
Abstract
Uncovering the genetic basis of agronomic traits in sorghum landraces that have adapted to various agro-climatic conditions would contribute to sorghum improvement efforts around the world. To identify quantitative trait nucleotides (QTNs) associated with nine agronomic traits in a panel of 304 sorghum accessions collected from diverse environments across Ethiopia (considered to be the center of origin and diversity), multi-locus genome-wide association studies (ML-GWAS) were performed using 79,754 high quality single nucleotide polymorphism (SNP) markers. Association analyses using six ML-GWAS models identified a set of 338 significantly (LOD ≥ 3)-associated QTNs for nine agronomic traits of sorghum accessions evaluated in two environments (E1 and E2) and their combined dataset (Em). Of these, 121 reliable QTNs, including 13 for flowering time (DF), 13 for plant height (PH), 9 for tiller number (TN), 15 for panicle weight (PWT), 30 for grain yield per panicle (GYP), 12 for structural panicle mass (SPM), 13 for hundred seed weight (HSW), 6 for grain number per panicle (GNP), and 10 for panicle exertion (PE) were consistently detected by at least three ML-GWAS methods and/or in two different environments. Notably, Ethylene responsive transcription factor gene AP2/ERF, known for regulation of plant growth, and the sorghum Terminal flower1/TF1 gene, which functions in the control of floral architecture, were identified as strong candidate genes associated with PH and HSW, respectively. This study provides an entry point for further validation studies to elucidate complex mechanisms controlling important agronomic traits in sorghum.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sorghum (Sorghum bicolor (L.) Moench) is an annual C4 plant belonging to the botanical family Poaceae under the Andropogoneae tribe (Clifford et al. 1990). It is the 5th most important cereal crop globally (FAO 2019) and a dietary staple for over 750 million people in the semi-arid regions of the world (FAO 2018). Because of its ability to cope with unfavorable growing conditions, sorghum will continue to feed the world’s expanding populations under the changing climate (Paterson 2008). Therefore, continuous improvement of sorghum cultivars for high yield is one of the main goals of sorghum breeding programs.
Yield is a polygenic trait and is affected by many factors such as plant phenology, morphology, and other physiological indices (Nadolska-Orczyk et al. 2017). Uncovering the genetic basis of these traits is critical for their effective manipulation, thus making the crop more efficient and resilient under a changing climate (Cattivelli et al. 2008). During the last two decades, extensive efforts have been made to identify genomic regions/quantitative trait loci (QTLs) underlying traits of agronomic interest in sorghum through bi-parental linkage mapping studies (Crasta et al. 1999; Haussmann et al. 2002; Rama Reddy et al. 2014; Sanchez et al. 2002; Subudhi et al. 2000; Sukumaran et al. 2016; Tao et al. 2000; Tuinstra 1997; Xu et al. 2000). However, this approach provides low mapping resolution, limited allelic diversity, and population specificity of detected QTLs (Feltus et al. 2006; Gupta et al. 2005; Korte and Farlow 2013). These limitations thus partly contributed to the slow transfer of knowledge from bi-parental QTL studies to practical applications in plant breeding.
In recent years, genome-wide association study (GWAS) has been widely used to identify genomic regions controlling traits of interest. Albeit being prone to false positive results, its high resolution and broader allele coverage make GWAS an important addition to the toolkit for genetic dissection of complex traits (Fang et al. 2017; Li et al. 2012; Ma et al. 2018; Zhao et al. 2011; Zhu et al. 2008). Sorghum is an ideal crop for linkage mapping studies due to its moderate linkage disequilibrium and self-pollination system (Hamblin et al. 2005). Several studies in sorghum have recently used association mapping to uncover the genetic control of important traits. Traits are as follows: flowering time (Bouchet et al. 2017; Zhao et al. 2016); plant height, panicle length, panicle exertion, tiller number, and seed number (Shehzad and Okuno 2014; Zhao et al. 2016); culm length and number of panicle (Shehzad and Okuno 2014); inflorescence trait components (Morris et al. 2012); grain fill duration, panicle weight, and harvest index (Boyles et al. 2015); and grain yield (Boyles et al. 2016). However, most of these studies had various limitations: Firstly, most of these studies used germplasm that had gone through the sorghum conversion program (Morris et al. 2012; Zhao et al. 2016) reducing genomic diversity in regions targeted for selection and hence limited success to dissect underlying loci for various traits in sorghum (Morris et al. 2012). Secondly, they were based on single-locus GWAS (SL-GWAS) methods that are limited in detecting marginal effects quantitative trait nucleotides (QTNs) (Wang et al. 2016), and hence the multiple QTNs controlling complex traits could not be effectively identified in sorghum.
To overcome the major limitations of SL-GWAS, a series of multi-locus GWAS methods, including mrMLM (Wang et al. 2016), FASTmrMLM (Tamba et al. 2017), FASTmrEMMA (Wen et al. 2017), ISIS EM-BLASSO (Tamba et al. 2017), pLARmEB (Zhang et al. 2017), and pKWmEB (Ren et al. 2018) have emerged as a powerful tool for QTN detection and QTN effect estimation for complex traits (Wang et al. 2016; Li et al. 2017; Chang et al. 2018; Peng et al. 2018). The approach has already been successfully utilized to dissect the genetic basis of important traits in several crops, such as maize (Zhang et al. 2018), rice (Liu et al. 2020) and barley (Hu et al. 2018). In addition, Ethiopia is a center of origin and diversity of sorghum and has tremendous genetic diversity in the crop for various traits (Snowden 1936; Stemler et al. 1977). The availability of such diverse germplasm provides an opportunity for new insight into the genetic architecture of important traits, and applying this knowledge in sorghum breeding programs might advance efficient genetic improvement of this crop.
In this study, we utilized the advantageous multi-locus GWAS to investigate the genetic control of nine important agronomic traits in natural population of 304 sorghum accessions by using 79,754 high quality SNP markers. We aim to identify common QTNs via multiple methodologies and then deduce potential candidate genes that can be further validated and utilized in marker-assisted selection (MAS) to enhance the efficiency of cultivar development.
Materials and methods
Plant materials and phenotyping
A total of 304 diverse sorghum accessions were collected from farmers’ fields of major sorghum growing regions (Amhara, Oromia, Southern Nations, and Tigray) of Ethiopia. The complete list of accessions and relevant information are previously reported (Wondimu et al. 2021). These accessions were evaluated for important agronomic traits at two environments, Kobo (North Ethiopia, altitude: 1400 m) and Mieso (East Ethiopia, altitude: 1380 m) during the 2018 cropping season. The meteorological data for the two environments is given in Supplementary Table 1.
In brief, with an alpha lattice design, all accessions were sown at two field sites in two replications with a plot size of 4.5 m2 consisting of 2 rows with a spacing of 75 cm between rows. Fertilizer was applied at the rate of 100 kg/ha DAP at planting and 50 kg/ha urea at about 35 days after planting. Data were collected for nine agronomic traits following the standard sorghum descriptor (IBPGR and ICRISAT 1993). Days to 50% flowering (DF) was recorded as the number of days from emergence until 50% of the panicles in a plot were at mid-anthesis. Plant height (PH) was measured at the flowering stage from the ground surface to the tip of the main panicle; panicle exertion (PE) was measured as the length between the base of flag leaf and the base of the panicle; and number of tillers per plant (TN) was counted on the main stalk when the flower was in full bloom. At maturity, main panicles, from the ten random plants already earmarked, were cut and oven dried at 70 °C for 72 h. Before threshing, all panicles were weighed to get an average panicle weight (PWT), then the panicles were manually threshed and the weights of grain yield per panicle (GYP) and hundred seeds (HSW) were recorded. Structural panicle mass (SPM) was calculated as the difference between PWT and GYP, and grain number per panicle (GNP) was estimated as the ratio of GYP to HSW and multiplied by 100.
Phenotypic data analysis
Summary statistics were calculated for each trait at each environment. Phenotypic data from each environment were analyzed by a single environment linear mixed model with sorghum accessions fitted as fixed effects. The model was illustrated as:
where yijk is the random phenotypic effect of the genotype i at block j, in replication k; μ is the general mean; gi is the fixed effect of genotype i; rk is the random effect of replication k; bjk is the random effect of block j, in replication k; εijk is a random non-genetic effect, with εijk ~ N (0, σ2).
To assess the effects of genotype (G), environment (E), and G × E interaction for each trait, the two environments were combined, and the genetic effect associated with accessions was decomposed into two components, the genetic effect of accessions and the interaction effect between accessions and environment (G × E effect). The linear mixed model was:
In this case, the new terms Ei and Ei Gl are the random effects of environment and environment by genotype interaction, respectively. Fixed and random effects in the model were tested using the F-test and likelihood ratio test (Neyman and Pearson 1928), respectively. Variance components were estimated using a residual maximum likelihood method (Harville 1977). Broad-sense heritability (h2) value for all traits was then calculated using the formula given by Allard (1999). All mixed model analyses were performed using the REML (residual maximum likelihood) algorithm of SAS v9.2 (SAS Institute Inc 2008).
SNP genotyping
The 304 sorghum accessions were genotyped using genotyping-by-sequencing (GBS) methodology (Elshire et al. 2011), as briefly described in our previous work (Wondimu et al. 2021). The raw data for all accessions across 115,501 SNPs is publicly available at figshare ( https://doi.org/10.25387/g3.12813224). Data filtering using minor allele frequency (MAF > 5%) for the 304 samples yielded a total of 79,754 high quality SNP markers for the current genome-wide association study.
LD
Pairwise linkage disequilibrium (LD) as measured by the allele frequency correlations (r2) of each pair of SNPs was estimated separately for each chromosome and across the ten chromosomes in TASSEL 5.0 using a sliding window of 50 bp (Bradbury et al. 2007). The critical value of r2 of 0.1 was considered as LD decay criterion (Nordborg et al. 2002; Palaisa et al. 2003; Remington et al. 2001). LD decay curve for each chromosome and whole genome level was fitted using a non-linear regression model in R software (R Core team 2019), as described by Remington et al. (2001).
ML-GWAS
Multi-locus genome-wide association analysis (ML-GWAS) analyses were performed using three datasets: (i) Kobo-2018 (E1), (ii) Mieso-2018 (E2), and (iii) Kobo-2018 and Meiso-2018 combined dataset (Em). Best linear unbiased estimators (BLUEs) of the genotypic values for each of the above nine traits in two environments (E1 and E2) and their combined dataset (Em) were estimated using the REML algorithm, as described above. Marker-trait association analyses were performed using six ML-GWAS methods, including mrMLM (Wang et al. 2016), FASTmrMLM (Tamba and Zhang 2018), FASTmrEMMA (Wen et al. 2018), pLARmEB (Zhang et al. 2017), pKWmEB (Ren et al. 2018), and ISIS EM-BLASSO (Tamba et al. 2017) implemented in the “mrMLM.GUI” R package (https://cran.r-project.org/web/packages/mrMLM/index.html). Population structure for these accessions has been previously estimated as six subpopulations (Wondimu et al. 2021) using ADMIXTURE analysis (Alexander et al. 2009). The co-ancestry coefficient matrix (Q) of the 304 accessions is publicly available at figshare ( https://doi.org/10.25387/g3.12813224). Kinship matrix (K), an estimate of the level of relatedness among individuals, was internally calculated within mrMLM.GUI package. The population structure (Q) and kinship (K) matrices were then included in all the tested models to minimize the identification of false-positive associations and increase the statistical analysis power. All parameters in GWAS were set at default values. The critical threshold for significantly associated QTNs was set at LOD ≥ 3.0 for all the six multi-locus models, as described in previous studies (Tamba et al. 2017). The resulting -log10 (P) values from the ML-GWAS approaches were used to draw the Manhattan and Q-Q plots using the mrMLM.GUI package in R software (R Core team 2019).
Identification of reliable/stable QTNs and candidate genes
We considered a QTN reliable when it is detected by at least three multi-locus GWAS methods and/or in at least two situations (E1, E2, and Em). Additionally, QTNs that are consistently detected across at least two situations (E1, E2, and Em) were further regarded as stable QTNs and followed in this study. To determine the regions of interest for selection of potential candidate genes, the average LD decay in which flanking SNP markers had strong LD (r2 > 0.1) was used. All the genes present in the association region with known putative functions were extracted from the most recently annotated sorghum reference genome v3.1 (McCormick et al. 2017) available at phytozome (https://phytozome.jgi.doe.gov). By comprehensive analysis of gene annotation information promising candidate genes for each trait were further mined.
Results
Phenotypic variation
The distributions of the nine agronomic traits measured in sorghum accessions evaluated in this work are depicted graphically using histograms (Fig. 1). Two-way ANOVA showed significant (p < 0.05) differences among the genotypes (G) and genotype by environment (G × E) interaction effects for all the traits studied (Table 1), suggesting the wide genetic variability among the Ethiopian sorghum accessions, which provides opportunities for effective selection. As for heritability estimates, the traits DF, PH, PE, and HSW presented relatively high heritability values (h2 > 0.5), while TN, PWT, GYP, SPM, and GNP had moderate heritability estimates (Table 1).

Histogram showing the distribution of the nine agronomic traits evaluated in two different environments. DF, days to flowering (days); PH, plant height (cm); TN, number of tillers per plant (no.); PWT, panicle weight (g); GYP, grain yield per panicle (g/panicle); SPM, structural panicle mass (g); HSW, hundred seed weight (g); GNP, grain number per panicle (no.); PE, panicle exertion (cm). Environments, E1, Kobo-2018; E2, Mieso-2018
Comparing the mean performance of the accessions in each of the environments (Table 1), mean days to flowering (DF) was slightly earlier in E1 (96 days) than E2 (99 days); however, mean plant height (PH, 311.45 cm) and mean panicle exertion (PE, 9.08 cm) were relatively higher in E1 than the 270.24 and 6.56 cm observed in E2 (Table 1).
Structural panicle mass (SPM) and grain number per panicle (GNP) had greater variation in E2 than in E1. However, the remaining traits displayed more consistent variation between the two environments. The complete phenotypic data of all accessions in two environments (E1 and E2) and their combined data (Em) are provided in Supplementary Table 2.
Whole genome patterns of LD
Characterizing patterns of LD is critical for the design of association studies (Mather et al. 2007) and interpretation of association peaks (Huang et al. 2010). In general, there was a rapid LD decay with increasing physical distance along the 10 sorghum chromosomes (Fig. 2 and Supplementary Fig. S1). At a threshold value of 0.1, LD decays within 60–80 kb on chromosomes 5, 6, 7, and 9 but 80–100 kb on chromosomes 1, 2, 3, 4, 8, and 10 (Supplementary Fig. S1). On average, LD decays to background levels (r2 < 0.1) within 100 kb (Fig. 2).

Genome-wide LD (r2) decay in the 304 Ethiopian sorghum accessions. Average r2 (squared allele frequency correlation between pairs of SNPs) were plotted against the corresponding genetic distance between markers. The vertical solid green line represents the average genome-wide LD decay (i.e., LD decay = 64,550 base pairs) point
This LD decay estimate is higher than previously reported values in sorghum of 10–30 kb (Wang et al. 2013) and 10–15 kb (Hamblin et al. 2005). This difference may be due to the low coverage of the genome by the markers and the small number of genotypes in previous studies. Since sorghum is largely self-pollinated, we expect higher levels of LD than in outcrossing species (Flint-Garcia et al. 2003). Accordingly, the extent of LD in sorghum is similar to that of rice (∼65–150 kb) (Mather et al. 2007), another self-pollinated crop, but much greater than maize (∼2 kb) (Yan et al. 2009), which is an out-crosser. Although we expect mapping resolution to range widely across the genome depending on the chromosome, the overall modest LD decay rate (< 100 kb) makes this Ethiopian collection suitable for GWAS.
QTNs identified by ML-GWAS
To explore the genetic factors associated with nine agronomic traits, we conducted ML-GWAS based on a total of 79,754 high quality SNP markers (The genomic distribution of the SNP markers used in this study is shown in Fig. 3), and BLUEs from three datasets (E1, E2, and Em). Using six ML-GWAS models, a total of 338 QTNs distributed on 10 chromosomes were identified that are significantly associated with nine agronomic traits based on a LOD score threshold of ≥ 3 in three situations/environments (E1, E2, and Em), as summarized in Table 2. A full list of the QTNs significantly associated with the phenotypes in each environment (E1 and E2) and the combined dataset (Em) is presented in Supplementary Table 3, while the Manhattan and Q-Q plots of the ML-GWAS results are reported in Supplementary Figs. S2 and S3. Of the identified QTNs, 66, 110. and 162 were identified in E1, E2, and Em situations, respectively (Table 2). Among the ML-GWAS models, mrMLM resulted in the greatest number of significant QTNs identified (192), whereas the FASTmrEMMA had the lowest number of QTNs (78). Chromosome 1 had the highest number of the identified QTNs (49), followed by chromosome 9 (41), and chromosome 3 (39). Overall, the LOD value ranged from 3.01 to 8.37, and the proportion of phenotypic variance explained (r2) by each QTN ranged from 0.45 to 25.92% (Table 2).

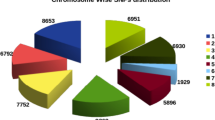
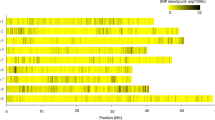
Genomic distribution of the 79,754 high quality SNP markers across the ten sorghum chromosomes and their corresponding density
To obtain accurate results, only QTNs showing repeatability (i.e., detected by at least three different ML-GWAS models and/or in two different situations/ environments) were considered reliable. Using these criteria, we identified a total of 121 reliable QTNs significantly associated with nine agronomic traits, as presented in Supplementary Table S4. The 121 QTNs identified each explained a low percentage of phenotypic variation (PVE): DF (n = 13, PVE = 0.60–21.60%), PH (n = 13, PVE = 2.04–16.28%), TN (n = 9, PVE = 1.01–25.92%), PWT (n = 15, PVE = 1.54–15.65), GYP (n = 30, PVE = 0.79–13.64%), SPM (n = 12, PVE = 2.20–11.97%), HSW (n = 13, PVE = 0.01–16.54%), GNP (n = 6, PVE = 1.85–11.33%), and PE (n = 10, PVE = 1.86–17.76%). Additionally, a total of 29 QTNs were significantly associated with more than one trait (Supplementary Table S4). For instances, the traits DF and PH shared a common QTN (S10_13295281) mapped on chromosome 10 that on average explained ~ 4.50% of the variation for the traits, whereas GYP and GNP had seven common QTNs (S1_22881870, S1_28143445, S2_58161802, S3_12356222, S7_63176270, S9_38639556, and S10_47554177) on chromosomes 1, 2, 3, 7, 9, and 10, and accounting for 2.04–13.64% of the total phenotypic variance for these traits. The traits PWT, GYP, and GNP also shared four common QTNs (S1_70244848, S8_6755616, S8_48609940, and S9_438623) mapped on chromosomes 1, 8, and 9 (Supplementary Table S4).
Identification of stable QTNs and candidate genes
A total of 46 QTNs consistently detected in at least two environments (E1, E2, and Em) were regarded as stable QTNs (Table 3). All these stable QTNs were distributed on the 10 sorghum chromosomes, with chromosome 10 showing the lowest number of associations, while chromosome 8 showing the highest number of associations (10 QTNs associated with seven traits).
Among the 46 stable QTNs detected in at least two environments, 7, 9, and 13 were detected by three, four, and five ML-GWAS methods, respectively (Table 3). Moreover, 7 QTNs (S1_56717177, S1_56748133, S7_42021189, S8_43981111, S8_6755616, S9_57542210, and S10_13295281) were identified by six ML-GWAS methods to be associated with five agronomic traits in at least two environments, with LOD score values ranging from 3.05 to 11.42 (Table 3). Interestingly, 2 QTNs (S8_43981111for DF and S9_48893285 for PE) with moderate effects (r2 = ~ 6%) were consistently detected across all situations/environments (E1, E2, and Em). The region containing one stable QTN (S8_6755616, LOD = 3.14–7.75; r2 = 1.45–5.21%) on chromosome 8 was significantly associated with PWT and GYP in two environments (E1 and Em).
To further understand the genetic basis of agronomic traits, we detected several candidate genes surrounding 100 kb upstream and downstream of the above 46 stable QTN position, as suggested by the LD decay analysis in this study (Fig. 2). The complete list of candidate genes in proximity of the stable QTNs is reported in Supplementary Table S5). For instances, two putative candidate genes, Sobic.001G266200 and Sobic.007G193300 surrounding significant QTNs associated with DF have annotations as F-box and MADS-box family proteins, respectively, that are involved in multiple developmental processes in plants (Saha et al. 2015).
Two candidate genes (Sobic.001G013800 and Sobic.003G324400) were also identified for PH on chromosomes 1 and 3, respectively, with the first gene encoding Ser/Thr protein phosphatase family protein and the other encoding Ethylene responsive transcription factor (AP2/ERF) family protein (Supplementary Table S5). Interestingly, several candidate genes including, Sobic.009G075400 (Protein RALF-like 4), Sobic.008G102200 (Photosystem II reaction center protein), Sobic.004G053400 (similar to Auxin responsive protein-like), and Sobic.008G037300 (similar to Terminal flower1/TF1), and Sobic.009G237900 (Plastocyanin-like domain protein) were identified adjacent to the stable QTNs associated with TN, PWT, SPM, HSW, and PE, respectively. Further examples are given in Fig. 4 and Supplementary Table S5.

Linkage groups and chromosomal positions of stable QTNs and candidate genes identified for sorghum agronomic traits. The stable QTNs and candidate genes are labeled on the right side of chromosomes, and trait name abbreviations display different traits. QTNs and candidate genes on each chromosome are highlighted with colors. The intervals between adjacent loci in chromosomes denote the physical distance in mega bases
Discussion
Although several studies already identified the genetic basis of important agronomic traits in sorghum using GWAS (Bouchet et al. 2017; Boyles et al. 2016; Morris et al. 2012; Zhao et al. 2016), panels composed exclusively of sorghum accessions from the center of origin and diversity had not been sufficiently explored (Girma et al. 2019). Moreover, very few studies have implemented the ML-GWAS approach to identify genetic variants in sorghum. The use of ML-GWAS has become a powerful means to identify genomic regions underlying traits of interest, particularly for complex traits controlled by multiple genes of small effect (Wen et al. 2018; Zhang et al. 2019). Hence, associated genomic regions reported herein provide valuable knowledge that could be further investigated for advancing understanding of the genetic control of traits of economic and adaptive importance.
In this study, we identified a total of 121 reliable QTNs detected by at least three ML-GWAS models and/or in two different environments (Supplementary Table 4). A comparison of the six ML-GWAS methods revealed that mrMLM was more powerful and robust than the other five models in the detection of reliable QTNs for agronomic traits. Most of the QTNs identified in this study were observed in only one environment, supporting our observation of the presence of significant genotype by environment (G × E) interaction effects for all the traits studied (Wondimu et al. 2020; Table 1). The presence of the G × E interaction is one of the main challenges in selecting QTNs in breeding programs, as gene expression of these QTNs depend on the evaluation environments (Wu et al. 2020). On the other hand, the stable QTNs identified herein provide great prospects for future genetic improvement of the traits evaluated in this study through the accumulation of favorable alleles. Genetic correlations between traits can be ascribed to gene linkage and/or pleiotropy (Saltz et al. 2017). In this study, a total of 29 pleiotropic QTNs were detected associated with more than one trait (Supplementary Table S4). Among these, one QTN (S10_13295281) on chromosome 10 was associated with DF and PH. Another four pleiotropic QTNs (S1_70244848, S8_6755616, S8_48609940, and S9_438623) mapped on chromosomes 1, 8, and 9 were associated with PWT, GYP, and GNP. The presence of pleiotropic effects of these QTNs controlling different agronomic traits has previously been suggested by our phenotypic correlation analysis (Wondimu et al. 2020).
As most of the agronomic traits studied are controlled by polygenes, the effects of most of the QTNs identified in this study were small, confirming the quantitative nature of the traits (Gupta et al. 2020). Nonetheless SL-GWAS methods have been widely adopted; they are limited in detecting marginal effects QTNs (Wang et al. 2016), and hence the use of ML-GWAS methods can mitigate the above limitation and estimate the effects of all markers at the same time (Cui et al. 2018).
To explore the spectra of candidate genes, we focused on physical intervals supported by the LD decay information (i.e., 100 kb upstream and downstream of associated QTNs; Fig. 2). One of the stable QTN discovered in this study is S7_62550036 that explained ~ 4.0% of the variation in flowering time (DF) (Supplementary Table 5). This marker is in close proximity to Sobic.007G193300 gene, which encodes a MADS transcription factor family protein. MADS family members widely take part in the key regulatory pathways of plant growth and reproduction, including flower formation (Callens et al. 2018). In rice, the OsMADS family is involved in controlling flowering time and development of flower organs (Yu et al. 2014). Another QTN (S3_65025755) with important effect on plant height (PH) is located near the Sobic.003G324400 gene that encodes Ethylene responsive transcription factor (AP2/ERF) family protein, which has been reported to limit internode elongation by down regulating gibberellin biosynthesis genes in rice (Qi et al. 2011). The candidate gene, Sobic.008G102200, associated with panicle weight (PWT) encodes Photosystem II reaction center protein, which is important for light harvesting during photosynthesis (Pietrzykowska et al. 2014). Thus, its possible role in photosynthesis might in theory explain its association with panicle weight, as panicle yield can be determined by factors regulating photosynthetic rate (Ramamoorthy et al. 2017). The QTN (S8_3450030) associated with hundred seed weight (HSW) is very close to a gene, Sobic.008G037300 (Terminal flower1/TF1), which functions in the control of flowering time and floral architecture (Alvarez et al. 1992). Mutations in TFL1 accelerate flowering time and resulted in higher seed weight in Arabidopsis (Hanano and Goto 2011). Another gene, Sobic.009G237900, encoding plastocyanin-like domain (Cu_bind_like) protein was found near S9_57542210 associated with PE (Supplementary Table S5). Previous studies have indicated that phytocyanin gene family is involved in key plant activities, including apical bud organ development in plants (Fedorova et al. 2002).
Other candidates emerging from our search include genes putatively involved in biotic and abiotic stress responses, kinase activity, transport, and signal transduction (Supplementary Table 5). For instance, Sobic.001G266700 (zinc finger domain; C3HC4 zinc finger), and Sobic.004G028600 (Leucine-rich repeat receptor-like protein kinase/LRR-RLKs) were located near QTNs (S1_50707856 and S4_2182692, respectively) significantly associated with DF and TN. Previous studies identified C3H4-type zinc finger member, as the gene most strongly upregulated by various abiotic stresses including drought (Ali-Benali et al. 2012). It has also been proposed that LRR-RLKs might be involved in early responses to drought and ABA perception (Osakabe et al. 2005).
Conclusions
This study involved field-based phenotyping and genotyping-by-sequencing of Ethiopian sorghum landrace collection, representing a wide range of genetic variation that has evolved under diverse environmental conditions. This approach helped identified valuable loci and potential candidate genes underlying genetic variation in nine important agronomic traits of sorghum. Here, we presented a list of important QTNs and candidate genes that offer opportunities for identifying specific genes associated with complex traits and elucidating underlying biological functions. Furthermore, functional validation of these newly discovered candidate genes is important to confirm the association results observed in the present study and perhaps providing a foundation for engineering alternative alleles with still-greater value. Overall, the results reported herein advance our understanding of the genetic mechanisms underlying complex traits and further support the development of new DNA marker tools for efficient genetic improvement of this crop through molecular breeding.
Data availability
The data generated during the current study are included in the manuscript and its supplementary materials as Tables S1, S2, S3, S4, and S5. The list of 304 accessions and raw SNP datasets used during the current study are available in the figshare repository ( https://doi.org/10.25387/g3.12813224). The seeds of sorghum accessions used in this study are deposited in Jimma University repository under APFS Project accession number JU0330. Please contact the corresponding author for availability.
Abbreviations
- GBS:
-
Genotyping-by-sequencing
- ML-GWAS:
-
Multi-locus genome-wide association studies
- SL-GWAS:
-
Single- locus genome-wide association studies
- DF :
-
Days to flowering
- PH :
-
Plant height
- PE :
-
Panicle exertion
- TN :
-
Tiller number
- PWT :
-
Panicle weight
- GYP :
-
Grain yield per panicle:
- SPM :
-
Structural panicle mass
- GNP :
-
Grain number per panicle
- HSW :
-
Hundred seed weight
- SNP :
-
Single-nucleotide polymorphism
- QTL:
-
Quantitative trait locus
- QTN:
-
Quantitative trait nucleotide
- MAF:
-
Minor allele frequency
- LD:
-
Linkage disequilibrium
- ANOVA:
-
Analysis of variance
- LOD :
-
Logarithm of odds
- mrMLM:
-
Multi locus random-SNP-effect MLM
- FASTmrMLM:
-
Fast mrMLM
- ISIS EM-BLASSO:
-
Iterative modified-sure independence screening expectation–maximization-Bayesian least absolute shrinkage and selection operator
- pKWmEB:
-
Integration of Kruskal–Wallis test with empirical Bayes
- FASTmrEMMA:
-
Fast multi-locus random-SNP-effect efficient mixed model analysis
- pLARmEB:
-
Polygenic-background-control-based least angle regression plus empirical Bayes
- r 2(%):
-
the proportion of total phenotypic variance explained by each QTN
- r 2 :
-
The squared correlation coefficient
References
Alexander DH, Novembre J, Lange K (2009) Fast model-based estimation of ancestry in unrelated individuals. Genome Res 19(9):1655–1664
Ali-Benali MA, Badawi M, Houde Y, Houde M (2012) Identification of oxidative stress-responsive C2H2 zinc fingers associated with Al tolerance in near isogenic wheat lines. Plant Soil 366(1–2):199–212
Allard RW (1999) Principles of Plant Breeding. John Wiley and Sons, New York. p. 485
Alvarez J, Guli CL, Yu X-H, Smyth DR (1992) Terminal flower: a gene affecting inflorescence development in Arabidopsis thaliana. Plant J 2:103–116
Bouchet S, Olatoye MO, Marla SR, Perumal R, Tesso T, Yu J et al (2017) Increased power to dissect adaptive traits in global sorghum diversity using a nested association mapping population. Genetics 206(2):573–585
Boyles RE, Cooper EA, Myers MT, Brenton Z, Rauh BL, Morris GP, Kresovich S (2016) Genome-wide association studies of grain yield components in diverse sorghum germplasm. Plant Genome. https://doi.org/10.3835/plantgenome2015.09.0091
Boyles RE, Pfeiffer BK, Cooper EA, Rauh BL, Zielinski KJ, Myers MT et al (2016) Genetic dissection of sorghum grain quality traits using diverse and segregating populations. Theor Appl Genet 130(4):697–716
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 1;23(19):2633–5. https://academic.oup.com/bioinformatics/article/23/19/2633/185151
Callens C, Tucker MR, Zhang D, Wilson ZA (2018) Dissecting the role of MADS-box genes in monocot floral development and diversity. J Exp Bot 69(10):2435–2459. https://doi.org/10.1093/jxb/ery086
Cattivelli L, Rizza F, Franz-W B, Mazzucotelli E, Mastrangelo AM, Francia E et al (2008) Drought tolerance improvement in crop plants: an integrated view from breeding to genomics. Field Crop Res 105(1–2):1–14
Chang F, Guo C, Sun F, Zhang J, Wang Z, Kong J et al (2018) Genome-wide association studies for dynamic plant height and number of nodes on the main stem in summer sowing soybeans. Front Plant Sci 9:1184. https://doi.org/10.3389/fpls.2018.01184
Clifford HT, Clayton WD, Renvoize SA (1990) Genera Graminum. Grasses of the World Kew Bulletin 45(1):208
Crasta OR, Xu WW, Rosenow DT, Mullet J, Nguyen HT (1999) Mapping of post-flowering drought resistance traits in grain sorghum: association between QTLs influencing premature senescence and maturity. Mol Gen Genet MGG 262(3):579–588
Cui Y, Zhang F, Zhou Y (2018) The application of multi-locus GWAS for the detection of salt-tolerance loci in rice. Front Plant Sci 9:1464. https://doi.org/10.3389/fpls.2018.01464
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES et al (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. Orban L, editor. PLoS ONE 6(5):e19379
Fang C, Ma Y, Wu S, Liu Z, Wang Z, Yang R et al (2017) Genome-wide association studies dissect the genetic networks underlying agronomical traits in soybean. Genome Biol 18(1). https://doi.org/10.1186/s13059-017-1289-9
FAO (2018) World Food and Agriculture - Statistical Pocketbook. https://doi.org/10.4060/CA1796EN
FAO (2019) World Food and Agriculture - Statistical Pocketbook. https://doi.org/10.4060/ca6463en
Fedorova M, VandeMortel J, Matsumoto PA, Cho J, Town CD, VandenBosch KA et al (2002) Genome wide identification of nodule-specific transcripts in the model legume Medicago truncatula. Plant Physiol 130:519–537. https://doi.org/10.1104/pp/006833
Feltus FA, Hart GE, Schertz KF, Casa AM, Kresovich S, Abraham S et al (2006) Alignment of genetic maps and QTLs between inter- and intra-specific sorghum populations. Theor Appl Genet 112(7):1295–1305
Flint-Garcia SA, Thornsberry JM, Buckler ES (2003) Structure of linkage disequilibrium in plants. Annu Rev Plant Biol 54(1):357–374.
Girma G, Nida H, Seyoum A, Mekonen M, Nega A et al (2019) A large–scale genome-wide association analyses of Ethiopian sorghum landrace collection reveal loci associated with important traits. Front Plant Sci 10:691. https://doi.org/10.3389/fpls.2019.00691
Gupta PK, Rustgi S, Kulwal PL (2005) Linkage disequilibrium and association studies in higher plants: present status and future prospects. Plant Mol Biol 57(4):461–485
Gupta N, Zargar SM, Singh R, Nazir M, Mahajan R, Salgotra RK (2020) Marker association study of yield attributing traits in common bean (Phaseolus vulgaris L.). Mol Biol Rep 47:6769–6783. https://doi.org/10.1007/s11033-020-05735-6
Hamblin MT, Salas Fernandez MG, Casa AM, Mitchell SE, Paterson AH, Kresovich S (2005) Equilibrium processes cannot explain high levels of short- and medium-range linkage disequilibrium in the domesticated grass Sorghum bicolor. Genetics 171(3):1247–1256
Hanano S, Goto K (2011) Arabidopsis TERMINAL FLOWER1 is involved in the regulation of flowering time and inflorescence development through transcriptional repression. Plant Cell 23:3172–3184
Harville DA (1977) Maximum likelihood approaches to variance component estimation and to related problems. Source J Am Stat Assoc 72:320–338
Haussmann B, Mahalakshmi V, Reddy B, Seetharama N, Hash C, Geiger H (2002) QTL mapping of stay-green in two sorghum recombinant inbred populations. Theor Appl Genet 106(1):133–142
Huang XX, Wei T, Sang Q, Zhao Q, Feng Y, Zhao C, Li C, Zhu T, Lu Z, Zhang M, Li D, Fan Y, Guo A, Wang L, Wang L, Deng W, Li Y, Lu Q, Weng KL (2010) Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet 42(11):961–967
Hu X, Zuo J, Wang J, Liu L, Sun G, Li C et al (2018) Multi-locus genome-wide association studies for 14 main agronomic traits in Barley. Front Plant Sci 871(November):1–14. https://doi.org/10.3389/fpls.2018.01683
Korte A, Farlow A (2013) The advantages and limitations of trait analysis with GWAS: a review. Plant Methods 9(1):29
Li H, Peng Z, Yang X, Wang W, Fu J, Wang J et al (2012) Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat Genet 45(1):43–50
Li H, Zhang L, Hu J, Zhang F, Chen B, Xu K et al (2017) Genome-wide association mapping reveals the genetic control underlying branch angle in rapeseed (Brassica napus L.). Front Plant Sci 8:1054. https://doi.org/10.3389/fpls.2017.01054
Liu S, Zhong H, Meng X, Sun T, Li Y, Pinson SRM et al (2020) Genome-wide association studies of ionomic and agronomic traits in USDA mini core collection of rice and comparative analyses of different mapping methods. BMC Plant Biol 20(1):441. https://doi.org/10.1186/s12870-020-02603-0
Ma F, Xu Y, Ma Z, Li L, An D (2018) Genome-wide association and validation of key loci for yield-related traits in wheat founder parent Xiaoyan 6. Mol Breeding 38(7). https://doi.org/10.1007/s11032-018-0837-7
Mather KA, Caicedo AL, Polato NR, Olsen KM, McCouch S, Purugganan MD (2007) The extent of linkage disequilibrium in rice (Oryza sativaL.). Genetics 177(4):2223–32
McCormick RF, Truong SK, Sreedasyam A, Jenkins J, Shu S, Sims D et al (2017) The Sorghum bicolor reference genome: improved assembly, gene annotations, a transcriptome atlas, and signatures of genome organization. Plant J 93(2):338–354
Morris GP, Ramu P, Deshpande SP, Hash CT, Shah T, Upadhyaya HD et al (2012) population genomic and genome-wide association studies of agroclimatic traits in sorghum. Proc Natl Acad Sci 110(2):453–458
Nadolska-Orczyk A, Rajchel IK, Orczyk W, Gasparis S (2017) Major genes determining yield-related traits in wheat and barley. Theor Appl Genet 130(6):1081–1098
Neyman J, Pearson ES (1928) On the use and interpretation of certain test criteria for purposes of statistical inference. Biometrika 20A: 175–240
Nordborg M, Borevitz JO, Bergelson J, Berry CC, Chory J, Hagenblad J et al (2002) The extent of linkage disequilibrium in Arabidopsis thaliana. Nat Genet 30(2):190–193
Osakabe Y, Maruyama K, Seki M, Satou M, Shinozaki K, Yamaguchi-Shinozaki K (2005) Leucine-rich repeat receptor-like kinase1 is a key membrane-bound regulator of abscisic acid early signaling in Arabidopsis. Plant Cell 17(4):1105–1119
Palaisa KA, Morgante M, Williams M, Rafalski A (2003) Contrasting effects of selection on sequence diversity and linkage disequilibrium at two phytoene synthase loci. Plant Cell 15(8):1795–1806
Paterson AH (2008) Genomics of Sorghum. Int J Plant Genomics 2008:16
Peng B, Li Y, Wang Y, Liu C, Liu Z, Tan W et al (2018) QTL analysis for yield components and kernel-related traits in maize across multi-environments. Theor Appl Genet 122:1305–1320. https://doi.org/10.1007/s00122-011-1532-9
Pietrzykowska M, Suorsa M, Semchonok DA, Tikkanen M, Boekema EJ, Aro E-M et al (2014) The light-harvesting chlorophyll a/b binding proteins Lhcb1 and Lhcb2 play complementary roles during state transitions in Arabidopsis. Plant Cell 26(9):3646–3660
Qi W, Sun F, Wang Q, Chen M, Huang Y, Feng Y-Q et al (2011) Rice ethylene-response AP2/ERF factor OsEATB restricts internode elongation by down-regulating a gibberellin biosynthetic gene. Plant Physiol 157(1):216–228
R Core Team (2019) R: a language and environment for statistical computing. R Core Team, Vienna
Rama Reddy N, Ragimasalawada M, Sabbavarapu M, Nadoor S, Patil J (2014) Detection and validation of stay-green QTL in post-rainy sorghum involving widely adapted cultivar, M35–1 and a popular stay-green genotype B35. BMC Genomics 15(1):909
Ramamoorthy R, Vishal B, Ramachandran S, Kumar PP (2017) The OsPS1-F gene regulates growth and development in rice by modulating photosynthetic electron transport rate. Plant Cell Rep 37(2):377–385
Remington DL, Thornsberry JM, Matsuoka Y, Wilson LM, Whitt SR, Doebley J et al (2001) Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc Natl Acad Sci 98(20):11479–11484
Ren WL, Wen YJ, Dunwell JM, Zhang YM (2018) PKWmEB: integration of Kruskal-Wallis test with empirical Bayes under polygenic background control for multi-locus genome-wide association study. Heredity (edinb) 120(3):208–218. https://doi.org/10.1038/s41437-017-0007-4
Saha G, Park J-I, Jung H-J, Ahmed NU, Kayum MA, Chung M-Y et al (2015) Genome-wide identification and characterization of MADS-box family genes related to organ development and stress resistance in Brassica rapa. BMC Genomics 6:178. https://doi.org/10.1186/s12864-015-1349-z
Saltz JB, Hessel FC, Kelly MW (2017) Trait correlations in the genomics era. Trends Ecol Evol 32:279–290. https://doi.org/10.1016/j.tree.2016.12.008
Sanchez AC, Subudhi PK, Rosenow DT, Nguyen HT (2002) Mapping QTLs associated with drought resistance in sorghum (Sorghum bicolor L. Moench). Plant Molecular Biology 48(5/6):713–26
SAS Institute (2008) Statistical Analytical Systems SAS/STAT User’s Guide. Version 8(2). SAS Institute Inc., Cary
Shehzad T, Okuno K (2014) QTL mapping for yield and yield-contributing traits in sorghum (Sorghum bicolor (L) Moench) with genome-based SSR markers. Euphytica 203(1):17–31
Snowden JD (1936) The cultivated races of sorghum. 1936. London: Adlard and Son
Stemler ABL, Harlan JR, de Wet JMJ (1977) The sorghums of Ethiopia. Econ Bot 31(4):446–460
Subudhi PK, Rosenow DT, Nguyen HT (2000) Quantitative trait loci for the stay green trait in sorghum (Sorghum bicolor L. Moench): consistency across genetic backgrounds and environments. Theoretical and Applied Genetics 101(5–6):733–41
Sukumaran S, Li X, Li X, Zhu C, Bai G, Perumal R et al (2016) QTL Mapping for grain yield, flowering time, and stay-green traits in sorghum with genotyping-by-sequencing markers. Crop Sci 56(4):1429–1442
Tamba CL, Ni YL, Zhang YM (2017) Iterative sure independence screening EM-Bayesian LASSO algorithm for multi-locus genome-wide association studies. PLoS Comput Biol 13:e1005357. https://doi.org/10.1371/journal.pcbi.1005357
Tao YZ, Henzell RG, Jordan DR, Butler DG, Kelly AM, McIntyre CL (2000) Identification of genomic regions associated with stay green in sorghum by testing RILs in multiple environments. Theor Appl Genet 100(8):1225–1232
Tuinstra MR, Grote EM, Goldsbrough PB, Ejeta G (1997) Genetic analysis of post-flowering drought tolerance and components of grain development in sorghum. Mol Breeding 3(6):439–448
Wang Y-H, Upadhyaya HD, Burrell AM, Sahraeian SME, Klein RR, Klein PE (2013) Genetic structure and linkage disequilibrium in a diverse, representative collection of the C4 model plant, Sorghum bicolor. G3: Genes|Genomes|Genetics;3(5):783–93. https://doi.org/10.1534/g3.112.004861
Wang SB, Feng JY, Ren WL, Huang B, Zhou L, Wen YJ et al (2016) Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology. Sci Rep 6:19444. https://doi.org/10.1038/srep19444
Wen YJ, Zhang H, Ni YL, Huang B, Zhang J, Feng JY et al (2017) Methodological implementation of mixed linear models in multilocus genome-wide association studies. Brief Bioinformatics 19:700–712. https://doi.org/10.1093/bib/bbx028
Wen YJ, Zhang H, Ni YL, Huang B, Zhang J, Feng JY et al (2018) Methodological implementation of mixed linear models in multi-locus genome-wide association studies. Brief Bioinform 19:700–712. https://doi.org/10.1093/bib/bbw145
Wondimu Z, Bantte K, Paterson A. H, Worku W. 2020. Agro-morphological diversity of Ethiopian sorghum [Sorghum bicolor (L.) Moench] landraces under water limited environments. Genet Resour Crop Evol 2149–2160. https://doi.org/10.1007/s10722-020-00968-7.
Wondimu Z, Dong H, Paterson AH, Worku W, Bantte K. (2021) Genetic diversity, population structure, and selection signature in Ethiopian sorghum [Sorghum bicolor L. (Moench)] germplasm. G3 Genes|Genomes|Genetics; 11(6). https://doi.org/10.1093/g3journal/jkab087
Wu J, Wang L, Fu J, Chen J, Wei S, Zhang S et al (2020) Resequencing of 683 common bean genotypes identifies yield component trait associations across a north–south cline. Nat Genet 52:118–125. https://doi.org/10.1038/s41588-019-0546-0
Xu W, Subudhi PK, Crasta OR, Rosenow DT, Mullet JE, Nguyen HT (2000) Molecular mapping of QTLs conferring stay-green in grain sorghum (Sorghum bicolor L. Moench). Genome 43(3):461–9
Yan J, Shah T, Warburton ML, Buckler ES, McMullen MD, Crouch J (2009) Genetic characterization and linkage disequilibrium estimation of a global maize collection using SNP markers. PLoS ONE 4(12):e8451
Yu C, Su S, Xu Y, Zhao Y, Yan A, Huang L et al (2014) The effects of fluctuations in the nutrient supply on the expression of five members of the AGL17 clade of MADS-Box genes in rice. PLoS One 9:e105597. https://doi.org/10.1371/journal.pone.0105597
Zhang J, Feng JY, Ni YL, Wen YJ, Niu Y, Tamba CL et al (2017) PLARmEB: integration of least angle regression with empirical Bayes for multilocus genome-wide association studies. Heredity 118:517–524. https://doi.org/10.1038/hdy.2017.8
Zhang YM, Jia Z, Dunwell JM (2019) Editorial: the applications of new multi-locus gwas methodologies in the genetic dissection of complex traits. Front Plant Sci 10:100. https://doi.org/10.3389/fpls.2019.00100
Zhang Y, Liu P, Zhang X, Zheng Q, Chen M, Ge F et al (2018) Multi-locus genome-wide association study reveals the genetic architecture of stalk lodging resistance-related traits in maize. Front Plant Sci 9:611. https://doi.org/10.3389/fpls.2018.00611
Zhao J, Mantilla Perez MB, Hu J, Salas Fernandez MG (2011) Genome-wide association study in sorghum. The Plant Genome; 9(2)
Zhao J, Mantilla Perez MB, Hu J, Salas Fernandez MG (2016) Genome-wide association study for nine plant architecture traits in sorghum. The Plant Genome. https://doi.org/10.3835/plantgenome2015.06.0044
Zhu C, Gore M, Buckler ES, Yu J (2008) Status and prospects of association mapping in plants. The Plant Genome 1(1):5–20
Acknowledgements
We also thank the staff of Genomics and Bioinformatics Core Facility at the University of Georgia, USA for their support in GBS and bioinformatics services. The institutional support provided by Melkassa and Sirinka Agricultural Research Centers during the field experiments is highly appreciated.
Funding
This study was financially supported by the United States Agency for International Development’s (USAID) Feed the Future Laboratory for Climate Resilient Sorghum.
Author information
Authors and Affiliations
Contributions
Kassahun Bantte and Andrew Paterson conceived and designed the research work. Hongxu Dong performed GBS and bioinformatics analysis. Zeleke Wondimu analyzed the data and drafted the manuscript. Kassahun Bantte, Andrew Paterson, Hongxu Dong, and Walelign Worku critically reviewed the manuscript and provided comments. All the authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wondimu, Z., Dong, H., Paterson, A.H. et al. Genome-wide association study reveals genomic loci influencing agronomic traits in Ethiopian sorghum (Sorghum bicolor (L.) Moench) landraces. Mol Breeding 43, 32 (2023). https://doi.org/10.1007/s11032-023-01381-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-023-01381-5