
Abstract
Blush, the proportion of red overcolor on the skin surface of fruit, is highly variable in peach breeding germplasm and is important in the marketing of peach fruit. The fresh market peach industry demands a high level of blush to entice consumers, while the processing peach industry requires minimal blush. Therefore, blush is a major selection criterion in breeding programs. The use of DNA-based information could improve breeding efficiency and accuracy for fruit blush coverage, but a predictive DNA test is required. The objective of this study was to develop a DNA test for the prediction of blush coverage by targeting the major locus, R f , associated with blush variation. Initially, haplotypes were developed based on five SNP markers associated with variation in blush coverage. To convert the 5-SNP haplotype test into a single, simple PCR-based assay, 11 simple sequence repeat markers were designed and used to screen individuals representing all SNP haplotypes. The most informative assay, named Ppe-Rf-SSR, was chosen to screen 200 individuals of the RosBREED peach reference germplasm set that incorporated germplasm from four breeding programs. Ppe-Rf-SSR accurately differentiated individuals with high-, medium-, and low-blush coverage in most lineages. Outcomes highlighted that DNA tests can be quite predictive for some breeding programs or specific germplasm sets, while for others the predictiveness can falter. Therefore, the confirmation of genotype effects for any DNA test is recommended in new germplasm before routine use. The prediction accuracy and breeding utility of Ppe-Rf-SSR in the University of Arkansas breeding program were subsequently confirmed by screening 443 seedlings, independent of the initial DNA test development process, derived from 18 cross-combinations of 28 parents. Ppe-Rf-SSR can be used to efficiently and accurately predict fruit blush coverage, especially in fresh market germplasm, and has been deployed for routine use in the University of Arkansas peach breeding program.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The peach, Prunus persica L. Batsch, has been cultivated and enjoyed worldwide for centuries (Janick 2005), ranking in the top 10 in total world fruit production in 2013, with US annual production of almost one million tons (FAO 2015). The allure of the fruit and consumers’ initial fruit purchase is based primarily on external appearance and aroma (Olmstead et al. 2015). A high degree of blush, the red pigmentation on the fruit skin surface, entices consumers, and contributes to the flavor and nutritional quality of fruit because of the associated anthocyanin compounds (Frett et al. 2014; Rahim et al. 2014). For retailers, blush, along with size and firmness, have historically been the principal components of fruit quality (Crisosto and Costa 2008). Furthermore, skin color is an important factor in determining fresh market peach prices at the producer (Parker et al. 1991) and retail levels (Jordan et al. 1986). In the USA, the USDA has established specific standards for evaluating and differentiating quality grades of fresh market peaches and the proportion of fruit skin “blushed, pink or red” color is grade-defining; a minimum of one-third surface coverage is required for USA. Fancy and one-fourth surface coverage on 50% of fruit in any lot is required for US Extra No. 1, the two highest quality grades (USDA 2004).
The proportion of blush on fruit of standard peach breeding germplasm is independent of skin ground color (yellow or white) (Bassi and Monet 2008) and can range from negligible to complete coverage (Fig. 1). Although extreme variation is easily observed, standard fresh market cultivars produce fruit with a high proportion of blush coverage while the processing types produce fruit with minimal blush coverage. Processing peach breeders have targeted development of new cultivars with minimal blush coverage, as red pigmentation causes flesh staining and peeling problems during fruit processing (Gradziel and McCaa 2008). In contrast, US and European fresh market breeders have primarily focused on developing cultivars with considerable levels of blush (Scorza and Sherman 1996; Sansavini et al. 2006; Okie et al. 2008) because of the preference of European and US consumers for high (∼80%) skin coverage (Byrne et al. 2012). However, even with this targeted focus, low blush levels, along with poor texture, low sweetness, and small size are likely responsible for the recent stagnancy in US fresh market peach consumption (Crisosto and Costa 2008; Olmstead et al. 2015). Development of fresh market peach cultivars with elevated blush levels could enhance fruit appearance, flavor, and health properties, thereby encouraging increased peach consumption (Frett et al. 2014).

Variation in proportion of blush coverage observed within a single segregating family of the University of Arkansas peach breeding program
Early research (Blake 1932; Blake 1940; Weinberger 1944) studied the inheritance of blush, but no firm conclusions were made (Scorza and Sherman 1996). More recently, estimates of blush heritability have ranged dramatically, with de Souza et al. (1998) reporting a high heritability (∼70%) only 12 years after Hansche (1986) reported the heritability to be three times lower. The greater genetic variability and improved evaluation and estimation methodology of de Souza et al. (1998) likely provided for a more accurate estimation, yet the inconsistencies in estimation and observed influence on blush development of environmental factors such as light exposure and nutrient availability (Crisosto and Costa 2008; Luchsinger et al. 2002; Byrne et al. 2012), uncontrolled in genetic studies, provide evidence for the proposed moderate heritability, and polygenic control of the trait (Byrne et al. 2012). Additionally, there are several known lineage-specific exceptions to the quantitative nature of blush, including the recessive complete blush coverage phenotype associated with the dominant allele of the “full red” (Fr) locus found in “UF Queen” and related University of Florida germplasm (Beckman and Sherman 2003) and the complete suppression of blush caused by the recessive allele of the “highlighter” (h) locus found in South American “criollo” and Italian germplasm (Beckman et al. 2005).
In standard US peach breeding germplasm, blush is a quantitative trait that is expressed during the final stage of fruit development and is maximized in high light environments (Bassi and Monet 2008; Frett et al. 2014). Since the early 2000s, at least seven independent studies have reported QTLs associated with peach blush (Yamamoto et al. 2001; Verde et al. 2002; Quilot et al. 2004; Yamamoto et al. 2005; Cantín et al. 2010; Eduardo et al. 2011; Frett et al. 2014). Although multiple QTLs were reported on linkage groups 2, 3, 4, 6, and 7, Frett et al. (2014) described a major effect QTL, labeled Blush.Pp.ZC-3.1, on peach chromosome 3 that explained up to 70% of blush phenotypic variance in an F2 population of “Zin Dai” × “Crimson Lady.” The candidate skin color gene, PprMYB10 (Lin-Wang et al. 2010), was located within this QTL region, and its activation is likely the major physiological determinant of red skin color in peach (Ravaglia et al. 2013; Rahim et al. 2014; Zhou et al. 2014). The orthology of PprMYB10 to MdMYB1 and MdMYB10 underlying the “red fruit” (R f ) locus of apple (Malus × domestica Borkh.), associated with red flesh and foliage as well as red skin overcolor (Takos et al. 2006; Chagné et al. 2007; Espley et al. 2007), and PavMYB10 underlying the R f locus of sweet cherry (Prunus avium L.), associated with red skin overcolor (Sooriyapathirana et al. 2010; Sandefur et al. 2016), provides strong evidence that extant genetic variation in MYB1/10 is a major causal agent of observed fruit color phenotypic variation within many crops across the Rosaceae family (Arús et al. 2006; Lin-Wang et al. 2010). Therefore, R f was adopted herein as the locus name, as this is the accepted name of the orthologous locus in apple and sweet cherry (Sandefur et al. 2016) to replace the QTL label of Blush.Pp.ZC-3.1 from Frett et al. (2014).
Based on a single SNP (SNP_IGA_341962) located upstream to PprMYB10, a cleaved amplified polymorphic sequence (CAPS) marker, CAPS_341962, was developed for use in marker-assisted selection (Frett et al. 2014). The developed CAPS marker was shown to differentiate three levels of blush within the 93 progeny used in the initial QTL discovery, and the predictiveness of the marker was confirmed in modern peach germplasm (Gasic et al. 2014). However, CAPS markers are incompatible with multiplexing and genotyping with CAPS markers requires additional steps (restriction enzyme digestion and incubation) and higher quality DNA extracts than do simple PCR markers, which can limit their use for routine breeding applications (Ramkumar et al. 2015). Nonetheless, this work showed that the R f locus containing PprMYB10 is a promising target for the development of a DNA test for the prediction of peach fruit skin blush coverage.
Over recent decades, peach breeders in the USA and internationally have released approximately 100 new cultivars annually (Sansavini et al. 2006; Byrne et al. 2012). While many of these cultivars have been widely adopted, traditional breeding is challenged by long generation cycles, large land requirements, and intensive phenotyping (Scorza and Sherman 1996). For most breeding programs, generation of new seedling families is easy, but their management, evaluation, and selection limit cultivar development (Byrne et al. 2012). In addition, establishing a parent’s genetic potential to produce high- or low-blush offspring has traditionally required progeny testing (Byrne et al. 2012), which is highly resource intensive because of the protracted juvenility period and associated phenotyping. The addition of DNA-based information to traditional breeding operations (i.e., marker-assisted selection) in horticultural crops has been discussed since the early 1990s as a means to overcome such obstacles. However, the lack of DNA tests for breeding-relevant traits (Byrne 2007), lack of understanding of the technology (Bliss 2010), and the perceived high cost of genotyping (Luby and Shaw 2001; Edge-Garza et al. 2015) have limited its broad application in horticultural breeding programs (Ru et al. 2015). One of the major limitations is the conceptual gap between genomics researchers and plant breeders, resulting in a failure to ensure that QTL information is converted into robust DNA tests (Collard and Mackill 2008; Iezzoni et al. 2010; Ru et al. 2015). DNA tests have been developed for several peach breeding-relevant traits such as pubescence (Vendramin et al. 2014), flat shape (Picañol et al. 2013), slow ripening (Eduardo et al. 2015), flesh color (Falchi et al. 2013), texture (Peace and Norelli 2009), and acidity (Eduardo et al. 2014), and the development and deployment of DNA tests for skin blush in particular has been successful in apple (Yuan et al. 2014) and sweet cherry (Sandefur et al. 2016).
A DNA test to predict peach fruit skin blush coverage could eliminate the need for extensive progeny testing and improve selection efficiency of this breeding-relevant trait. For wide use in breeding, such a genetic test must be simple to deploy and reliably predictive across diverse germplasm. The objective of this study was to develop a DNA test targeting the R f locus that can be used routinely in peach breeding for the prediction of fruit skin blush coverage.
Materials and methods
Germplasm
Three sets of germplasm were used to develop a DNA test to predict peach fruit blush coverage (Fig. 2 and below).

Experimental approach to develop a trait-predictive, locus-specific DNA test for routine breeding use. A three-step process established an association with SNP markers, converted the association to a single PCR assay, and then confirmed the DNA test’s utility in a target breeding program
Association set
The germplasm phenotyped and SNP-genotyped (n = 622) and used to establish the SNP haplotype-blush coverage association, termed the “Association set,” was part of the RosBREED US Peach Crop Reference (CR) Set (n = 308) and Breeding Pedigree (BP) Sets (n = 314) that used material from the University of Arkansas, University of California—Davis, Clemson University, and Texas A&M University peach breeding programs (Fig. 2; Online Resource 1). The sets were originally chosen to contain a diversity of important cultivars, ancestors, founders, and progeny representative of North American breeding germplasm (Peace et al. 2014). In total, 175 germplasm individuals (50 from the CR Set and 125 from BP Sets) were historically selected for commercial performance, while 447 (258 from the CR Set and 189 from BP Sets) were unselected seedlings in numerous small bi-parental families (8–63 seedlings per family). BP Set individuals were evaluated along with other germplasm but are not specified because this germplasm is privately held. Individuals were maintained as described by Frett et al. (2012).
Conversion set
A subset, termed “Conversion set” (n = 200), of the Association set (n = 114 CR Set individuals and n = 86 individuals of the BP Sets) for which DNA extracts from the original SNP genotyping (Verde et al. 2012) were available and that represented all haplotypes observed was used for marker conversion (Fig. 2; Online Resource 2). The conversion set consisted of 35 germplasm individuals (23 from the CR Set and 12 from BP Sets) historically selected for commercial performance and 165 (91 from the CR Set and 74 from BP Sets) unselected seedlings in numerous small bi-parental families.
Confirmation set
To confirm allele effects in the target breeding program, a different germplasm set was used. This “Confirmation set” (Fig. 2) comprised 443 seedlings from 18 cross-combinations among 28 parents made in 2010 at the University of Arkansas Fruit Research Station, Clarksville, AR.
Phenotypic data
All germplasm sets were phenotyped following the protocol of Frett et al. (2012). Blush coverage was determined by visual estimation on a 0–5 scale (0 = no blush, 1 = 1–20%, 2 = 21–50%, 3 = 51–80%, 4 = 81–99%, 5 = 100%) on each of five fruits, with the average per individual recorded. Phenotypic data were collected on the Association set (including the Conversion set) in 2010 and 2011. However, only 46% (n = 143) of individuals phenotyped in 2011 (n = 308) were phenotyped in 2010 and additional individuals were phenotyped in 2011. Because of the high congruence between 2010 and 2011 data (r 2 = 0.78) considering the expected heritability of the trait (∼70%; de Souza et al. 1998) and the substantially greater amount of data available in 2011, only phenotypic data collected in 2011 (Online Resource 1) was used in this study. The Confirmation set was independently phenotyped in 2014.
To facilitate later estimation of the general effects of SNP haplotypes, SNP diplotypes, SSR alleles, and SSR genotypes on blush coverage (below), the original 0–5 rating scale was converted into a 1–3 scale: 1 = 0–20% coverage (scores of 0 and 1 on the previous 0–5 scale), 2 = 21–80% coverage (scores of 2 and 3 on 0–5 scale), 3 = 81–100% coverage (scores of 4 and 5 on 0–5 scale).
Association
SNP haplotype construction
All association set individuals were genotyped using the International Peach SNP Consortium 9 K SNP array v1 (Verde et al. 2012). SNP array data was used to confirm the parentage of the Association set families (L. Antanaviciute, L. Cai, and C. Peace, manuscript in preparation). Subsequently, five SNP markers were chosen for haplotype construction because they flanked three peach MYB10 homologs, MYB10.1 (ppa026640m; PprMYB10 candidate gene), MYB10.2 (ppa016711m), and MYB10.3 (ppa020385) (Zhou et al. 2014) (Fig. 3; Online Resource 1). The MYB10 homologs and the five SNPs were located in the P. persica Whole Genome Sequence v1.0 (Verde et al. 2013) using the GBrowse tool on the Genome Database for Rosaceae (Jung et al. 2014; www.rosaceae.org). For all individuals, haplotypes were initially constructed using FlexQTL™ software (Bink et al. 2008) and subsequently confirmed based on inheritance analysis (allelic flow through pedigreed individuals) in the association set. A chi-squared test of independence was performed to examine the relation between the 5-SNP haplotypes and proportion of blush coverage and between the 5-SNP diplotypes and proportion of blush coverage (P < 0.05).

Graphical representation of Prunus persica L. Batsch chromosome 3 with the enlarged location of the ∼700 kb flanking the three PpMYB10 transcription factors, including the 5-SNP markers used for haplotyping and Ppe-Rf-SSR. Physical locations were sourced from the Prunus persica Whole Genome Sequence v1.0 (Verde et al. 2013) housed on the Genome Database for Rosaceae (Jung et al. 2014). Two flanking markers and their estimated genetic positions based on the Prunus-TE-F2 reference map are included in gray font
Haplotype and diplotype effects
The general effect of each haplotype and diplotype was expressed as “high” (H) (mode of 3 on 1–3 phenotypic scale), “medium” (M) (mode of 2 on 1–3 phenotypic scale), or “low” (L) (mode of 1 on 1–3 phenotypic scale). As each individual carried two haplotypes, each haplotype’s general effect was influenced by the frequency of the specific diplotype observed. Therefore, when a haplotype was principally observed in combination with just one or a few others, this was noted and the likely impact on the blush association was reported. Diplotype effects were calculated using the same method described above to provide an estimate of the associated effect of paired haplotypes.
The non-parametric Kruskal-Wallis test with post hoc Kruskalmc multiple comparisons test (Giraudoux 2015) (P < 0.05) was used to evaluate differences among haplotypes and diplotypes for their proportion of blush coverage, with all analyses conducted using a modified R script (R Core Team 2013). Six seedlings from University of California—Davis and one from Clemson University were not included in the analyses because of the DNA source used for SSR genotyping not matching that used for SNP haplotyping based on observations of SSR alleles not matching parentage.
Conversion
Candidate assays
The five SNP markers were converted to a single PCR-based simple sequence repeat (SSR) marker assay that best captured the desired allelic information by the following process.
Using the BatchPrimer3 SSR screening tool (You et al. 2008; http://probes.pw.usda.gov/batchprimer3/), microsatellite motifs were detected in the ±100 kb flanking sequence around peach MYB10.1 in the P. persica Whole Genome Sequence v1.0 (Verde et al. 2013) accessed via the Genome Database for Rosaceae (Jung et al. 2014). As 90 microsatellites were detected within this interval, only the 11 microsatellites that were closest to the MYB10 homologs had the largest number of tandem repeats and were well-spaced throughout the 200 kb region were targeted. Primer design followed criteria described by Stegmeir et al. (2014) using Primer3Plus software (Untergasser et al. 2012; www.primer3plus.com). All primer pairs were verified for specific amplification in only the genomic region of the R f locus using the NCBI BLAST software on the Prunus persica Whole Genome Sequence v1.0 (www.rosaceae.org/tools/ncbi_blast). Suitable primer pairs were considered candidate assays (Online Resource 3).
For candidate assays, PCRs were performed in a final mix total volume of 10.0 μl containing 1 μl DNA (∼20 ng μl−1), 5.70 μl of autoclaved double-distilled water, 2.0 μl Taq PCR buffer [×5; GoTaq® buffer (Promega Corp., Madison, WI)], 0.20 μl deoxyribonucleotide triphosphates (dNTP) (10 mmol−1 for each dNTP), 0.50 μl of each forward and reverse primer (10 mmol−1; Integrated DNA Technologies, Coralville, IA), and 0.10 μl Taq polymerase [5 U μl−1; GoTaq® (Promega Corp., Madison, WI)]. PCR conditions were as follows: 95 °C for 5 min followed by 35 cycles of 95 °C for 30 s, 60 °C for 45 s, and 72 °C for 1.5 min, followed by a final extension at 72 °C for 7 min.
Visualization of PCR results for candidate assays was completed with fragment separation on denaturing gels containing 4% acrylamide and 7.5 M urea in 1× TBE buffer run on a 50-cm Sequi-Gen GT system (BioRad, Hercules, CA) for ∼2 h at 85 W. Gels were stained with Promega Silver Sequence™ Staining System (Promega Corp., Madison, WI) as per manufacturer’s instructions. Amplified fragments from each primer pair for 20 individuals representing the most common SNP haplotypes were compared, and primer pairs producing fragments matching SNP haplotype patterns were shortlisted for subsequent use on a high-throughput platform to check suitability for routine breeding use. Visualization of shortlisted assay outcomes and the chosen candidate DNA test were completed with fragment separation on an ABI Prism 3730xl DNA Analyzer (Applied Biosystems by Life Technologies, Grand Island, NY) and size calling with GeneMarker® software (SoftGenetics, LLC, State College, PA). An internal size standard (Integrated DNA Technologies, Coralville, IA) and control samples with known allele sizes were used to ensure accurate size-calling.
Chosen assay
The Ppe-Rf-SSR DNA test (SSR#2) that was subsequently developed had a forward primer sequence of 5′ CGG ACA ATA TTC TTG TGA TGC 3′ (melting temperature 58.1 °C and GC content 43%) and a reverse primer sequence of 5′ GAT CTG AGC CAT TAC ACA GTG C 3′ (melting temperature 59.8 °C and GC content 50%).
DNA test genotypic effects
A chi-squared test of independence was performed to examine the relation between the Ppe-Rf-SSR alleles and genotypes and the proportion of blush coverage (P < 0.05). To represent the general effect of each allele and genotype, an associated phenotype was calculated using the same method as that described above for calculating SNP haplotype and diplotype general effects. The non-parametric Kruskal-Wallis test with post hoc Kruskalmc multiple comparisons test (Giraudoux 2015) (P < 0.05) was used to evaluate phenotypic differences between Ppe-Rf-SSR alleles and genotypes, with all analyses conducted using a modified R script (R Core Team 2013).
Confirmation
To confirm the genotypic effects of the DNA test developed in the target breeding program, the Confirmation set was screened with Ppe-Rf-SSR. DNA extraction was conducted for the “Confirmation set” (n = 443) according to the method of Edge-Garza et al. (2014).
Visualization of the PCR outcomes and calculation of DNA test genotypic effects were conducted as described above for the conversion process.
Results
Association
Phenotypic variation
Individuals with extreme phenotypes in the Association set (n = 622) were rare: only 4% of germplasm (n = 25) had no blush (score of 0) and only 2% (n = 12) had complete blush coverage (score of 5). Germplasm with no blush consisted of two selections and 23 unselected seedlings from the CA breeding program and germplasm with complete blush coverage were all unselected seedlings from either the AR or TX breeding programs. Frequencies of intermediately blushed individuals (scores of 1 to 4) were higher (1 = 28%, 2 = 16%, 3 = 30%, 4 = 20%). The mode of the Association set was 3, indicating the germplasm set was slightly skewed toward higher proportions of blush coverage. The 1–3 scaling classified 32% of individuals as low (0–20% coverage), 46% as medium (21–80% coverage), and 21% as high (81–100% coverage).
SNP haplotypes
Among the 622 individuals of the Association set, 11 unique haplotypes were observed, with frequencies ranging from 2% (for H1 and H8) to 37% (for H7) (Table 1; Online Resource 1). In germplasm having undergone historical selection for commercial performance (cultivars and selections), haplotype frequencies ranged from 1% (for H2) to 24% (for H7). In unselected germplasm (seedlings), haplotype frequencies ranged from 2% (for H1) to 39% (for H7). All haplotypes, including the rarest (H2), were derived from more than one pedigree-traceable cultivar or selection (Online Resource 4).
Haplotypes H1, H2, H3, H5, and H6 were associated with low blush (mode of 1 on 1–3 scale), haplotypes H4, H7, H8, H9, H10, and H11 were associated with medium blush (mode of 2 on 1–3 scale), and no haplotypes were associated with high blush (mode of 3 on 1–3 scale) (Table 1). The above low blush haplotypes, when grouped, had a mean blush level of 1.3 ± 0.8 and mode of 1 while the medium blush haplotype group had a mean blush level of 2.9 ± 1.1 and mode of 3. The low-blush haplotypes H1, H3, and H5 had a significantly lower blush coverage than each of the medium-blush haplotypes; however, haplotypes H2 and H6 were not significantly different from H4, possibly owing to H4 being paired with an allele associated with low blush in approximately 70% of individuals. The medium-blush haplotypes H7, H9, H10, and H11 had a significantly higher blush coverage than each of the low-blush haplotypes. Although haplotypes H8 and H10 were associated with medium blush, they were not significantly different from the assumption of allele-proportion of blush independence (chi-square; P < 0.05). However, haplotype H10 had a significantly higher proportion of blush coverage than each of the low-blush haplotypes and haplotype H8 had a significantly higher proportion of blush coverage than each of the low-blush haplotypes except for H2 and H6. For H10, approximately half of the sources [“Carson,” “China Pearl,” “Greensboro,” and a single breeding program selection (D62-193)] were associated with low blush, while the other sources, which included several University of Arkansas and Texas A&M selections, were associated with medium blush. The only other haplotype to be inconsistently associated with a simplified blush phenotype was H7, where all but a single source, a P. persica × Prunus davidiana hybrid (P × D), were associated with medium blush, whereas P × D was associated with low blush. However, due to the low frequency of the P × D source of the H7 haplotype, this haplotype had a significantly higher blush coverage than each of the low blush haplotypes.
SNP diplotypes
In total, 38 of 66 possible diplotypes were observed in the Association set (Online Resource 5). Diplotype frequencies ranged from <1% (for 13 diplotypes) to 21% (for H7 | H7), with a mean of 2.6%. For all common L | L diplotypes (which consisted of two low-blush haplotypes, H1, H2, H3, H5, and H6 on the 1–3 scale), represented by a minimum of six individuals, their association with a low proportion of blush coverage matched the expected association based on the haplotype combination. All remaining diplotypes (L | M and M | M), except for H3 | H10 and H5 | H10, were associated with a medium blush level. When averaged, the L | M haplotype combinations had a higher frequency of individuals scored as 1 than as 3 and the M | M haplotype combinations had a higher frequency of individuals scored as 3 than as 1 (Online Resource 5). The three diplotype groups, L | L, L | M, and M | M, each had significantly different levels of blush coverage. As 95% of H3 | H10 and 90% of H5 | H10 individuals’ H10 haplotypes came from the low-blush sources of the haplotype (‘China Pearl’ and D62-193) instead of the medium-blush sources, the H3 | H10 and H5 | H10 diplotypes were associated with a low proportion of blush coverage. The only L | M diplotypes significantly different from M | M diplotypes were those where the M haplotype was H4. In all other cases, only L | L and M | M diplotypes were significantly different. When all diplotypes were grouped using the 1–3 scale as L | L, L | M, or M | M and H10 was considered L or M depending on haplotype source, M | M had an average blush level of 3.3 ± 0.8 and mode of 3, L | M had an average of 2.2 ± 1.0 and mode of 2, and L | L had an average of 1.1 ± 0.6 and a mode of 1 based on the original 0–5 rating scale.
Conversion
Candidate assays
A total of 24 di-nucleotide microsatellite motifs, each with at least 10 repeats, and 15 tri-nucleotide motifs and nine tetra-nucleotide motifs, each with at least four repeats, were detected in the ±100 kb flanking sequence around peach MYB10.1. The di-nucleotide microsatellites contained up to 26 repeats, whereas the tri- and tetra-nucleotide microsatellites each contained only four repeats. Eleven di-nucleotide microsatellites were subsequently chosen as targets for primer design (Online Resource 3). After screening of the 20 of individuals representing the most common SNP haplotypes, six of these 11 candidate assays for R f (#2, #3, #4, #6, #7, and #9; Online Resource 3) were deemed polymorphic. Of these, only SSR#2, located 36 kb upstream of peach MYB10.2 and 76 kb upstream of peach MYB10.1, was able to clearly differentiate the two haplotype groups of medium and low proportion of blush coverage based on the 1–3 scale. This SSR was chosen for additional evaluation and subsequently named Ppe-Rf-SSR.
Ppe-Rf-SSR alleles and effects
Ppe-Rf-SSR (SSR#2) was observed to have four unique SSR alleles (395, 397, 399, and 401 bp in length; Fig. 4) within the 200 Conversion set individuals screened (Table 1; Online Resource 2). A fifth allele, 403, was observed only in one University of Arkansas breeding program selection and 31 seedlings derived from this selection in the Confirmation set. Among the 200 individuals of the Conversion set, allele frequency in germplasm having undergone historical selection for commercial performance (n = 70) ranged from 7% (for 397) to 56% (for 395) (Online Resource 6). In unselected germplasm, the trend was similar, with 10% of alleles being 397 and 52% being 395.

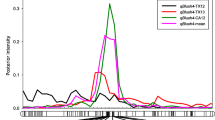
High-throughput ABI output of Ppe-Rf-SSR genotypes: a 395 | 395, b 397 | 397, c 399 | 399, d 401 | 401, e 395 | 397, f 395 | 399, g 395 | 401, h 397 | 399, i 397 | 401, j 399 | 401. Numbers (e.g., 395) represent base pairs with the height of each peak relative to its fluorescence intensity
The majority, four of five (H2, H3, H5, and H6), of the 5-SNP haplotypes associated with low blush coverage on the 1–3 simplified phenotype scale were represented by the 399 or 401 Ppe-Rf-SSR alleles, while most, four of the five (H7, H9, H10, and H11), haplotypes associated with medium blush coverage on the same scale were represented by the 395 or 397 allele in the Conversion set (Table 1). All sources of the H1 allele were represented by the 399 or 401 allele except for P × D which was represented by the 395 allele. All individuals with the H10 and H7 haplotypes including those from the low-blush sources had the 395 or 397 allele. Both the H4 and H8 alleles, which were associated with medium blush on the 1–3 scale, were represented by the 399 allele. All sources of the H1, H4, and H9 haplotypes were able to be investigated for association with Ppe-Rf-SSR alleles. One source of the H2, H3, and H8 haplotypes, two sources of the H10 and H11 haplotypes, three sources of the H5 and H6 haplotypes, and five sources of the H7 haplotype were not robustly tested because of access to too few individuals that carried these haplotypes. Although these sources of H2, H3, H5, and H6 were not tested, the other sources of these haplotypes were observed to be associated with the 399 or 401 allele. Similarly, the untested sources of H7, H10, and H11 were likely associated with the 395 or 397 as all other sources of these haplotypes were associated with these two alleles (Table 1).
Among the 200 individuals of the Conversion set, alleles 395 and 397 were associated with a medium proportion of blush coverage (mode of 2 on 1–3 scale) and alleles 399 and 401 were associated with a low proportion of blush coverage (mode of 1 on 1–3 scale) (Fig. 5a; Online Resource 6). The 395 allele had a significantly higher proportion of blush coverage than the 397 allele, and both 395 and 397 had a significantly higher proportion of blush coverage than the 399 and 401 alleles, which were not significantly different. When 395 and 397 were grouped as medium blush and 399 and 401 were grouped as low blush, the two allele groups were significantly different, with the mean blush level of the medium group being 3.1 ± 1.1 with mode of 3 and the mean blush level of the low group being 1.3 ± 0.8 with a mode of 1 (Online Resource 6). Although the 397 allele was observed to have a medium proportion of blush coverage (on 1–3 scale), it was not significantly different from the assumption of allele-proportion of blush independence (chi-square; P < 0.05) when considering all individual carrying it. When the source of the 397 allele was considered, two of the sources, “Carson” and D62-193, were associated with a low level of blush coverage, while all other sources were associated with a medium blush coverage (on 1–3 scale). The individuals with the “Carson” or D62-193 source of 397 were observed to have the form of the H10 allele associated with low blush. When these individuals were removed from the analysis, the frequency of individuals carrying the 397 allele and scored as 1 (on 1–3 scale) decreased from 34 to 0%, and 397 no longer had a significantly lower blush coverage than 395. The only other allele to be inconsistently associated with a simplified blush phenotype was 395, where all but a single source, P × D, were associated with a medium level of blush coverage. The individuals with the P × D source of 395 all carried the H1 allele that was associated with the low-blush phenotype. The medium-blush allele group, on average and with the inconsistent sources accounted for, had a blush rating, on the 0–5 scale, 2.5 times greater than the average of the low-blush allele group (Online Resource 6).

Proportion of individuals rated as low (0–1), medium (2–3), or high (4–5) blush coverage, and 0–5 scale mean ± standard deviation for each Ppe-Rf-SSR allele (a and c) and genotype (b and d) within the Conversion (a and b; n = 200) and Confirmation (c and d; n = 443) sets. Individuals recorded with a 395 or 397 allele but with the low-blush source of the allele were not removed to mimic a situation where haplotype and pedigree information were unknown
When considering the 1–3 scale, the phenotypic effects of Ppe-Rf-SSR genotypes matched those expected based on additivity of allele effects, although the 395 | 397 and 397 | 399 genotypes had a higher than expected frequency of individuals scored as low blush (Fig. 5b). Of the individuals scored as 395 | 397, 25% had one or two alleles from the P × D source of 395 or the “Carson” and D62-193 source of 397, whereas 92% of individuals scored as 397 | 399 had 397 from either “Carson” or D62-193. When these sources were considered as different alleles, the mean of the 395 | 397 group increased from 2.5 to 3.1 on the 0–5 scale and the single 397 | 399 individual was rated as a 4 on the 0–5 scale in comparison to the 11 397b | 399 individuals that had a mean of 1.2, much more aligned with the other L | L allele combination groups. With this change, the mean of the generalized M | M group increased by 0.15, L | M increased by 0.34, and L | L decreased by 0.02, and the standard deviation of each group decreased (Online Resource 7). With and without the discrepant forms of 395 and 397 removed, the L | L, L | M, and M | M genotype groups had significantly different proportionate levels of blush coverage.
Confirmation
From the screening of the Confirmation set (n = 443) with Ppe-Rf-SSR, allele frequency ranged from 4% (for 403) to 75% (for 395) (Online Resource 8). The 403 allele was not detected in the Conversion set and was only observed in a single University of Arkansas peach selection and 32 seedlings derived from this selection. More than half of all seedlings had the 395 | 395 genotype which was associated with a high blush coverage having a mean of 3.7 and a mode of 4 (Fig. 5d). No seedlings had genotypes of 399 | 399, 399 | 403, or 403 | 403. The two alleles associated with a medium degree of blush coverage in the Conversion set, 395 and 397, were also associated with a medium (1–3 scale) level in the Confirmation set (Fig. 5c). The 399 allele, associated with a low degree of blush coverage in the Conversion set, was associated with a medium (1–3 scale) level in the Confirmation set, but 399 was always paired with either 395 or 397 in the Confirmation set, thereby skewing the effect of this allele toward a higher level of blush coverage (Fig. 5c; Online Resource 8). The 403 allele had a similar effect to the 399 allele, and although it was associated with a medium (1–3 scale) proportion of blush coverage, it was always paired with the 395 allele. Both the 399 and 403 alleles increased the frequency of individuals scored as low blush coverage when paired with 395 or 397, in contrast to 395 | 395, 395 | 397, and 397 | 397 (Fig. 5d; Online Resource 8). Using the 1–3 scale, the 395 and 397 alleles had a significantly higher blush coverage than the 399 and 403 alleles. When the alleles associated with medium blush (395 with 397) were grouped (M) and those associated with a lower degree of blush were grouped (L) (399 with 403), the medium-blush group had a mean blush level of 3.4 ± 0.9, 0.7 higher than the low-blush group (2.7 ± 0.8), with the groups being significantly different. On average, the M | M genotypic group had a mean blush level of 3.6 ± 0.8, 0.9 higher than the L | M group (2.7 ± 0.8) (Fig. 5d; Online Resource 8), with the M | M group being significantly higher than L | M on all phenotypic scales.
Discussion
A trait-predictive, locus-specific DNA test for peach fruit skin blush coverage, Ppe-Rf-SSR, was successfully developed by converting the haplotypic information of five SNPs at the R f locus into a single PCR-based assay. Ppe-Rf-SSR was able to differentiate alleles associated with low, medium, and high blush coverage in a diverse set of individuals representing North American breeding germplasm and was confirmed to differentiate breeding-relevant blush levels in University of Arkansas peach breeding germplasm. The quantitative nature of peach skin blush was confirmed and an additive genetic effect detected, as in most cases individuals carrying two high-blush alleles had a significantly higher proportion of blush coverage than those carrying only a single high-blush allele. Three particular germplasm sources, P × D, “Carson,” and D62–193, introduced ambiguity into the DNA test’s predictions. However, when parentage from these three sources was considered, Ppe-Rf-SSR was completely predictive of peach fruit skin blush coverage determined by the R f locus.
The association of SNP haplotypes and SSR alleles with low, medium, and high levels of blush coverage observed within this study validated the utility in peach breeding germplasm of the major effect fruit skin color QTL that Frett et al. (2014) previously identified in this region. Many sources for each SNP haplotype were observed (3–33); however, many of the sources of the same haplotype likely trace to identical founding cultivars, although this hypothesis cannot easily be tested because of limited pedigree records that are often inaccurate owing to pollen contamination, labeling, and record-keeping errors (Doerksen and Herbinger 2010; Munoz et al. 2014). Because the phenotypic variance observed for proportion of blush coverage was greater than that explained by the R f locus alone, other loci associated with fruit skin blush coverage might be responsible for the within-group differences observed within the germplasm studied here. Such loci could be those proposed by Cantín et al. (2010), Eduardo et al. (2011), and Frett et al. (2014), with a major locus reported on chromosome 4 and several minor loci reported on chromosomes 6 and 7. This hypothesis could be tested with experimental controls to minimize environmental effects (e.g., differential exposure of developing fruit to sunlight) on blush development.
The 5-SNP haplotype and Ppe-Rf-SSR allele distributions within representative germplasm of four breeding programs used in this study reflects the target market of each program. Within the University of Arkansas, Clemson University, and Texas A&M University germplasm, Ppe-Rf-SSR alleles associated with a medium and high blush represented approximately 93% of alleles detected, consistent with their fresh market target. In contrast, 75% of alleles within the University of California—Davis program’s germplasm were low-blush alleles. This breeding program is the only US program focused on developing cultivars for the processing peach market, for which cultivars with very little blush coverage are desired. The large proportion of market-targeting alleles in each breeding program is consistent with directional selection for favorable skin color alleles and/or minimal use of new parents with unfavorable alleles.
Ppe-Rf-SSR can determine an individual’s zygosity at the R f locus, differentiating those with the genetic potential to produce fruit with a low or high proportion of blush coverage. By selecting for homozygous high blush or heterozygous genotypes, high or medium blush coverage, respectively, could be targeted in breeding. With the CAPS marker developed by Frett et al. (2014), only three genotypes are possible to be distinguished, in contrast to the nine genotypes for Ppe-Rf-SSR. While predictiveness of the CAPS marker was confirmed in 69 peach cultivars (Gasic et al. 2014), Ppe-Rf-SSR was confirmed in 200 individuals representative of the North American peach breeding germplasm as well as more than 400 seedlings from 18 cross-combinations of 28 important parents of the University of Arkansas peach breeding program. Although CAPS marker alleles can be separated using regular agarose electrophoresis (Gasic et al. 2014), they cannot be readily multiplexed, reducing their utility for routine application in breeding programs. Therefore, compared to the previously available CAPS marker, the new DNA test, Ppe-Rf-SSR, is a robust genetic screening tool for peach skin blush coverage that is more technically amenable to routine breeding use and provides greater phenotypic resolution. Additionally, by establishing the SNP-based haplotypes and their association with fruit skin blush coverage across a diverse germplasm set, the use of the SNP set spanning the R f locus could have broad applicability in peach breeding programs where SNP-based genotyping is preferred over PCR-based SSR assays.
The single source of the 395 bp Ppe-Rf-SSR allele that was associated with a low degree of blush coverage instead of a medium degree of blush coverage, P × D, was unique among the germplasm evaluated as it is an interspecific hybrid of commercial peach (P. persica) and its close wild relative P. davidiana. Using an advanced backcross breeding population derived from a cross of P. persica and P. davidiana, Quilot et al. (2004) found a QTL for red skin coloration on linkage group 5 that, in common peach breeding germplasm, is likely fixed for the red skin allele and therefore was not detected in other QTL mapping efforts that only used P. persica parents (Cantín et al. 2010; Eduardo et al. 2010; Frett et al. 2014). Therefore, the finding that the 395 allele of P × D and its offspring was apparently associated with low blush, rather than medium blush as in the majority of germplasm studied, could be due to the effect of the linkage group 5 locus segregating within this interspecific material. In addition, the “highlighter” attribute, representing a complete suppression of blush that was discovered in South American “criollo” and Italian germplasm, might already be present in processing peach germplasm (Beckman et al. 2005). Two of the three sources that did not match the expected medium-blush phenotype based on carrying the medium-blush allele were processing-type peaches with low blush levels that could be a result of an epistatic interaction between the “highlighter” and R f loci. The hypothesis that such a second locus modifies the R f locus effect is similar to that proposed by Sandefur et al. (2016) for sweet cherry fruit color and Decroocq et al. (2014) for apricot Plum Pox Virus resistance. A caveat regarding the R f locus is that if individuals related to P × D, “Carson,” or other such germplasm sources are used in the breeding program, pedigree information would be required to ensure appropriate interpretation of genotypic results. Continued investigation of Ppe-Rf-SSR genotype-phenotype associations on more diverse germplasm will help to ascertain the scope of the predictive power of this test across peach breeding germplasm.
The three germplasm sources with Ppe-Rf-SSR genotypes that did not have the expected phenotype compared to all other allele sources highlighted the need to confirm any DNA test’s prediction accuracy in a target breeding program’s germplasm prior to routine deployment. Independent confirmation of the predictive power of Ppe-Rf-SSR was successfully conducted here for the University of Arkansas peach breeding program, where the homozygous medium-blush allele group of seedlings had a higher proportion of blush coverage than the heterozygous seedlings, with both a low- and medium-blush allele. The observations of a low proportion (15%) of low-blush alleles and no parental combinations used that could have resulted in homozygous low-blush offspring provides insight into how Ppe-Rf-SSR could be effectively deployed in the Arkansas program. With parent selection using R f locus information, all offspring produced could be homozygous for medium-blush alleles, thereby eliminating the need to screen seedlings with this DNA test to increase the overall cultivar potential of seedlings produced.
The DNA test developed herein, Ppe-Rf-SSR, is based on a single microsatellite marker that uses a standard PCR protocol and is expected to be effective with low quality and quantity DNA extracts because of the simplicity of the PCR reaction. In addition, Ppe-Rf-SSR can be readily multiplexed with other DNA tests and its amplicons can be visualized on a range of genotyping platforms. This test has many potential uses, including determining if any medium-blush parent has the genetic potential to produce low-blush offspring (i.e., detecting R f heterozygotes), predicting genetic potential for blush coverage in juvenile seedlings to facilitate early selection/culling, and enabling efficient sorting of seedlings into low-blush processing or high-blush fresh market groups to simplify within-group evaluation. Although Ppe-Rf-SSR can be directly applied in breeding programs using germplasm similar to that evaluated in this report, it is recommended that the test’s prediction accuracy be confirmed in a target breeding program’s germplasm prior to routine deployment. The University of Arkansas Peach Breeding Program has already adopted Ppe-Rf-SSR into routine breeding operations. In early spring 2015, ∼350 peach seedlings were screened with this DNA test combined in multiplexed PCR reactions with DNA tests for other fruit quality and disease resistance traits. The successful adoption of Ppe-Rf-SSR demonstrates the breeding utility of this trait-predictive DNA test and the potential impact of such DNA-based diagnostic technologies in fruit breeding.
References
Arús P, Yamamoto T, Dirlewanger E, Abbott AG (2006) Synteny in the Rosaceae. In: Janick J (ed) Plant Breeding Reviews Volume 25. Wiley, New York, pp 175–211
Bassi D, Monet R (2008) Botany and taxonomy. In: Layne DR, Bassi D (eds) The peach, botany, production and uses. CAB International Press, Wallingford, UK, pp 1–36
Beckman TG, Sherman WB (2003) Probable qualitative inheritance of full red skin color in peach. Hortscience 38:1184–1185
Beckman TG, Alcazar JR, Sherman WB, Werner DJ (2005) Evidence for qualitative suppression of red skin color in peach. Hortscience 40:523–524
Bink MCAM, Boer MP, ter Braak CJF, Jansen J, Voorrips RE, van de Weg W (2008) Bayesian analysis of complex traits in pedigreed plant populations. Euphytica 161:85–96
Blake MA (1932) The J.H. Hale as a parent in peach crosses. Proc Amer Soc Hort Sci 29:131–136
Blake MA (1940) Some results of crosses of early ripening varieties of peaches. Proc Amer Soc Hort Sci 37:232–241
Bliss F (2010) Marker-assisted breeding in horticultural crops. Acta Hort 859:339–350
Byrne DH (2007) Molecular marker use in perennial plant breeding. Acta Hort 751:163–167
Byrne D, Bassols M, Bassi D, Piagnani M, Gasic K, Reighard G, Moreno M, Pérez S (2012) Peach. In: Badenes M, Byrne D (eds) Fruit Breeding. Springer, New York, pp 505–570
Cantín CM, Crisosto CH, Ogundiwin EA, Gradziel T, Torrents J, Moreno MA, Gogorcena Y (2010) Chilling injury susceptibility in an intraspecific peach [Prunus persica (L.) Bastch] progeny. Postharvest Biol Technol 58:79–87
Chagné D, Carlisle CM, Blong C, Volz RK, Whitworth CJ, Oraguzie NC, Crowhurst RN, Allan AC, Espley RV, Hellens RP, Gardiner SE (2007) Mapping a candidate gene (MdMYB10) for red flesh and foliage colour in apple. BMC Genomics 8:212
Collard BCY, Mackill DJ (2008) Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Phil Trans R Soc Lond B Biol Sci 363:557–572
Crisosto CH, Costa G (2008) Preharvest factors affecting peach quality. In: Layne DR, Bassi D (eds) The peach, botany, production and uses. CAB International Press, Wallingford, UK, pp 536–549
de Souza VAB, Byrne DH, Taylor JF (1998) Heritability, genetic and phenotypic correlations, and predicted selection response quantitative traits in peach. II. An analysis of several fruit traits. J Amer Soc Hort Sci 123:604–611
Decroocq S, Chague A, Lambert P, Roch G, Audergon JM, Geuna F, Chiozzotto R, Bassi D, Dondini L, Tartarini S, Salava J, Krska B, Palmisano F, Karayiannis I, Decroocq V (2014) Selecting with markers linked to the PPVres major QTL is not sufficient to predict resistance to Plum Pox Virus (PPV) in apricot. Tree Genet Genomes 10:1161–1170
Doerksen TK, Herbinger CM (2010) Impact of reconstructed pedigrees on progeny-test breeding values in red spruce. Tree Genet Genomes 6:591–600
Edge-Garza D, Rowland T, Haendiges S, Peace C (2014) A high-throughput and cost-efficient DNA extraction protocol for the tree fruit crops apple, sweet cherry, and peach relying on silica beads during tissue sampling. Mol Breed 34:2225–2228
Edge-Garza D, Luby J, Peace C (2015) Decision support for cost-efficient and logistically feasible markerassisted seedling selection in fruit breeding. Mol Breed 35:223
Eduardo I, Pacheco I, Chietera G, Bassi D, Pozzi C, Vecchietti A, Rossini L (2011) QTL analysis of fruit quality traits in two peach intraspecific populations and importance of maturity date pleiotropic effect. Tree Genet Genomes 7:323–335
Eduardo I, López-Girona E, Batlle I, Reig F, Iglesias I, Howad W, Arús P, Aranzana MJ (2014) Development of diagnostic markers for selection of the subacid trait in peach. Tree Genet Genomes 10:1695–1709
Eduardo I, Picañol R, Rojas E, Batlle I, Howad W, Aranzana MH, Arús P (2015) Mapping of a major gene for the slow ripening character in peach: co-location with the maturity date gene and development of a candidate gene-based diagnostic marker for its selection. Euphytica 205:627–636
Espley RV, Hellens RP, Putterill J, Stevenson DE, Kutty-Amma S, Allan AC (2007) Red colouration in apple fruit is due to the activity of the MYB transcription factor, MdMYB10. Plant J 49:414–427
Falchi R, Vendramin E, Zanon L, Scalabrin S, Cipriani G, Verde I, Vizzotto G, Morgante M (2013) Three distinct mutational mechanisms acting on a single gene underpin the origin of yellow flesh in peach. Plant J 76:175–187
Food and Agriculture Organization of the United Nations (2015) FAOSTAT. FAOSTAT (Database). http://faostat3.fao.org/browse/Q/QC/E. Accessed 5 Nov 2015
Frett TJ, Reighard GL, Okie WR, Gasic K (2014) Mapping quantitative trait loci association with blush in peach [Prunus persica (L.) Batsch]. Tree Genet Genomes 10:367–381
Frett TJ, Gasic K, Clark JR, Byrne D, Gradziel T, Crisosto C (2012) Standardized phenotyping for fruit quality in peach [Prunus persica (L.) Batsch]. J Amer Pom Soc 66:214–219
Gasic K, Quick R, Abdelghafar A, Frett TJ, Rauh B, Reighard G (2014) Marker assisted breeding for red skin coloration in peach. XXII Plant and Animal Genome conference, January 11–15, San Diego, p 435
Giraudoux P (2015) Pgirmess: data analysis in ecology. R package version 1.6.2. http://CRAN.R-project.org/package=pgirmess. Accessed 28 Sept 2015
Gradziel TM, McCaa JP (2008) Processing peach cultivar development. In: Layne DR, Bassi D (eds) The peach, botany, production and uses. CAB International Press, Wallingford, UK, pp 175–192
Hansche PE (1986) Heritability of fruit quality traits in peach and nectarine breeding stocks dwarfed by dw gene. Hortscience 21:1193–1195
Iezzoni A, Weebadde C, Luby J, Yue CY, van de Weg E, Fazio G, Main D, Peace CP, Bassil N, McFerson J (2010) RosBREED: enabling marker-assisted breeding in Rosaceae. Acta Hort 859:389–394
Janick J (2005) The origins of fruits, fruit growing, and fruit breeding. In: Janick J (ed) Plant breeding reviews, volume 25. Wiley & Sons, Oxford, UK, pp 255–320
Jordan JL, Shewfelt RL, Prussia SE (1986) The value of peach quality characteristics in the postharvest system. Acta Hort 203:175–182
Jung S, Ficklin S, Lee T, Cheng CH, Blenda A, Zheng P, Yu J, Bombarely A, Cho L, Ru S, Evans K, Peace C, Abbott A, Mueller L, Olmstead MA, Main D (2014) The genome database for Rosaceae (GDR): year 10 update. Nucl Acids Res 42:D1237–D1244
Lin-Wang KL, Bolitho K, Garryn K, Kortstee A, Karunairetnam S, McGhie TK, Espley RV, Hellens RP, Allan AC (2010) An R2R3 MYB transcription factor associated with regulation of the anthocyanin biosynthetic pathway in Rosaceae. BMC Plant Biol 10:50
Luby J, Shaw D (2001) Does marker-assisted selection make dollars and sense in a fruit breeding program? Hortscience 36:872–879
Luchsinger L, Ortin P, Reginato G, Infante R (2002) Influence of canopy fruit position on the maturity and quality of Angelus peaches. Acta Hort 592:515–521
Munoz PR, Resende MFR, Huber DA, Quesada T, Resende MDV, Neale DB, Wegrzyn JL, Kirst M, Peter GF (2014) Genomic relationship matrix for correcting pedigree errors in breeding populations: impact on genetic parameters and genomic selection accuracy. Crop Sci 53:1115–1123
Okie WR, Bacon T, Bassi D (2008) Fresh-market cultivar development. In: Layne DR, Bassi D (eds) The peach, botany, production and uses. CAB International Press, Wallingford, UK, pp 139–174
Olmstead MA, Gilbert JL, Colquhoun TA, Clark DG, Kluson R, Moskowitz HR (2015) In pursuit of the perfect peach: consumer-assisted selection of peach fruit traits. Hortscience 50:1202–1212
Parker D, Zilberman D, Moulton K (1991) How quality relates to price in California fresh peaches. Calif Agric 45:14–16
Peace C, Norelli J (2009) Genomics approaches to crop improvement in the Rosaceae. In: Folta KM, Gardiner SE (eds) Genetics and genomics of Rosaceae. Springer, NY, pp 19–54
Peace CP, Luby JJ, van de Weg WE, Bink MCAM, Iezzoni AF (2014) A strategy for developing representative germplasm sets for systematic QTL validation, demonstrated for apple, peach, and sweet cherry. Tree Genet Genomes 10:1679–1694
Picañol R, Eduardo I, Aranzana MJ, Howad W, Batlle I, Iglesias I, Alonso JM, Arús P (2013) Combinging linkage and association mapping to search for markers linked to the flat fruit character in peach. Euphytica 190:279–288
Quilot B, Wu BH, Kervella J, Génard M, Foulongne M, Moreau K (2004) QTL analysis of quality traits in an advanced backcross between Prunus persica cultivars and the wild relative species P. davidiana. Theor Appl Genet 109:884–897
R Core Team (2013) R: a language and environment for statistical computing. R Foundation for Statistical Computing. Vienna, Austria. http://www.R-project.org/. Accessed 28 Sept 2015
Rahim MA, Busatto N, Trainotti L (2014) Regulation of anthocyanin biosynthesis in peach fruits. Planta 240:913–929
Ramkumar G, Prahalada GD, Hechanova SL, Vinarao R, Jena KK (2015) Development and validation of SNP-based functional codominant markers for two major disease resistance genes in rice (O. sativa L.). Mol Breed 35:129
Ravaglia D, Espley R, Hendry-Kirk RA, Andreotti C, Ziosi V, Hellens R, Cost G, Allan A (2013) Transcriptional regulation of flavonoid biosynthesis in nectarine (Prunus persica) by a set of R2R3 MYB transcription factors. BMC Plant Biol 13:68
Ru S, Main D, Peace C (2015) Current applications, challenges, and perspectives of marker-assisted seedling selection in Rosaceae tree fruit breeding. Tree Genet Genomes 11:1–12
Sandefur P, Oraguzie N, Peace C (2016) A DNA test for routine prediction in breeding of sweet cherry fruit color, Pav-Rf-SSR. Mol Breed 36:33
Sansavini S, Gamberini A, Bassi D (2006) Peach breeding, genetics and new cultivar trends. Acta Hort 713:23–48
Scorza R, Sherman WB (1996) Peaches. In: Janick J, Moore JN (eds) Fruit Breeding. Wiley, New York, pp 325–440
Sooriyapathirana SS, Khan A, Sebolt AM, Wang D, Bushakra JM, Lin-Wang K, Allan AC, Gardiner SE, Chagné D, Iezzoni AF (2010) QTL analysis and candidate gene mapping for skin and flesh color in sweet cherry fruit (Prunus avium L.). Tree Genet Genomes 6:821–832
Stegmeir T, Schuster M, Sebolt A, Rosyara U, Sundin G, Iezzoni A (2014) Cherry leaf spot resistance in cherry (Prunus) is associated with a quantitative trait locus on linkage group 4 inherited from P. canescens. Mol Breed 34:927–935
Takos AM, Jaffe FW, Jacob SR, Bogs J, Robinson SP, Walker AR (2006) Light-induced expression of a MYB gene regulates anthocyanin biosynthesis in red apples. Plant Physiol 142:1216–1232
United State Department of Agriculture (2004) United States standards for grades of peaches. http://www.agmrc.org/media/cms/peaches_4EFAEFAA6947E.pdf. Accessed 21 Oct 2015
Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG (2012) Primer3—new capabilities and interfaces. Nucleic Acids Res 40:e115
Vendramin E, Pea G, Dondini L, Pacheco I, Dettori MT, Gazza L, Scalabrin S, Strozzi F, Tartarini S, Bassi D, Verde I, Rossini L (2014) A unique mutation in a MYB gene cosegregates with the nectarine phenotype in peach. PLoS One 9(3):e90574
Verde I, Quarta R, Cedrola C, Dettori MT (2002) QTL analysis of agronomic traits in a BC1 peach population. Acta Hort 592:291–295
Verde I, Bassil N, Scalabrin S, Gilmore B, Lawley CT, GAsic K, Micheletti D, Rosyara UR, Cattonaro F, Vendramin E, Main D, Aramini V, Blas AL, Mockler TC, Bryant DW, Wilhelm L, Troggio M, Sosinski B, Aranzana MJ, Arús P, Iezzoni A, Morgante M, Peace C (2012) Development and evaluation of a 9 k SNP array for peach by internationally coordinated SNP detection and validation in breeding germplasm. PLoS One 7(4):e35668
Verde I, Abbott AG, Scalabrin S, Jung S, Shu S, Marroni F, Zhebentyayeva T, Dettori MT, Grimwood J, Cattonaro F, Zuccolo A, Rossini L, Jenkins J, Vendramin E, Meisel LA, Decroocq V, Sosinski B, Prochnik S, Mitros T, Policriti A, Cipriani G, Dondini L, Ficklin S, Goodstein DM, Xuan P, Del Fabbro C, Aramini V, Copetti D, Gonzalez S, Horner DS, Ralchi R, Lucas S, Mica E, Maldonado J, Lazzari B, Bielenberg D, Pirona R, Miculan M, Barakat A, Testolin R, Stella A, Tartarini S, Tonutti P, Arús P, Orellana A, Wells C, Main D, Vizzotto G, Silva H, Salamini F, Schmutz J, Morgante M, Rokhsar DS (2013) The high-quality draft of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nature Genet 45:487–494
Weinberger JH (1944) Characteristics of the progeny of certain peach varieties. Proc Amer Soc Hort Sci 45:233–238
Yamamoto T, Shimada T, Imai T, Yaegaki H, Haji T, Matsuta N, Yamaguchi M, Hayashi T (2001) Characterization of morphological traits based on a genetic linkage map in peach. Breed Sci 51:271–278
Yamamoto T, Yamaguchi M, Hayashi T (2005) An integrated genetic linkage map of peach by SSR, STS, AFLP, and RAPD. J Jap Soc Hort Sci 74:204–213
You FM, Huo H, Gu YQ, Luo MC, Ma Y, Hane D, Lazo GR, Dvorak J, Anderson OD (2008) BatchPrimer3: a high throughput web application for PCR and sequencing primer design. BMC Bioinformatics 9:253
Yuan K, Wang C, Wang J, Xin L, Zhou G, Li L, Shen G (2014) Analysis of the MdMYB1 gene sequence and development of new molecular markers related to apple skin color and fruit-bearing traits. Mol Gen Genomics 289:1257–1265
Zhou Y, Zhou H, Lin-Wang K, Vimolmangkang S, Espley RV, Wang L, Allan AC, Han Y (2014) Transcriptome analysis and transient transformation suggest an ancient duplicated MYB transcription factor as a candidate gene for leaf red coloration in peach. BMC Plant Biol 14:338
Acknowledgments
The authors thank Alejandra Salgado, Jonathan Fresnedo, Tim Hartmann, David Byrne, Tom Gradziel, Andrew Jecmen, Terry Rowland, and Daniel Edge-Garza for their technical support and suggestions and thank all reviewers for their comments. This work was partially supported by the USDA National Institute of Food and Agriculture—Specialty Crop Research Initiative projects, “RosBREED: Enabling marker-assisted breeding in Rosaceae” (2009-51181-05808) and “RosBREED: Combining disease resistance with horticultural quality in new rosaceous cultivars” (2014-51181-22378) and with USDA Hatch funds provided to the Department of Horticulture, Washington State University.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Online Resource 1
(PDF 605 kb)
Online Resource 2
(PDF 211 kb)
Online Resource 3
(PDF 17 kb)
Online Resource 4
(PDF 112 kb)
Online Resource 5
(PDF 209 kb)
Online Resource 6
(PDF 141 kb)
Online Resource 7
(PDF 158 kb)
Online Resource 8
(PDF 136 kb)
Rights and permissions
About this article

Cite this article
Sandefur, P., Frett, T., Clark, J. et al. A DNA test for routine prediction in breeding of peach blush, Ppe-Rf-SSR. Mol Breeding 37, 11 (2017). https://doi.org/10.1007/s11032-016-0615-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-016-0615-3