the branches of the tree are the number of replicates taken in bootstrap analysis. Some sequences of known R-genes from rice were also included in the analysis for comparison
Some of the recombinant clones (EcPSiPs) showed higher similarity with PSiPs in rice than RGHs and formed a separate cluster. EcRGH21 formed a cluster by itself both at the nucleotide and amino acid levels. The amino acid sequences of EcRGHs were further compared with the previously cloned plant disease resistance genes (Fig. 3). The EcRGHs formed separate groups with some similarity to other NBS-LRR genes from monocots but not from dicots. All the 31 NBS sequences contained the characteristic NBS motifs of kinase-2 and kinase-3a of plant R-genes confirming that the clones characterized in the present study belong to the NBS-LRR gene superfamily. Amino acid sequences of the six clusters (A to F) have a tryptophan residue (W) at the end of the kinase-2 motif, which is characteristic of the non-TIR sub-class of R-genes (Fig. 4). Logos were constructed based on the conserved residues in the kinase-2 and kinase-3a motifs of the NBS domain of EcRGHs (Fig. 5a, b).

Alignment of EcRGHs proteins showing the conserved motifs like kinase-2 and kinase-3a of the NBS domain of R-protein. All the amino acid sequences are aligned and the kinase-2 and kinase-3a domains are marked using CLC Workbench software

a Logo of kinase-2 motif of NBS domain of EcRGHs showing the highly conserved residues. b Logo of kinase-3 motif of NBS domain of EcRGHs showing the highly conserved residues
Amino acid sequences of each class were aligned and six oligonucleotide primers were designed to the conserved regions of the EcRGHs (Table 4). All the six EcRGH-specific primer combinations showed a DNA fragment in the size range of 400 bp (Fig. 6) when the cDNA from finger millet was used as a template. The amplicons were further verified by colony PCR and three clones from each of the six ligation events were sequenced and the sequence data was used for homology searches.

Agarose gel electrophoresis pattern of finger millet DNA. RGHs amplified by RT-PCR with EcRGH-specific primers. Lanes 1–6 DNA fragments amplified with 6 different RGH-specific primers, lane M 1-kb DNA ladder, lane 7 RNA used as control template (for no DNA contamination), lane B no DNA (blank). PCR was carried out using gene-specific primers and amplified product was run on 1.5% agarose gel in TAE, stained with ethidium bromide and visualized on a UV transilluminator. Control reactions were performed with RNA and cDNA using primers to check the genomic DNA contamination
The ratios of non-synonymous to synonymous nucleotide substitution (dN/dS) were calculated for the four different EcRGH clusters and were considered to be indicators of evolutionary pressure on a class of genes. The values were found to be less than one (Table 5) in all the classes of EcRGHs.
Discussion
Many classes of R-genes are present in plant species and arise from a combination of mainly three protein domains, namely nucleotide binding sites (NBS), leucine-rich repeats (LRR) and kinase. These together or separately form component(s) of a signal transduction pathway in plants to elicit the right kind of defence response to counteract invasion of the different pathogens. Among the different classes of R-genes reported so far, the majority belong to the NBS-LRR class (Ayliffe and Lagudah 2004; Hulbert et al. 2001). The NBS domain has conserved amino acid sequences, which has given rise to the candidate gene approach that uses the conserved motifs among the NBS-LRR class resistance genes in plant species for large-scale isolation of resistance gene analogues from any plant species of interest using PCR with degenerate oligonucleotide primers (Shen et al. 1998; Madsen et al. 2003). The LRR region, on the other hand, is mostly involved in protein–protein interaction which is important in pathogen recognition by R-genes (Lehmann 2002).
The candidate gene approach was tried in one of the important coarse cereal crops, finger millet, in which little molecular work has been carried out so far. Sequence analysis indicated that the recombinant clones generated from one ligation event were not all similar but were different in terms of the actual nucleotide sequence and the length of the fragments amplified. The BLAST search indicated that cloned putative RGHs of finger millet showed homology to NBS-LRR-type disease resistance protein homologues from some of the monocots and also to other plant species like tomato, ginger and sunflower, suggesting the highly conserved nature of NBS-LRR domains in different crop species.
The percentage identity between clusters was less than 50%, and within each cluster there was more than 70% identity. Such phylogenetic grouping was further supported by high bootstrap values. Other approaches used to classify RGH sequences include cross-hybridization, but the results vary with the stringency of post-hybridization washes (Shen et al. 1998) and this approach was not used to classify EcRGHs. The use of degenerate forward primer with normal reverse primer can be used to amplify very divergent RGHs from different plant species, as has been demonstrated with peanut and finger millet in our laboratory (Srinivas Reddy 2008, unpublished data). The RGHs amplified with S2/AS3 and LM637/LM638 primers showed maximum homology with each other and were considered to belong to the same cluster, which could be due to the primer sequence similarity. Although many of the sequences showed high levels of similarity, a few nucleotide differences that were present might have functional importance. For example, an NBS region in potato associated with virus resistance line had a difference of only 12 nucleotides from the homologous regions found in all of the susceptible lines (Sorri et al. 1999).
It is well established that the protein-level search and comparisons showed more homology with the NBS-LRR region of many RGHs than at the nucleotide level. Thus, pair-wise comparison at the amino acid level is more reliable than at the nucleotide level for establishing the identity of RGHs (Totad et al. 2005). Hence, nucleotide sequences were translated into polypeptides and the comparison of amino acid sequences of EcRGHs showed similarity with NBS region of well-characterized R-genes of rice, RPS2, RPS5 and RPP8 of A. thaliana. Five of the recombinant clones (EcPSiPs) had higher homology with PSiP (Moutinho et al. 2001) of rice and lacked the kinase-2 and kinase-3a motifs characteristic of NBS type of R-genes. These were eliminated from cluster analysis of NBS sequences at the amino acid level.
The highly conserved kinase-2 and kinase-3a motifs were seen in all the EcRGHs isolated from finger millet. The NBS-LRR genes are usually grouped into two different subfamilies (Meyers et al. 1999; Pan et al. 2000); subfamily I contains the TIR element (Toll-interleukin-1 receptor-like domain) and has been found only in dicots, whereas the subfamily II which lacks the TIR domain (called non-TIR) was found in both monocots and dicots. The presence of a tryptophan (W) residue in the kinase-2 (FXXXXW) motif of the NBS domain in all the EcRGHs cloned indicates that these belong to the non-TIR-NBS-LRR subclass of R-genes as reported for most monocots (Meyers et al. 1999). The last amino acid residue of the kinase-2 domain can be used to predict with 95% accuracy whether an R-gene belongs to the TIR-NBS-LRR class or the non-TIR-NBS-LRR family; conservation of tryptophan (W) at this location is tightly linked to the non-TIR class of R-genes (RPS2, RPS5 and RPP8) of A. thaliana, whereas conservation of aspartic acid reside (D) or its uncharged derivative asparagine (N) is characteristic of the TIR class of R-genes (N and L6) (Meyers et al. 1999; Pan et al. 2000; Jeong et al. 2001; Penuela et al. 2002). In the TIR-NBS-LRR group there is a characteristic consensus motif FXXXXF and a highly conserved glycine that is present between kinase-1a and kinase-2 motifs, whereas the non-TIR-NBS-LRR group contains the consensus sequence FXXXXW (Pan et al. 2000).
In plant genomes, NBS-LRR-type genes are abundant, with approximately 150 described in the Colombia ecotype of Arabidopsis (Meyers et al. 2003), more than 500 in the rice genome (Bai et al. 2002; Goff et al. 2002; Monosi et al. 2004; Yang et al. 2006) and only about 50% in the sorghum genome in comparison to those present in rice (Paterson et al. 2009). The NBS-LRR genes of rice are very similar to the non–Toll-interleukin-1 receptor homologue region (non-TIR) class R-genes of Arabidopsis but none showed similarity to the TIR class genes. In monocots, non-TIR-NBS types of R-genes are the major class and the present work also indicated that a large number of R-gene homologues present in the finger millet genome belong to the major non-TIR-NBS-LRR class of R-genes. Prior to this report, no RGHs of finger millet were available in the literature or in GenBank. Consequently, the present study represents the first report on RGHs in finger millet.
The EcPSiPs have neither the FXXXXF characteristic of TIR-NBS-LRR nor the FXXXXW characteristic of the non-TIR-NBS-LRR group. PSiP is a signalling protein specific to pollen tissue and is involved in the control of polarity. The perturbation of its function with ODNs (oligodeoxynucleotides) suggests its fundamental role in the maintenance of transduction mechanisms operating in pollen tubes. The analysis of a PSiP amino acid sequence indicated the presence of a NBS motif A (also known as P-loop) for binding ATP or GTP, and LRRs predicting a role in signalling pathways. LRRs have been suggested to function downstream as mediators of protein–protein interactions in signal transduction pathways (Suzuki et al. 1990). The sequence resemblance of PSiP to disease resistance proteins evokes the possibility that in plants AC (adenylyl cyclase) enzymes might be disguised under the broad spectra of large gene families, like the R-gene family. Plant disease-resistance genes like the RPS2 gene contain leucine zipper, P-loop domains and LRRs, an interaction domain similar to that of yeast AC (Bent et al. 1994). The intimacy between AC and stress signalling proteins in plants has been suggested (Bolwell 1992), and the cellular responses driven by pathogens elicit a response similar to the one operating in pollen tubes.
Expression studies of RGHs in a particular tissue are not yet studied in detail; there are also very few reports on R-gene expression (Aswati Nair and Thomas 2007; Hammond-Kosack and Jones 1997; Hulbert et al. 2001; Yoshimura et al. 1998; Wang et al. 1999; Fourmann et al. 2001; Rivikin et al. 1999; Vincente and King 2001). Elucidation of finger millet RGH expression was carried using primers representative of each class of RGHs. These RGHs have potential to be used as probes for further R-gene isolation studies.
Rates of non-synonymous to synonymous nucleotide substitution quantify selection pressure by comparing the rate of substitution at silent sites (dS, KA), which are presumed natural, to the rate of substitutions at non-silent sites (dN, KS), which possibly experience selection (Rocha et al. 2006: Mustonen and Lassig 2007). A ratio of KA/KS greater than one indicates diversifying selection and less than one indicates purifying selection (Michelmore and Meyers 1998). Lower KA/KS values in the TIR than the non-TIR sequences in various plant species and in A. thaliana were observed, suggesting greater functional constraints in the TIR subfamily (Cannon et al. 2008). The NBS region is predicted to be the signalling component that activates plant defences and has been found to be highly conserved across different plant species. Therefore, it is expected that there are few non-synonymous to synonymous changes in this region of R-genes in finger millet. The KA/KS ratio of finger millet RGHs was found to be less than one, suggesting purifying selection. However, the LRR region of these genes is predicted to interact (directly or indirectly) with the pathogen controlling specificity and is the most divergent region of this class of resistance genes. Hence, it is important to identify and sequence the LRR regions of the isolated RGHs, and only then the predictions made on this class of R-genes on the basis of NBS region can be further validated. We agree with the conclusions drawn by Michelmore and Meyers (1998) while studying the NBS-LRR gene superfamily in various species, that it is under purifying selection whereas the LRR domain is under divergent selection, as indicated by the high KA/KS ratio. Further, the slow evolution of R-genes versus the faster evolution of avirulence genes of pathogens leading to new races needs to be considered for durable resistance in crop plants.
A number of resistance genes to rice blast like Pi-ta, Pi36 and Pi37 have an NBS-LRR domain that recognizes the fungal pathogen, M. grisea, in a race-specific manner (Bryan et al. 2000; Lin et al. 2007). Similarly, the Pi2 gene is a member of a gene cluster comprising nine gene members (named Nbs1-Pi2 to Nbs9-Pi2) and encodes a protein with NBS and LRR domains that confer (Zhou et al. 2006) resistance to rice blast pathogen. Though a genetic linkage map is available for finger millet (Dida et al. 2007), it is far from saturation, and the inclusion of more markers in the map will make it possible to draw comparisons between rice and finger millet blast in terms of the identification of the resistance genes and their significance.
Efforts are already underway to map the RGHs for use in marker-assisted selection and also for the isolation of full-length R-genes. NBS profiling is another method of isolating more RGHs from finger millet and this has been applied in other crop species (van der Linden et al. 2004). The availability of cloned disease resistance genes will permit studies of resistance gene structure, function and evolution. The molecular cloning of disease resistance genes will uncover new ways to deploy these genes against many of the biotic constraints, which will have a major impact in agriculture. Information on more R-gene sequences is necessary to delineate more structural domains which are the basis for the search for RGHs in any crop plant (Totad et al. 2005). Sequencing of more plant genomes will make it possible to assess the evolutionary significance of the NBS-LRR class of resistance genes in host–pathogen interaction at the molecular level.
References
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Anonymous (1995) Package of practices for high yielding varieties, UAS, Bangalore
Anonymous (1996) Annual Report for 1995–96. All India Coordinated Small Millets Improvement Project, UAS, ICAR, Bangalore-65
Aswati Nair R, Thomas G (2007) Isolation, characterization and expression studies of resistance gene candidates (RGCs) from Zinger spp. Theor Appl Genet 116:123–134
Ayliffe MA, Lagudah ES (2004) Molecular genetics of disease resistance in cereals. Ann Bot 94:765–773
Bai J, Pennil LA, Ning J, Lee SW, Ramalingam J, Webb CA, Zhao B, Sun Q, Nelson JC, Leach JE, Hulbert SH (2002) Diversity in nucleotide binding site-leucine- rich repeat genes in cereals. Genome Res 12:1871–1884
Baker B, Zambryski P, Staskawicz B, Dinesh-Kumar SP (1997) Signaling in plant–microbe interactions. Science 276:726–733
Barbeau WE, Hilu KW (1993) Protein, calcium, iron and amino acid content of selected wild and domesticated cultivars of finger millet. Plant Foods Hum Nutr 43:97–104
Bent AF, Kunkel BN, Dahlbeck D, Brown KL, Schmidt R, Giraudat J, Leung J, Staskawicz BJ (1994) RPS2 of Arabidopsis thaliana: a leucine-rich repeat class of plant disease resistance genes. Science 265:1856–1860
Bolwell GP (1992) A role for phosphorylation in the down-regulation of phenylalanine ammonia-lyase in suspension-cultured cells of French bean. Phytochemistry 31:4081–4086
Bryan GT, Wu K-S, Farral L, Jia Y, Hershey HP, McAdams HP, Faulk KN, Donaldson GK, Tarchini R, Valent B (2000) A single amino acid difference distinguishes resistant and susceptible alleles of rice blast gene Pi-ta. Plant Cell 12:2033–2046
Cannon SB, Zhu H, Baumgarten AM, Spangler R, May G, Cook DR (2008) Diversity, distribution, and ancient taxonomic relationships within the TIR and non-TIR NBS-LRR resistance gene subfamilies. J Mol Evol 54:548–562
Dida MM, Srinivasachary, Ramakrishnan S, Bennetzen JL, Gale MD, Devos KM (2007) The genetic map of finger millet, Eleusine coracana. Theor Appl Genet 114:321–332
Dixon MS, Hatzixanthis K, Jones DA, Harrison K, Jones JD (1998) The tomato Cf-5 disease resistance gene and six homologues show pronounced allelic variation in leucine-rich repeat copy number. Plant Cell 10:1915–1925
Flor HH (1956) The complementary genetic systems in flax and flax rust. Adv Genet 8:29–54
Foote T, Roberts M, Kurata N, Sasaki T, Moore G (1997) Detailed comparative mapping of cereal chromosome regions corresponding to the Phi locus in wheat. Genetics 147:801–807
Fourmann M, Chaariot F, Froger N, Delourne R, Brunel D (2001) Expression mapping and genetic viability of Brassica napus disease resistance gene analogues. Genome 44:1083–1099
Gassmann W, Hinsch ME, Staskawicz BJ (1999) The bacterial-resistance gene is a member of the TIR-NBS-LRR family of disease-resistance genes. Plant J 20:265–277
Goff SA, Ricke D, Lan TH, Presting G, Wang R, Dunn M et al (2002) A draft sequence of the rice genome (Oryza sativa L. ssp japonica). Science 296:79–92
Govindu HC, Shivanandappa N, Renfro BL (1970) Observations on diseases of Eleusine coracana with special reference to host resistance to the helminthosporium disease. In plant disease problems. Ind Phytopath Soc, New Delhi, pp 415–424
Grant MR, Godiard L, Straube E, Ashfield T, Lewald J, Sattler A, Innes RW, Dangl JL (1995) Structure of the Arabidopsis RPM1 gene enabling dual specificity disease resistance. Science 269:843–846
Hammond-Kosack KE, Jones JDG (1997) Plant disease resistance genes. Ann Rev Plant Physiol Mol Biol 48:575–607
Hulbert SH, Webb CA, Smith SM, Sun Q (2001) Resistance gene complexes; evolution and utilization. Annu Rev Phytopathol 39:285–312
ICRISAT and FAO (1996) The World sorghum and millet economies: facts, trends and outlook
Jeong SC, Hayes AJ, Biyashev RM, Saghai Maroof MA (2001) Diversity and evolution of a non-TIR-NBS sequence family that clusters to a chromosomal ‘hotspot’ for disease resistance genes in soybean. Theor Appl Genet 103:406–414
Johal GS, Briggs SP (1992) Reductase activity encoded by the HM1 disease resistance gene in maize. Science 258:985–987
Jones DA, Thomas CM, Hammond-Kosack KE, Balint-Kurti PJ, Jones JDG (1994) Isolation of the tomato Cf-9 gene for resistance to Cladosporium fulvum by transposon tagging. Science 266:789–793
Kanazin V, Marek LD, Shoemaker RC (1996) Resistance gene analogs are conserved and clustered in soybean. Proc Natl Acad Sci USA 93:11746–11750
Lawrence GJ, Finneagan EJ, Ayliffe MA, Ellis JG (1995) The L6 gene for flax rust resistance is related to the Arabidopsis bacterial resistance gene RPS2 and the tobacco viral resistance gene N. Plant Cell 7:1195–1206
Lehmann P (2002) Structure and function of plant disease resistance genes. J Appl Genet 43:403–414
Leister D, Ballvora A, Salamini F, Gebhardt C (1996) A PCR-based approach for isolating pathogen resistance genes from potato with potential for wide application in plants. Nature Genet 14:421–429
Lin F, Chen S, Que Z, Wang L, Liu X, Pan Q (2007) The blast resistance gene Pi37 encodes a nucleotide binding site leucine-rich repeat protein and is a member of a resistance gene cluster on rice chromosome. Genetics 177:1871–1880
Madsen IH, Colters NC, Rakwalska M, Backer G, Sendal L, Krussel I (2003) Barley disease resistance gene analogs of the NBS-LRR class–identification and mapping. Mol Genet 261:150–161
Meyers BC, Dickerman AN, Michelmore RW, Sivaramakrishnan S, Sobral BW, Young ND (1999) Disease resistance gene encodes members of an ancient and diverse protein family within the nucleotide-binding superfamily. Plant J 20:317–332
Meyers BC, Kozik A, Griego A, Kuang H, Michelmore RW (2003) Genome-wide analysis of NBS-LRR-encoding genes in Arabidopsis. Plant Cell 15:809–834
Michelmore RW (1995) Molecular approaches to manipulation of disease resistance genes. Annu Rev Phytopathol 15:393–427
Michelmore RW (1996) Flood warning–resistance genes unleashed. Nature Genet 14:376–378
Michelmore RW, Meyers BC (1998) Clusters of resistance genes in plants evolve by divergent selection and a birth and death process. Genome Res 8:1113–1130
Monosi B, Wisser RJ, Pennill L, Hulbert SH (2004) Full-genome analysis of resistance gene homologues in rice. Theor Appl Genet 109:1434–1447
Moutinho A, Hussey PJ, Trewavas AJ, Malho R (2001) cAMP acts as a second messenger in pollen tube growth and reorientation. PNAS 98:10481–10486
Mustonen V, Lassig M (2007) Adaptation to fluctuating selection in Drosophila. Proc Natl Acad Sci USA 104:2277–2282
Pan Q, Wendel J, Fluhr R (2000) Divergent evolution of plant NBS-LRR resistance gene homologues in dicot and cereal genomes. J Mol Evol 50:203–213
Parker JE, Coleman MJ, Szabo V, Frost LN, Schmidt R, Van Der Biezen EA, Moores T, Dean C, Daniels MJ, Jones JDG (1997) The Arabidopsis downy mildew resistance gene Rpp5 shares similarity to the Toll and Interleukin-1 receptors with N and L6. Plant Cell 9:879–894
Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H et al (2009) The sorghum genome and the diversification of grasses. Nature 457:551–556
Penuela S, Danesh D, Young ND (2002) Targeted isolation, sequence analysis, and physical mapping of non-TIR NBS-LRR genes in soybean. Theor Appl Genet 104:261–272
Porebski S, Bailey LG, Baum BR (1997) Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol Biol Rep 15:8–15
Rivikin MI, Vallejos CE, McClean PE (1999) Disease-resistance related sequences in common bean. Genome 42:41–47
Rocha EPC, Smith JM, Hurst LD, Holden MTG, Cooper JE (2006) Comparison of dS/dN are time dependent for closely related bacterial genomes. J Theor Biol 239:226–235
Rossman AY, Howard RJ, Valent B (1990) Pyricularia grisea, the correct name of the rice blast disease fungus. Mycologia 82:509–512
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Seah S, Sivasithamparam K, Karakousis A, Lagudah ES (1998) Cloning and characterization of a family of disease resistance gene analogs from wheat and barley. Theor Appl Genet 97:937–945
Shen KA, Meyers BC, Islam-Faridi N, Stelly DM, Michelmore RW (1998) Resistance gene candidates identified using PCR with degenerate oligonucleotide primers map to resistance gene clusters in lettuce. Mol Plant Microbe Interac 11:815–823
Sivaramakrishnan S, Seetharama N (2001) Identification and isolation of disease resistance genes in crop plants. J Plant Biol 28:25–38
Sorri VA, Watanabe KN, Valkonen JPT (1999) Predicted kinase-3a motif of a resistance gene analogue as a unique marker for virus resistance. Theor Appl Genet 99:164–170
Suzuki N, Choe HR, Nishida Y, Yamawaki-Kataoka Y, Ohnishi S, Tamaoki T, Kataoka T (1990) Leucine-rich repeats and carboxyl terminus are required for interaction of yeast adenylate cyclase with RAS proteins. Proc Natl Acad Sci USA 87:8711–8715
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Totad AS, Fakrudin B, Kuruvinashetti MS (2005) Isolation and characterization of resistance gene analogs (RGAs) from sorghum (Sorghum bicolor L. Moench). Euphytica 143:179–188
Vadivoo AS, Joseph R, Ganesan NM (1998) Genetic variability and diversity for protein and calcium contents in finger millet (Eleusine coracana (L.) Gaertn) in relation to grain colour. Plant Foods Hum Nutr 52:353–364
Van der Linden CG, Wouters DC, Mihalka V, Kochieva EZ, Smulders MJ, Vosman B (2004) Efficient targeting of plant disease resistance loci using NBS profiling. Theor Appl Genet 109:384–393
Vincente J, King G (2001) Characterisation of disease resistance gene like sequences in Brassica oleracea L. Theor Appl Genet 102:555–563
Vishwanath S, Gowda SS, Seetharam A, Gowda SBT (1986) Reaction to blast disease of released and pre-released varieties of finger millet from different states. Intl Millet Newsl 5:31
Wang Z, Yano M, Yamanouchi U, Iwamoto M, Monna L, Hayasaka H, Katayose Y, Sasaki T (1999) The Pib gene for rice blast resistance belongs to nucleotide binding and leucine rich repeat class of plant disease resistance genes. Plant J 19:55–64
Whitham S, Dinesh-Kumar SP, Choi D, Hehl R, Corr C, Baker B (1994) The product of the tobacco mosaic virus resistance gene N: similarity to toll and the interleukin-1 receptor. Cell 78:1101–1115
Yang S, Feng Z, Zhang X, Jiang K, Jin X, Hang Y, Chen JQ, Tian D (2006) Genome-wide investigation on the genetic variations of rice disease resistance genes. Plant Mol Biol 62:181–193
Yoshimura S, Yamanouchi U, Katayose Y, Toki S, Wang Z, Kono I, Kurata N, Yano M, Iwata N, Sasaki T (1998) Expression of Xa1 a bacterial blight-resistance gene in rice is induced by bacterial inoculation. Proc Natl Acad Sci USA 95:1663–1668
Young ND (2000) The genetic architecture of resistance. Curr Opin Plant Biol 3:285–290
Yu YG, Buss GR, Saghai-Maroof MA (1996) Isolation of a super family of candidate disease-resistance genes in soybean based on a conserved nucleotide-binding site. Proc Natl Acad Sci USA 93:11751–11756
Zhou B, Qu S, Liu G, Dolan M, Sakai H, Lu G, Bellizzi M, Wang GL (2006) The eight amino-acid differences within three leucine-rich repeats between Pi2 and Piz-t resistance proteins determine the resistance specificity to Magnaporthe grisea. Mol Plant Microbe Interac 19:1216–1228
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bheema Lingeswara Reddy, I.N., Srinivas Reddy, D., Lakshmi Narasu, M. et al. Characterization of disease resistance gene homologues isolated from finger millet (Eleusine coracana L. Gaertn). Mol Breeding 27, 315–328 (2011). https://doi.org/10.1007/s11032-010-9433-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11032-010-9433-1