
Abstract
Flower color is considered an important appealing signal to pollinators and also a marker trait in Brassica crop breeding. However, the genetic basis of orange flower trait remains poorly understood in Brassica rapa. In this study, we conducted a genetic analysis of orange flower trait and fine mapped the underlying gene in B. rapa. Two populations, BC1F1 and BC1F2 with 478 and 443 individuals, respectively, were constructed from a cross between 94C9 (orange flower) and 92S105 (yellow flower). Genetic analysis showed that a single recessive gene, BrOF, controlled the orange flower trait. Using Indel and dCAPS markers developed from whole-genome resequencing data of 94C9 and 92S105, BrOF was mapped to a 41.5-kb region on chromosome A09 delimited by InDel409 and dCAPS425 containing six putative genes. Among them, only Bra037124 and Bra037125, which encode an AP2 domain–containing transcription factor and an SEC-C motif–containing protein/OTU-like cysteine protease family protein, respectively, were successfully cloned. The sequence analysis revealed two SNPs resulting in amino acid residue changes in the coding region of Bra037124, as well as seven SNPs and one insertion leading to amino acid residue mutations in the coding region of Bra037125, between 94C9 and 92S105. The reliability of a co-segregating marker InDel314 in marker-assisted selection (MAS) was confirmed by testing different yellow/orange flower Chinese cabbage lines. These results provide a good foundation to identify BrOF and facilitate our understanding of the genetic basis underlying the development of orange flowers in Chinese cabbage.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In insect-pollinated plants, flower color, determined by the content and composition of pigments, is considered one of the most important attractants to pollinators (Kevan and Baker 1983). Among the three primary pigments (carotenoids, flavonoids, and betalains) in plants, carotenoids are responsible for organ color from yellow to red (DellaPenna and Pogson 2006; Grotewold 2006). Carotenoids (C40 isoprenoids synthesized in plastids) are widely distributed in plants and microorganisms (Britton et al. 2004).
Carotenoids have different functions in various plant organs. In chloroplasts, carotenoids are involved in the construction of the photosynthetic reaction center and antenna complexes, and play roles in harvesting light energy and protecting photosynthetic tissues from photooxidative damage (Niyogi 2000; Walter and Strack 2011). Carotenoids accumulate in chromoplasts to provide distinct colors to flowers, which then attract pollinators (Kevan and Baker 1983). In addition, carotenoids serve as precursors for the biosynthesis of plant hormones, such as abscisic acid and strigolactones (Dun et al. 2009; Walter and Strack 2011).
Most steps of the carotenoid biosynthesis pathway have been elucidated in model plants at the molecular level (Ruiz-Sola and Rodrıguez-Concepcion 2012) enormously facilitating the study of this pathway in other plants. Recently, several key genes leading to changes in flower and fruit colors have been cloned. For example, in tomato, yellow flowers were converted to white and beige flowers with mutations in β-ring carotene hydroxylase 2 (CHY2) and zeaxanthin epoxidase (ZEP), respectively (Galpaz et al. 2006, 2008). In pepper, fruit color was changed from red to orange by a single-nucleotide mutation of CHY2 (Borovsky et al. 2013). The loss of carotenoid cleavage dioxygenase 4 (CCD4) function produced a yellow flower phenotype in chrysanthemum (Ohmiya et al. 2012; Yoshioka et al. 2012) and B. napus (Zhang et al. 2015a). Moreover, xanthophyll esterification and the development of chromoplasts also affect flower colors. In cauliflower, mutation of the Or gene was responsible for orange curd tissues (Li et al. 2001; Paolillo et al. 2004). Ariizumi et al. (2014) found that the functional disruption of pale yellow petal (PYP1) involved in xanthophyll esterification resulted in pale yellow petal phenotype in tomato.
The flower colors in the Brassica genus are diverse, including yellow, yellowish, white, orange, etc. (yellow being the most common color), and genetic analysis has been performed for some flower colors. The white and tangerine flower traits are controlled by a single recessive gene in B. rapa (Mohammad et al. 1942; Zhang et al. 2003). In B. napus, two recessive genes conferred the tangerine flower trait (Li et al. 1999) while the white flower trait was controlled by a single dominant gene (Zhang et al. 2015a). Currently, only a few genes controlling flower color have been cloned, such as the pale yellow flower gene carotenoid isomerase (BrCRTISO) in B. rapa (Lee et al. 2014) and the white flower gene BnaC3.CCD4 in B. napus (Zhang et al. 2015a). However, the orange flower gene in Brassica has not been reported and its formational molecular mechanism has not been elucidated.
In this study, two segregating populations, BC1F1 and BC1F2, were constructed with two pure lines, the orange flower line 94C9 and the yellow flower line 92S105. Molecular markers covering the entire genome of B. rapa were developed based on the genome resequencing data of the two parental lines, and then fine mapping of the candidate gene conferring the orange flower trait was performed. The cDNA sequences of the candidate genes within the mapping region were cloned, and the co-segregating markers were tested using different yellow/orange flower lines of Chinese cabbage. This study contributes to marker-assisted selection of the orange flower trait in B. rapa breeding and provides new information on flower color variation, especially orange flowers in B. rapa.
Materials and methods
Plant materials
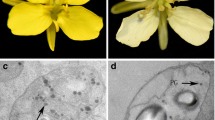
The Chinese cabbage research group in Northwest A&F University, Yangling, China, provided the eight Chinese cabbage pure lines used in this study: 94C9, 15S1014, and 94B6, all with orange flowers; 92S105, 15S1006, 94C6, 94B4, and 09Q5, all with yellow flowers. Here we used 94C9 and 92S105 (Fig. 1) as parents to study the inheritance pattern of flower color and performed fine mapping of the BrOF gene, where the line 94C9 was first crossed with 92S105 to produce the hybrid F1; then, the F1 plants were used as the male parent and backcrossed with 94C9 to produce the BC1F1 population with 478 individuals (Table 1). One heterozygous yellow flower individual from the BC1F1 population was self-pollinated to yield the BC1F2 population with 443 individuals (Table 1). Another six lines were used to validate the derived co-segregation markers.

Flower colors of the two parental lines. a 94C9, b 92S105
Seedlings of all materials were grown in a greenhouse and naturally vernalized, then transplanted to the vegetable experimental field of the Northwest A&F University in 2015 and 2018. The flower color of each individual was assessed based on two observations of at least ten flowers during the flowering stage with an 8-day interval.
DNA and RNA extraction, first-strand cDNA synthesis, and polymerase chain reaction
Genomic DNA was extracted from fresh leaves using the cetyl trimethylammonium bromide (CTAB) method following Porebski et al. (1997). Total RNA was extracted from the petals of opening flowers with the MiniBEST Plant RNA Extraction Kit (Takara, Dalian, China), and first-strand cDNA synthesis was performed with the PrimeScript™ II 1st Strand cDNA Synthesis Kit (Takara, Dalian, China). BC1F2 individuals, 10 yellow-flowered and 10 orange-flowered, were randomly chosen to construct yellow-flowered and orange-flowered DNA pools, respectively.
The PCR were conducted in 10-μL volumes containing 1 μL of genomic DNA (60 ng/μL), 0.5 μL of each forward and reverse primer (10 μM), and 5 μL of Master Mix (Vazyme, Nanjing, China) (Supplementary Table S1). The PCR amplification procedure was performed as follows: 94 °C for 4 min, 27–33 cycles of 94 °C for 30 s, 50–53 °C for 30 s, and 72 °C for 30 s, followed by a final extension at 72 °C for 5 min. The amplified products of insertion/deletion (InDel) markers were separated on 9% or 12% non-denaturing polyacrylamide gel. For derived cleaved amplified polymorphic sequences (dCAPS) markers, the amplified products were dissected with the restriction enzymes followed by 9% non-denaturing polyacrylamide gel (Pan et al. 2015).
Development of InDel and dCAPS markers
To develop more polymorphic markers, the 94C9 and 92S105 parental lines were resequenced with HiSeq X Ten (Macrogen, Shenzhen, China) at sequencing depths of approximately 100-fold. The resequencing data of the two parental lines were aligned onto the NCBI B. rapa reference genome (http://www.ncbi.nlm.nih.gov/genome/?term=Brassica+rapa), the genomic variants were called with Isaac Variant Caller (IVC), and the physical location of each genomic variant was anchored onto the corresponding chromosomes or scaffolds. Only insertions/deletions with more than 3 bp were applied when designing the InDel primers with Primer Premier 5.0 (http://www.premierbiosoft.com/primerdesign/) using the corresponding flanking sequences in the B. rapa reference genome. For the single-nucleotide polymorphisms (SNPs), dCAPS markers were developed with the website software “dCAPS Finder 2.0” (http://helix.wustl.edu/dcaps/dcaps.html). In this study, all the designed primers were synthesized by Sangon Biotech Co., Ltd. (Shanghai, China).
Identification of recombination events on SNP loci
To clone the DNA fragments containing SNP loci in the recombinants, PCR primers were designed based on the B. rapa reference genome. The PCR-amplified products were separated on a 1.5% agarose gel and purified with a DNA Gel Extraction Kit (Tiangen, Beijing, China). At least three purified products were sequenced by Aoke Biotech Co., Ltd. (Beijing, China). The DNA Star Lasergene (http://www.dnastar.com) and Chromas (http://technelysium.com.au/wp/chromas/) software packages were used to analyze the nucleotide sequences.
Fine mapping of the BrOF gene
All the developed InDel and dCAPS markers were screened with the genomic DNA of the two parental lines, 94C9 and 92S105, and the resulting polymorphic markers were used to detect the allelic diversity of individuals in the segregating populations. Using the segregation data of the polymorphic InDel and dCAPS markers and flower color phenotypic data of all population individuals, the genetic linkage map was constructed with the JoinMap 4.0 software using a logarithm of the odds (LOD) threshold score of 6.0. When there was only one recombinant plant between a marker and the target gene, the marker was considered as the nearest flanking marker for the target gene. Based on the B. rapa reference genome in BRAD (Brassica database, http://brassicadb.org/brad), the physical positions of mapped markers were used to construct the physical map of the BrOF gene. Genes located in the final delimited chromosome region were identified according to the annotation data of the BRAD B. rapa reference genome.
Cloning of the potential candidate genes located in the mapping region
To clone the cDNA sequences of the potential candidate genes located in the fine mapping region, specific primers were designed based on the reference genome of B. rapa. The PCR-amplified products were separated on a 1.5% agarose gel and purified with a DNA Gel Extraction Kit (Tiangen, Beijing, China). The purified products were cloned into the pMD18-T Vector (Takara, Dalian, China) and then transformed into the Escherichia coli strain DH5α. At least three clones were sequenced at Aoke Biotech Co., Ltd. (Beijing, China). The DNA Star Lasergene software package was used to analyze the cDNA sequences and their deduced amino acid sequences.
Results
Orange flower was a recessive trait controlled by a single gene, BrOF
All the hybrid F1 plants derived from the cross between the orange flower line 94C9 and the yellow flower line 92S105 had yellow flowers, consistent with the flowers of 92S105 and indicating that yellow flower trait was dominant over orange flower. The flower color survey showed that there were 243 yellow flower individuals and 235 orange flower individuals in the BC1F1 population, while the BC1F2 population had 336 yellow flower individuals and 107 orange flower individuals (Table 1). The χ2 test indicated that the BC1F1 and BC1F2 segregation ratios fitted the expected ratios of 1:1 (χ2 = 0.134, df = 1, p > 0.05) and 3:1 (χ2 = 0.169, df = 1, p > 0.05), respectively. Therefore, these results indicated that the orange flower trait was controlled by a single recessive gene.
Preliminary mapping of BrOF on chromosome A09
The two parental lines, 94C9 and 92S105, were resequenced to develop polymorphic markers for the preliminary mapping of BrOF. The resequencing data were aligned onto the NCBI B. rapa reference genome; the results showed that the resequencing data of 94C9 and 92S105 covered 89.54% and 88.58% of the reference genome, respectively. After removing duplications and searching the variants with IVC, a large number of InDels and SNPs were detected between 94C9 and 92S105.
Based on the InDel locations distributed on each chromosome, 101 InDel markers distributed over 10 chromosomes were developed, with 56 InDel markers showing polymorphism between the two parental lines (Supplementary Table S1). These 56 polymorphic markers were tested with 96 plants (54 yellow flower plants + 42 orange flower plants) selected from the BC1F1 population. As a result, seven markers (InDel143, InDel151, InDel181, InDel182, InDel183, InDel186, and InDel190) from chromosome A09 linked with the BrOF gene were used to construct a preliminary genetic linkage map. The BrOF gene was preliminarily mapped to a region flanked by InDel181 and InDel182 on chromosome A09 with corresponding genetic and physical distances of 8.7 cM and 1.6 Mb, respectively (Fig. 2a). In addition, the linkage of markers InDel181 and InDel182 with the BrOF gene was also confirmed by screening yellow-flowered and orange-flowered DNA pools (Supplementary Fig. S1).

Fine mapping of the orange flower gene BrOF in Chinese cabbage. a Preliminary linkage map of BrOF based on 96 individuals from the BC1F1 population. b Genetic linkage map of BrOF based on 478 BC1F1 plants. c Fine genetic map of BrOF based on combining 478 BC1F1 plants with 443 BC1F2 plants. Values in the bracket represent the physical position of markers based on the B. rapa reference genome in BRAD
Fine mapping of the BrOF gene
To fine map the BrOF gene, 74 InDel markers were developed in the preliminary mapping region based on the resequencing data of the two parents. Of those 74 markers, 43 exhibited polymorphism in the parental lines (Supplementary Table S1). When InDel181and InDel182 were used to test 478 BC1F1 plants, a total of 50 recombinants were found in which 24 recombination events occurred between InDel181 and BrOF, while the other 26 recombination events occurred between InDel182 and BrOF. These 50 recombinants were tested with the 43 new polymorphic markers. As a result, the BrOF gene was narrowed to a region between InDel307 and InDel315, both with one recombination event. To further shorten the mapping interval, three dCAPS markers were developed on the side of InDel307, in which one polymorphic marker (dCAPS425) was employed to screen the two recombinants (Fig. 3). The results revealed that the dCAPS425 marker also showed one recombination event with BrOF, which was further confirmed by sequence alignment of the DNA fragments containing the SNP amplified by the primers OY424 (Supplementary Table S1, Fig. S2). The segregation data from the polymorphic markers described above and the phenotypic data of flower color for all the individuals in the BC1F1 population were used to construct a high-resolution genetic map, which included 42 markers around the BrOF gene, as well as four markers (InDel309, InDel310, InDel314, and InDel409) that co-segregated with the BrOF gene, where the physical distance of the two flanking markers, InDel315 and dCAPS425, was 44.7 kb (Fig. 2b). Furthermore, the two flanking markers and four co-segregating markers were used to identify recombinants in the BC1F2 population (Fig. 3). The BrOF gene was finally restricted to a genomic region between InDel409 and dCAPS425 (Fig. 2c), with a genetic distance of 0.4 cM and a physical distance approximately 41.5 kb, respectively (Fig. 4). Based on the gene annotations in the corresponding genomic region of BRAD, six genes were found in the mapping region (Table 2).

Genotyping of BC1F1 (a) and BC1F2 (b) mapping populations using tightly linked markers dCAPS425, InDel314, and InDel409. M, DL50 marker; P1, 92S105; P2, 94C9; F1, F1 individual; BC1F1 plants, individuals from the BC1F1 population; BC1F2 plants, individuals from the BC1F2 population; Y, yellow flower phenotype; O, orange flower phenotype

Identification of candidate gene for BrOF. BrOF is restricted to a 41.5-kb region on A09 flanked by InDel409 and dCAPS425. This region contains six annotated genes according to the BRAD B. rapa reference genome
Cloning and sequence analysis of potential candidate genes located in the mapping region
Six designed primers, including OY911, OY912, OY913, OY914, OY915, and OY916, were used to clone the cDNA sequences of the potential candidate genes contained in the mapping region from the two parental lines, 94C9 and 92S105 (Supplementary Table S1). Only the cDNA sequences of Bra037124 and Bra037125 could be cloned. Sequence alignment analysis revealed that three SNPs were found in the coding region of Bra037124 between 94C9 and 92S105 (Supplementary Fig. S3) resulting in two amino acid residue mutations (Supplementary Fig. S4); there were 22 SNPs and one 3-bp insertion in the coding region of Bra037125 leading to seven amino acid residue changes and the insertion of one amino acid residue in 94C9, respectively (Supplementary Figs. S3 and S4).
Validation of the co-segregating markers
To validate the accuracy of the co-segregating markers (InDel309, InDel310, and InDel314) in marker-assisted selection (Fig. 2c), linkage analysis was performed in eight yellow/orange flower Chinese cabbage lines. The results showed that the genotypes of marker InDel314 were completely consistent with the phenotypes of flower color; however, the genotypes of the other two markers incompletely fitted the flower color phenotypes (Supplementary Fig. S5), indicating that the InDel314 marker could be applied to MAS of the orange flower trait in Chinese cabbage.
Discussion
Because of the rich sources of variation, the flower colors of Brassica plants vary from white to red with many colors occurring in the rapeseed (Zhang 2015). Many studies on the genetic inheritance patterns of flower color indicated that the white flower trait was either dominant over yellow flower (B.napus, Quazi 1988, Zhang et al. 2015a; B. oleracea, Kianian and Quiros 1992, Han et al. 2015) or recessive to yellow flower (B. rapa, Mohammad et al. 1942; B. juncea, Singh and Chauhan 2011, Zhang et al. 2018). Some studies also revealed that the white flower trait was controlled by a single gene (Mohammad et al. 1942; Quazi 1988; Kianian and Quiros 1992; Han et al. 2015; Zhang 2015); however, others reported that the white flower trait was maintained by two major genes, such as in B. juncea (Singh and Chauhan 2011; Zhang et al. 2018). The above results indicate that the molecular mechanisms controlling a single flower color might vary across Brassica species. Furthermore, other investigations indicated that the yellow flower trait was dominant over other flower colors, such as pale yellow and orange in B. rapa (Alam and Aziz 1954; Cours 1977; Feng et al. 2012; Lee et al. 2014) and tangerine and golden yellow in B. napus (Li et al. 1999; Zhang et al. 2000). In this study, the inheritance patterns of orange flower trait in B. rapa were studied using the hybrid F1, 94C9×92S105, and its BC1F1 and BC1F2 populations. The results showed that orange flower trait was controlled by a single gene that was recessive to the yellow flower, consistent with previous studies (Cours 1977; Feng et al. 2012).
Although the release of the Chinese cabbage genome information (Wang et al. 2011) was convenient for the development of molecular markers, the rapid development of whole-genome sequencing technology could contribute to develop many polymorphic molecular markers for investigated materials. In this study, the genomes of two parental lines, 94C9 and 92S105, were resequenced at high sequencing depths. Many variations were obtained by comparing the resequencing data between the two parental line genomes and the B. rapa reference genome, and then, 175 InDel and 3 dCAPS markers were developed and screened, 100 of which exhibited amplification polymorphism between the two parental lines greatly facilitating the process of fine mapping the BrOF gene. Subsequently, the BrOF gene was rapidly mapped to a 41.5-kb region on chromosome A09. Therefore, combining whole-genome resequencing with the conventional gene mapping is an efficient strategy for fine mapping a gene.
Previous studies have indicated that functional disruption of the BrCRTISO gene, which is located on the bottom of chromosome A09, results in formation of the orange head leaf trait of Chinese cabbage (Zhang et al. 2013, 2015b; Li et al. 2015; Su et al. 2015). In this study, the candidate gene for orange flower trait was mapped to the top of chromosome A09. Together, these results suggested that there may not be a close linkage relationship between the orange head leaf and orange flower traits in Chinese cabbage. However, Feng et al. (2012) reported that the morphological marker, orange flowers, co-segregated with the orange head leaf in two Chinese cabbage F2 populations. Therefore, further clarification is needed to determine whether the orange flower trait is controlled by different genes on chromosome A09 of Chinese cabbage.
In the present study, the BrOF gene was restricted to a region of 41.5 kb on the B. rapa chromosome A09 containing six annotated genes. We found no reports on these six genes and their homologous genes in Arabidopsis involved in carotenoid metabolism (Table 2). Among the six genes present, Bra037126, Bra037127, Bra037128, and Bra037129 encoding proteins have a transferase domain, a RALF domain, a bHLH domain, and a lipase-3 domain, respectively; however, members of families that contain these domains were not associated with color formation based on previous studies (Beisson et al. 1997; D’Auria 2006; Gremski et al. 2007; Hong et al. 2008; Kim et al. 2008; Zheng et al. 2009; Atkinson et al. 2013; Bergonci et al. 2014; Schuster et al. 2015; Murphy et al. 2016). The homologous genes of Bra037126, Bra037127, and Bra037128 in Arabidopsis were associated with disease resistance (Zheng et al. 2009), lateral root initiation (Murphy et al. 2016), and gynoecium development (Gremski et al. 2007; Schuster et al. 2015), respectively. The Bra037129 gene encodes a lipase class 3 family protein and plays an important role in both lipase metabolism and stress responses (Beisson et al. 1997; Hong et al. 2008; Kim et al. 2008). In B. juncea, mutation of the Bra032956 gene encoding an acyltransferase family protein may lead to the production of the white flower phenotype, which was recessive to the yellow flower phenotype (Zhang et al. 2018). Moreover, the cDNA sequences of these four genes could not be cloned, possibly due to their low expression levels. We concluded that these may not be the target genes controlling the orange flower trait and focused attention on the two remaining genes. The Bra037124 gene, homologous with AT5G67190 in Arabidopsis, encodes an AP2/ERF family protein that is primarily involved in responses to abiotic or biotic stress (Haake et al. 2002; Tsutsui et al. 2009; Dong and Liu 2010; Mizoi et al. 2012; Amalraj et al. 2016) and floral development (Jofuku et al. 1994). However, Lee et al. (2012) reported that SlERF6, belonging to AP2/ERF gene family, was associated with carotenoid metabolism in tomato. The Bra037125 gene encodes a protein of unknown function that contains both an SEC-C motif and an ovarian tumor domain (OTU)–like cysteine protease domain. The functions of the SEC-C and OTU protein families are also uncharacterized in Arabidopsis. The sequence analysis indicated that two amino acid residue mutations were found in the deduced amino acid sequences of Bra037124 between 94C9 and 92S105, and there were seven amino acid residue changes and one amino acid residue insertion in the deduced amino acid sequence of Bra037125 in 94C9 (Supplementary Fig. S4). In addition, the detection of reliability of three co-segregating markers for MAS indicated that only the genotype of InDel314 showed completely consistent with the corresponding phenotype, and InDel314 was located in the intergenic region between Bra037124 and Bra037125. Taken together, we hypothesize that Bra037124 or Bra037125 might be the likely candidate for the BrOF gene (Table 2). Further research is needed to identify the BrOF gene and reveal the molecular mechanism behind formation of the orange flower phenotype.
The results from the fine genetic mapping of BrOF showed that InDel309, InDel310, and InDel314 co-segregated with this target gene in the segregating populations that were derived from the cross between the parental lines 94C9 and 92S105. These three markers were tested with 8 lines (O1, O2, O3, Y1, Y2, Y3, Y4, and Y5) (Supplementary Fig. S5), and only the genotypes of marker InDel314 were completely consistent with the phenotypes of flower color. Three yellow flower lines (Y2, Y3, and Y4) had the same genotypes of markers InDel309 and InDel310 as the orange flower lines (O1, O2, and O3), which made the suitability of these two markers for MAS inbreeding processes weaker than that of InDel314. This inconsistent result might be the reason that the mechanism underlying yellow flower formation is different, where other regulating alleles or genes may also be involved that are not located in the target region of BrOF. The formation of flower color can rely on complicated regulatory mechanisms, as in the white flower trait of B. juncea, which was controlled by two major genes (Singh and Chauhan 2011; Zhang et al. 2018). Therefore, we might infer that the yellow flowers of Y2, Y3, and Y4 were also affected by other genetic factors, which led to inconsistencies in markers InDel309 and InDel310 compared with marker InDel314. The relatively large genetic distances between the InDel309 and InDel310 markers and the BrOF target gene might also account for this inconsistent result.
In conclusion, our study indicated that a single recessive gene controlled the orange flower trait in Chinese Cabbage. The BrOF gene was mapped to a 41.5-kb region on chromosome A09 flanked by two markers, InDel409 and dCAPS425. Two potential candidate genes for the orange flower trait were identified in the target region, Bra037124 and Bra037125. The practical application of a co-segregating marker, InDel314, in MAS was validated. Therefore, our findings offer a good foundation for the functional identification of the BrOF gene and provide a molecular marker for selecting new orange flower lines of Chinese cabbage.
Change history
15 July 2019
The original article unfortunately contains an incorrect supplementary materials.
References
Alam Z, Aziz M (1954) Inheritance of flower colour in some self-fertile oleiferous Brassicae. Pak J Sci Res 6:27–36
Amalraj A, Luang S, Kumar MY, Sornaraj P, Eini O, Kovalchuk N, Bazanova N, Li Y, Yang NN, Eliby S, Langridge P, Hrmova M, Lopato S (2016) Change of function of the wheat stress-responsive transcriptional repressor TaRAP2.1L by repressor motif modification. Plant Biotechnol J 14:820–832
Ariizumi T, Kishimoto S, Kakami R, Maoka T, Hirakawa H, Suzuki Y, Ozeki Y, Shirasawa K, Bernillon S, Okabe Y, Moing A, Asamizu E, Rothan C, Ohmiya A, Ezura H (2014) Identification of the carotenoid modifying gene PALE YELLOW PETAL 1 as an essential factor in xanthophyll esterification and yellow flower pigmentation in tomato (Solanum lycopersicum). Plant J 79:453–465
Atkinson NJ, Lilley CJ, Urwin PE (2013) Identification of genes involved in the response of Arabidopsis to simultaneous biotic and abiotic stresses. Plant Physiol 162:2028–2041
Beisson F, Gardies AM, Teissere M, Ferte N, Noat G (1997) Anesterase neosynthesized in post-germinated sunflower seeds is related to a new family of lipolytic enzymes. Plant Physiol Biochem 35:761–765
Bergonci T, Ribeiro B, Ceciliato PH, Guerrero-Abad JC, SilvaFilho MC, Moura DS (2014) Arabidopsis thaliana RALF1 opposes brassinosteroid effects on root cell elongation and lateral root formation. J Exp Bot 65:2219–2230
Borovsky Y, Tadmor Y, Bar E, Meir A, Lewinsohn E, Paran I (2013) Induced mutation in β-CAROTENE HYDROXYLASE results in accumulation of β-carotene and conversion of red to orange color in pepper fruit. Theor Appl Genet 126:557–565
Britton G, Liaaen-Jensen S, Pfander HP (2004) Carotenoids handbook. Birkhauser Verlag, Basel
Cours BJ (1977) Genetic studies in Brassica campestris L. (M Sc thesis). Dissertation, University of Wisconsin
D’Auria JC (2006) Acyltransferases in plants: a good time to be BAHD. Curr Opin Plant Biol 9:331–340
DellaPenna D, Pogson BJ (2006) Vitamin synthesis in plants: tocopherols and carotenoids. Annu Rev Plant Biol 57:711–738
Dong CJ, Liu JY (2010) The Arabidopsis EAR-motif-containing protein RAP2.1 functions as an active transcriptional repressor to keep stress responses under tight control. BMC Plant Biol 10:47–61
Dun EA, Brewer PB, Beveridge CA (2009) Strigolactones: discovery of the elusive shoot branching hormone. Trends Plant Sci 14:364–372
Feng H, Li YF, Liu ZY, Liu J (2012) Mapping of or, a gene conferring orange color on the inner leaf of the Chinese cabbage (Brassica rapa L. ssp. pekinensis). Mol Breed 29:235–244
Galpaz N, Ronen G, Khalfa Z, Zamir D, Hirschberg J (2006) A chromoplast-specific carotenoid biosynthesis pathway is revealed by cloning of the tomato white-flower locus. Plant Cell 18:1947–1960
Galpaz N, Wang Q, Menda N, Zamir D, Hirschberg J (2008) Abscisic acid deficiency in the tomato mutant high-pigment 3 leading to increased plastid number and higher fruit lycopene content. Plant J 53:717–730
Gremski K, Ditta G, Yanofsky MF (2007) The HECATE genes regulate female reproductive tract development in Arabidopsis thaliana. Development 134:3593–3601
Grotewold E (2006) The genetics and biochemistry of floral pigments. Annu Rev Plant Biol 57:761–780
Haake V, Cook D, Riechmann JL, Pineda O, Thomashow MF, Zhang JZ (2002) Transcription factor CBF4 is a regulator of drought adaptation in Arabidopsis. Plant Physiol 130:639–648
Han FQ, Yang C, Fang ZY, Yang LM, Zhuang M, Lv HH, Liu YM, Li ZS, Liu B, Yu HL, Liu XP, Zhang YY (2015) Inheritance and InDel markers closely linked to petal color gene (cpc-1) in Brassica oleracea. Mol Breed 35:160–167
Hong JK, Choi HW, Hwang IS, Kim DS, Kim NH, Choi DS, Kim YJ, Hwang BK (2008) Function of a novel GDSL-type pepper lipase gene, CaGLIP1, in disease susceptibility and abiotic stress tolerance. Planta 227:539–558
Jofuku KD, Denboer BG, Vanmontagu M, Okamuro JK (1994) Control of Arabidopsis flower and seed development by the homeotic gene APETALA2. Plant Cell 6:1211–1225
Kevan PG, Baker HG (1983) Insects as flower visitors and pollinators. Annu Rev Entomol 28:407–453
Kianian SF, Quiros CF (1992) Trait inheritance, fertility, and genomic relationships of some n=9 Brassica species. Genet Resour Crop Evol 39:165–175
Kim KJ, Lim JH, Kim MJ, Kim T, Chung HM, Paek KH (2008) GDSL-lipase1 (CaGL1) contributes to wound stress resistance by modulation of CaPR-4 expression in hot pepper. Biochem Biophys Res Commun 374:693–698
Lee JM, Joung JG, McQuinn R, Chung MY, Fei Z, Tieman D, Klee H, Giovannoni J (2012) Combined transcriptome, genetic diversity and metabolite profiling in tomato fruit reveals that the ethylene response factor SlERF6 plays an important role in ripening and carotenoid accumulation. Plant J 70:191–204
Lee S, Lee SC, Byun DH, Lee DY, Park JY, Lee JH, Lee HO, Sung SH, Yang TJ (2014) Association of molecular markers derived from the BrCRISTO1 gene with prolycopene-enriched orange-colored leaves in Brassica rapa. Theor Appl Genet 127:179–191
Li M, Chen WJ, Yi DL (1999) Studies on the inheritance of CMS restorer R18 with red color flower in rapeseed (Brassica napus L). Sci Agric Sin 32:27–30
Li L, Paolillo DJ, Parthasarathy MV, Dimuzio EM, Garvin DF (2001) A novel gene mutation that confers abnormal patterns of β-carotene accumulation in cauliflower (Brassica oleracea var. botrytis). Plant J 26:59–67
Li PR, Zhang SJ, Zhang SF, Li F, Zhang H, Liu XY, Wu J, Wang XW, Sun RF (2015) Carotenoid identification and molecular analysis of carotenoid isomerase-encoding BrCRTISO, the candidate gene for inner leaf orange coloration in Chinese cabbage. Mol Breed 35:72–83
Mizoi J, Shinozaki K, Yamaguchi-Shinozaki K (2012) AP2/ERF family transcription factors in plant abiotic stress responses. Biochim Biophys Acta 1819:86–96
Mohammad A, Sikka SM, Aziz MA (1942) Inheritance of seed colour in some oleiferous Brassicae. Indian J Genet Plant Breeding 2:112–127
Murphy E, Vu DL, Vandenbroeck L, Lin Z, Ramakrishna P, Vandecotte B, Gaudinier A, Goh T, Slane D, Beeckman T, Inzé D, Brady SM, Fukaki H, De Smet I (2016) RALFL34 regulates formative cell divisions in Arabidopsis pericycle during lateral root initiation. J Exp Bot 67:4863–4875
Niyogi K (2000) Safety valves for photosynthesis. Curr Opin Plant Biol 3:455–460
Ohmiya A, Toyoda T, Watanabe H, Emoto K, Hase Y, Yoshioka S (2012) Mechanism behind petal color mutation induced by heavy-ion-beam irradiation of recalcitrant chrysanthemum cultivar. J Jpn Soc Hort Sci 81:269–274
Pan YP, Bo KL, Cheng ZH, Weng YQ (2015) The loss-of-function GLABROUS 3 mutation in cucumber is due to LTR-retrotransposon insertion in a class IV HD-ZIP transcription factor gene CsGL3 that is epistatic over CsGL1. BMC Plant Biol 15:302–316
Paolillo DJ, Garvin DF, Parthasarathy MV (2004) The chromoplasts of Or mutants of cauliflower (Brassica oleracea L. var. botrytis). Protoplasma 224:245–253
Porebski S, Bailey LG, Baum BR (1997) Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol Biol Report 15:8–15
Quazi MH (1988) Interspecific hybrids between Brassica napus L. and B. oleracea L. developed by embryo culture. Theor Appl Genet 75:309–318
Ruiz-Sola MA, Rodrıguez-Concepcion M (2012) Carotenoid biosynthesis in Arabidopsis: a colorful pathway. Arabidopsis Book 10:e015
Schuster C, Gaillochet C, Lohmann JU (2015) Arabidopsis HECATE genes function in phytohormone control during gynoecium development. Development 142:3343–3350
Singh KH, Chauhan JS (2011) Genetics of flower colour in Indian mustard (Brassica juncea L. Czern & Coss). Indian J Genet 71:377–378
Su TB, Yu SC, Wang J, Zhang FL, Yu YJ, Zhang DS, Zhao XY, Wang WH (2015) Loss of function of the carotenoid isomerase gene BrCRTISO confers orange color to the inner leaves of Chinese cabbage (Brassica rapa L. ssp. pekinensis). Plant Mol Biol Report 33:648–659
Tsutsui T, Kato W, Asada Y, Sako K, Sato T, Sonoda Y, Kidokoro S, Yamaguchi-Shinozaki K, Tamaoki M, Arakawa K, Ichikawa T, Nakazawa M, Seki M, Shinozaki K, Matsui M, Ikeda A, Yamaguchi J (2009) DEAR1, a transcriptional repressor of DREB protein that mediates plant defense and freezing stress responses in Arabidopsis. J Plant Res 122:633–643
Walter MH, Strack D (2011) Carotenoids and their cleavage products: biosynthesis and functions. Nat Prod Rep 28:663–692
Wang XW, Wang HZ, Wang J et al (2011) The genome of the mesopolyploid crop species Brassica rapa. Nat Genet 43:1035–1039
Yoshioka S, Aida R, Yamamizo C, Shibata M, Ohmiya A (2012) The carotenoid cleavage dioxygenase 4 (CmCCD4a) gene family encodes a key regulator of petal color mutation in chrysanthemum. Euphytica 184:377–387
Zhang B (2015) Development of chromosome segment substitution lines for QTL analysis of important agronomic traits and cloning the white-flowered gene in Brassica napus L. Dissertation, Huazhong Agricultural University
Zhang XX, Li RH, Chen L, Niu SL, Li Q, Xu K, Wen J, Yi B, Ma CZ, Tu JX, Fu TD, Shen JX (2018) Inheritance and gene mapping of the white flower trait in Brassica juncea. Mol Breed 38:20–29
Zhang JX, Li HX, Zhang MK, Hui MX, Wang Q, Li L, Zhang LG (2013) Fine mapping and identification of candidate Br-or gene controlling orange head of Chinese cabbage (Brassica rapa L. ssp. pekinensis). Mol Breed 32:799–805
Zhang B, Liu C, Wang YQ, Yao X, Wang F, Wu JS, King GJ, Liu KD (2015a) Disruption of a CAROTENOID CLEAVAGE DIOXYGENASE 4 gene converts flower colour from white to yellow in Brassica species. New Phytol 206:1513–1526
Zhang JF, Pu HM, Qi CK, Fu SZ (2000) Inheritance of flower color character in oilseed rape (Brassica napus L). Chin J Oil Crop Sci 22:1–4
Zheng ZY, Qualley A, Fan BF, Dudareva N, Chen ZX (2009) An important role of a BAHD acyl transferase-like protein in plant innate immunity. Plant J 57:1040–1053
Zhang JX, Yuan H, Fei ZJ, Pogson BJ, Zhang LG, Li L (2015b) Molecular characterization and transcriptome analysis of orange head Chinese cabbage (Brassica rapa L. ssp. pekinensis). Planta 241:1381–1394
Zhang DS, Zhang FL, Xu JB (2003) Hereditary characteristics of heading and flower colors on Chinese cabbage. Acta Agric Boreali Sin 18:81–84
Author contribution statement
L. Z. and N. Z. conceived and designed the experiments. N. Z. performed molecular marker development, the genetic map construction, and cloning of the genes contained in the mapped region, and wrote the paper. H. Z., Y. R., and L. C. performed phenotypic observation and DNA extraction, and J. Z. verified the accuracy of the co-segregating markers. L. Z. provided the B. rapa materials, revised the paper, and supervised the research.
Funding
This work was supported by the National Key Research and Development Program of China (2017YFD0101802) and the National Science and Technology Support Program of China (2014BAD01B0802).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 56 kb)
Rights and permissions
About this article

Cite this article
Zhang, N., Zhang, H., Ren, Y. et al. Genetic analysis and gene mapping of the orange flower trait in Chinese cabbage (Brassica rapa L.). Mol Breeding 39, 76 (2019). https://doi.org/10.1007/s11032-019-0984-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-019-0984-5