
Abstract
Cherry leaf spot (CLS), caused by the fungal pathogen Blumeriella jaapii (Rehm) Arx (telomorph Phloeosporella padi [Lib.] Arx), is a major disease in all humid cherry-growing regions worldwide causing leaf yellowing and defoliation. The diploid Prunus species, P. canescens, had previously been identified as a source of CLS resistance. Therefore, the objective of this study was to identify quantitative trait loci (QTL) for CLS resistance derived from P. canescens in both diploid sweet cherry (P. avium) and tetraploid sour cherry (P. cerasus). Because of the simpler genetics of diploid cherry, the initial investigation was done with P. canescens-derived materials from crosses with sweet cherry, followed by validation using P. canescens-derived plant materials from sour cherry. A major QTL controlling P. canescens-derived CLS resistance, named CLSR_G4, was identified on linkage group 4 in sweet cherry and validated in sour cherry. All CLS-resistant individuals had one P. canescens-derived allele for CLSR_G4. A second QTL may be necessary for CLS resistance as one-fifth–one-third of the progeny individuals with the P. canescens-derived allele for CLSR_G4 were susceptible.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cherry leaf spot (CLS), caused by the fungal pathogen Blumeriella jaapii (Rehm) Arx (telomorph Phloeosporella padi [Lib.] Arx), is a major disease in all humid cherry-growing regions worldwide. Infection starts from the deposition of the sexual ascospores on the newly expanding leaves in early spring. From these spores, small purplish spots appear from which the fungus grows and eventually produces asexual conidia, which are responsible for secondary infections that occur throughout the summer under favorable wet conditions. While sour cherries are generally more susceptible to infection of CLS and the resulting leaf yellowing and defoliation, sweet cherries are also affected. When not controlled by fungicides, CLS can cause early leaf defoliation which can result in fruit that are poorly colored, soft, and low in soluble solids (Keitt et al. 1937). Premature defoliation can also weaken the tree and reduce winter hardiness, which can lead to flower bud freeze damage and even tree death (Howell and Stackhouse 1973). Studies have shown that fruit have priority over other sinks in Prunus (Richards 1986), so fewer leaves would produce fewer storage carbohydrates for the following year’s growth.
As many as seven to eight fungicide applications are needed each growing season on sour cherry to manage CLS, resulting in substantial costs to growers and a significant amount of pesticides released into the environment. There is also the threat that the fungicides currently being used to control CLS may be removed from the market, jeopardizing the sustainability of the sour cherry industry. The pathogen, B. jaapii, has also been found to develop resistance to site-specific sterol demethylation inhibitor fungicides (DMIs), which have been used extensively to control CLS on cherry (Proffer et al. 2006). This fungicide resistance (to a major class of CLS controlling fungicides) increases the need for new cultivars that have genetic resistance to CLS. Many studies have shown that there are no sour cherry cultivars that have complete resistance to CLS (Budan et al. 2005; Schuster and Tobutt 2004; Sjulin et al. 1989); however, in all of these studies, there were some individuals that displayed moderate resistance, indicating some polygenic resistance.
The susceptibility of all current sour and sweet cherry cultivars to CLS warranted the need to examine wild Prunus species to find resistance. One promising species to donate CLS resistance was shown to be the wild diploid species P. canescens (Schuster and Tobutt 2004; Wharton et al. 2003; Wharton and Iezzoni 2005). As a result, P. canescens has been used in breeding for CLS resistance in both sweet and sour cherry (Schuster et al. 2013) with diploid and tetraploid populations, respectively, segregating for disease resistance. With the development of RosBREED’s (www.rosbreed.org) Illumina Infinium® cherry SNP array (Peace et al. 2012), it was possible to locate introgressed chromosome regions from P. canescens due to high marker coverage across all eight linkage groups and attempt to identify QTL for this resistance.
The objective of this study was to determine the inheritance of P. canescens-derived resistance to CLS in sweet cherry and identify QTL controlling this resistance. Because of the simpler genetics of diploid cherry, the initial investigation was done with P. canescens-derived materials from crosses with sweet cherry, followed by validation using P. canescens-derived plant materials from sour cherry. In this study, we used the Bayesian approach for QTL discovery, implemented in FlexQTL™ software (Bink et al. 2002; Rosyara et al. 2013). This allowed us to follow genotypic regions from multiple populations through generations by including parents and grandparents in the analysis.
Materials and methods
Plant materials
This study included both diploid sweet and tetraploid sour cherry plant materials and their parents developed from crosses with the diploid P. canescens. Two sweet cherry populations were developed and evaluated at the Julius Kühn-Institut (JKI) in Dresden, Germany. The first population consisted of 34 individuals from the cross ‘Namati × F5-18-167. ‘Namati,’ a sweet cherry cultivar released from the breeding program of H. Mihatsch at Naumberg, is an open-pollinated seedling of the old variety ‘Bopparder Kracker.’ F5-18-167 is a CLS-resistant F1 clone from P. avium M53 × P. canescens, which was from a rootstock breeding program of B. Wolfram at Dresden-Pillnitz. The second sweet cherry population consisted of 38 individuals which were ‘Namati’ open-pollinated. Also included in the analyses were the parents ‘Namati,’ F5-18-167 and the P. canescens grandparent (Supplementary Table S1). Presence of P. canescens ancestry was tested by S-allele genotyping using the S-RNase primer pair PaConsI-F/PaConsI-R2 according to Sonneveld et al. (2006) with fragments visualized using a Beckman capillary sequencer (GenomeLab GeXP).
The inheritance of CLS resistance in sour cherry was tested using 15 progeny individuals derived from crosses between the resistant P. canescens-derived tetraploid parent 23-23-13 and the susceptible sour cherry cultivars ‘Montmorency’ and ‘Újfehértói Fürtös’ (‘UF’) (Supplementary Table S2; Supplementary Fig. S1; P. canescens was the great grandparent in these populations). The resulting sour cherry selections and P. canescens disease-resistant selection (US Plant Introduction Q39515) used in this study were maintained at Michigan State University’s Clarksville Research Center in Clarksville, MI, USA.
Disease rating
The sweet cherry populations and the parent genotypes were grown without any chemical plant protection in a test orchard in Dresden, Germany. In 2008–2011, CLS severity was evaluated after natural leaf spot inoculation according to the following rating scale (Fig. 1):

Images of cherry leaf spot disease rating reaction types. a Reaction type 1, b Reaction type 3, and c reaction type 5
Reaction type on the leaf (R-type):
-
1.
no symptoms and green leaf (Fig. 1a)
-
2.
scattered small-pigmented lesions, chlorotic, or necrotic points
-
3.
larger lesions, partly with aerial mycelium and stunted sporulating acervuli (Fig. 1b)
-
4.
sporulating acervuli with chlorotic and necrotic lesions
-
5.
heavily sporulating acervuli (Fig. 1c).
For the sour cherry individuals, CLS was also not controlled in the orchard containing the plant material used for this analysis, resulting in severe disease pressure. In 2010, CLS severity was recorded using the same scale as with sweet cherry. In 2011, the trees were rated only as resistant or susceptible, with those considered resistant if infection did not result in conidia formation and trees did not defoliate (disease score of two or less). Trees were considered susceptible if lesions and conidia formation were observed on the leaves, and/or the trees were defoliated. For the genetic analysis, evaluated trees were divided into resistant and susceptible classes according to the following characteristics: resistant (R)—reaction types 1–2; susceptible (S)—reaction types 3–5.
SNP genotyping and confirmation of marker order
P. canescens (Q39515), ‘Namati,’ F5-18-167, and 72 seedlings (‘Namati’ × F5-18-167 and ‘Namati’ open pollinated) were genotyped using the RosBREED Illumina Infinium® cherry SNP array of 5,696 SNP markers (Peace et al. 2012). SNP genotypes were determined using the Genotyping Module of GenomeStudio Data Analysis software v2010.3 (Illumina 2010). A total of 2,949 polymorphic SNPs were found, from which 548 SNPs were selected manually to cover the eight Prunus linkage groups (Supplementary Table S3, un-highlighted markers). Markers for the ‘Namati’ × F5-18-167 map were selected to be spread across each chromosome as equally as possible based on peach physical map distances. The map used for QTL analysis was based on the peach physical map positions (Verde et al. 2013) where the physical map was scaled to approximate genetic map positions using a conversion factor of 1 Mb = 4 cM. The relative marker order and distances were verified by comparing expected and observed double cross-over frequencies using the informative meiosis function of FlexQTL™ (Bink et al. 2002).
For sour cherry, a separate GenomeStudio project was done where ‘UF,’ ‘Montmorency,’ 23-23-13, and 15 seedlings (from the crosses of ‘UF’ × 23-23-13 and ‘Montmorency’ × 23-23-13) and other sour cherry founders and populations (Table S4 in Peace et al. 2012) were genotyped as described in sweet cherry (384 individuals total). However, available SNP data from a diverse array of sweet cherry selections and seedlings (105 individuals) were included to aid in the determination of dosage by showing the two homozygous (AAAA and BBBB for sour cherry corresponding to AA and BB in sweet cherry) and balanced heterozygous (AABB for sour cherry corresponding to AB in sweet cherry) classes (Supplementary Fig. S2).
QTL analysis and resistant allele identification in sweet cherry
QTL mapping was done initially using the genome-wide set of 548 SNPs selected from those markers found to be polymorphic, followed by an analysis of a single chromosome with a dense map. After a QTL was located on linkage group 4 (G4), all available polymorphic SNPs found for that linkage group, i.e., 241 markers, were used in the QTL analysis (Supplementary Table S3; G4 markers highlighted and un-highlighted).
The phenotypic data used in the QTL analysis were the mean disease scores for 2008–2011 (Supplementary Table S1). The parents and the P. canescens grandparent of the populations were included in the analysis. QTL analysis was done using a Markov chain Monte Carlo (MCMC)-based Bayesian analysis method (Bink et al. 2002, 2008) implemented in the FlexQTL™ software as was done in Rosyara et al. (2013). In the analysis with all eight Prunus chromosomes, the simulation length used was 100,000 iterations with a thinning value of 10. In the analysis of just G4, the simulation length used was 200,000 iterations with a thinning value of 20.
The haplotypes for the QTL identified were manually constructed for both sweet and sour cherry using the SNP data where linkage phase of the markers could be determined. To determine the effects of the QTL alleles identified, Student’s t tests were performed with all sweet cherry individuals comparing the mean disease scores of the two groups and the presence/absence of the P. canescens haplotype at the QTL region. This was performed on all sweet cherry individuals and then for only those progeny that were from the cross ‘Namati’ × F5-18-167.
Simple sequence repeat (SSR) markers were designed to follow the P. canescens QTL allele in sour cherry when haplotypes were unable to be constructed based on ambiguous SNP calls. Four primer pairs were designed for the QTL region (Supplementary Table S4). Sequence used to develop the SSRs was from the peach genome v1.0 (Verde et al. 2013) available at www.rosaceae.org. Microsatellite repeats were found using Microsatellite Repeats Finder (sites.google.com/site/genetics20620/microsatellite). The criteria for SSR selection were as follows: 2 bp minimum repeat length and 5 bp maximum repeat length with a minimum number of 10 repeats. If multiple SSRs were found in the region of interest, those with the largest number of tandem repeats were selected. Primer pairs were designed using Primer3web (bioinfo.ut.ee/primer3/), with product size limited to 150–250 bp, and primers selected on either side of the repeated region with a CG clamp. Primers were then compared to the peach genome v1.0 scaffolds (Verde et al. 2013) using NCBI BLAST. Primers which co-located to multiple scaffolds with at least 75 % sequence similarity were not used. The PCR mixture used was as in Omstead et al. (2008). PCR conditions for CLS004 and CLS005 were as follows: 94 °C for 5 min followed by 35 cycles of 94 °C for 30 s, 54 °C for 45 s, 72 °C for 1 min, and an elongation step of 72 °C for 5 min. For CLS026 and CLS028, a touchdown PCR was used. Conditions were as follows: 94 °C for 5 min followed by 9 cycles of 94 °C for 30 s, 60 °C for 45 s (−1 °C per cycle), 72 °C for 1 min and then 24 cycles of 94 °C for 30 s, 55 °C for 45 s, 72 °C for 1 min with an elongation step of 72 °C for 5 min. The PCR fragments were separated in a 6 % polyacrylamide gel and visualized with silver staining.
Results
P. canescens-derived cherry leaf spot resistance in sweet cherry
S-allele genotyping supported P. canescens ancestry by the presence or absence of a unique S-allele derived from P. canescens, called S can1 . In the cross ‘Namati’ (S 1 S 4 ) × F5-18-167(S 4 S can1 ), all the progeny would have the P. canescens-derived allele S can1 from F5-18-167 as the S 4 allele would be incompatible in the ‘Namati’ style. All 34 progeny individuals from the ‘Namati’ × F5-18-167 population were confirmed to have P. canescens ancestry due to the presence of the S can1 allele, or in the case of two progeny where the S-allele genotype was not obtained, due to the presence of unique P. canescens SNP markers (data not presented). Conversely, none of the 38 progeny individuals from the ‘Namati’ × open-pollinated population had S can1 (Supplementary Table S1).
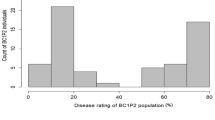
In sweet cherry, the frequency distribution of mean disease scores with all progeny individuals from both populations combined showed a continuous distribution (Supplementary Fig. S3). In the population of 34 individuals with P. canescens in their background (‘Namati’ × F5-18-167), and in the populations of 38 individuals without, a bi-modal and a continuous distribution were observed, respectively (Supplementary Fig. S3). The continuous distribution indicated that there may be multiple QTLs influencing disease resistance. However, in the population derived from P. canescens, the bi-modal distribution suggests one major dominant gene influencing disease score, and segregating in a 1:1 fashion for resistance:susceptibility (Supplementary Fig. S3). In susceptible infections, the CLS fungus was able to grow and produce conidia for secondary infection, giving scores of three or higher which was considered susceptible. Individuals with disease scores of <2 were considered to be resistant.
The map of the ‘Namati’ × F5-18-167 population had an average marker coverage of 1 marker every 1.2 cM, with the largest gap on G7 of 12.1 cM. The genome-wide QTL analysis showed positive evidence for only one QTL on G4 with a Bayes factor of 4.5 (Supplementary Table S5). Once a QTL was located on G4, all 241 polymorphic SNPs found for this linkage group were used in the FlexQTL™ analysis (Supplementary Table S3). When the QTL analysis was run using all of the polymorphic G4 markers, there was decisive evidence for one QTL (Bayes factor of 10.9 for a one QTL model vs. a zero QTL model). This QTL was located on the top of G4 between SNP markers ss490552323 and ss490552500 (between 4.0 and 13.8 cM, with a peach physical map distance between 1.00 and 3.46 Mb) (Fig. 2). Two additional replicates (with different random seeds) gave consistent results (data not shown). This QTL, named CLSR_G4 for CLS resistance found on G4, explained 45 % of the phenotypic variance for disease resistance.

CLS resistance QTL location on G4 between SNP markers ss490552323 and ss490552500 (between 4.0 and 13.8 cM), accompanied by the trace plot for the QTL region
Haplotype construction and QTL allele identification
To determine the effects of the different CLSR_G4 alleles, CLSR_G4 haplotypes were constructed. As P. canescens was homozygous at this region with several unique SNPs, this haplotype was easy to construct as linkage phase between the SNPs was known (Supplementary Figure S4). A comparison of mean disease scores showed that individuals with the P. canescens haplotype for CLSR_G4 had significantly lower disease scores (Table 1). The mean disease scores for all sweet cherry individuals were 2.3 and 3.2 for individuals with the P. canescens haplotype and those without it, respectively (Table 1). When considering only those 34 individuals from the cross ‘Namati’ × F5-18-167, those progeny with and without the CLSR_G4 P. canescens-derived R haplotype had an even larger difference in diseases scores, with a mean disease scores of 2.3 and 4.1 for those with the P. canescens CLSR_G4 haplotype and those without it, respectively (Table 1). However, not all individuals with the P. canescens CLSR_G4 haplotype were rated as resistant to CLS (disease score <2), as five of the 15 individuals with this haplotype were susceptible (Supplementary Table S6). No individuals in this family without the P. canescens CLSR-G4 haplotype for this QTL were resistant, indicating that while this QTL is necessary for CLS resistance, there may be other modifier or complementary genes involved.
To search for a possible second gene involved in CLS resistance, a bulked segregant analysis was done in sweet cherry using the genome-wide SNP data. The SNP genotypes were compared for individuals that had the CLSR_G4 P. canescens-derived R haplotype and were either resistant (n = 10) or susceptible (n = 5). No marker was found to be absent only in those individuals that were susceptible while present in the resistant individuals. Therefore, this analysis failed to identify a second possible chromosome region containing gene(s) contributing to CLS resistance.
P. canescens-derived cherry leaf spot resistance in sour cherry
For sour cherry, of the 15 23-23-13-derived seedlings screened, six had susceptible ratings for CLS, while the other nine had resistant ratings (Supplementary Table S2). Both ‘UF’ and ‘Montmorency’ were susceptible, while the P. canescens-derived parent 23-23-13 was resistant (Supplementary Table S2).
Of the 18 sour cherry individuals genotyped, haplotypes for the CLSR_G4 region could be determined for all parents (‘Montmorency,’ ‘UF,’ and the P. canescens-derived 23-23-13) and nine of the 15 progeny (Supplementary Fig. S5A, B). All of the progeny of the cross between the resistant parent 23-23-13 and either susceptible parent ‘Montmorency’ or ‘UF’ contained the P. canescens haplotype R that was associated with disease resistance in the sweet cherry mapping population (Supplementary Fig. S4). Haplotypes could not be determined for six sour cherry individuals due to ambiguous SNP calls for this region.
To verify that the P. canescens CLSR_G4 R haplotype was present in those sour cherry individuals where haplotypes were unable to be constructed, four SSR markers situated within the haplotype and spanning the QTL region between SNP markers ss490552323 (4.0 cM, 1.0 Mb) and ss490552500 (13.8 cM, 3.46 Mb) were used (Supplementary Table S4; Fig. S6). All four SSRs had a unique fragment representing the P. canescens chromosome, which was present in the resistant parent 23-23-13 and in 12 of the 15 seedlings (Fig. 3). This allowed the verification of the P. canescens chromosome at this QTL region, even when haplotypes were unable to be constructed. In those individuals with the P. canescens fragment, each individual had the P. canescens SSR fragment for all four SSRs indicating that there was no cross over in the CLSR_G4 region. The three seedlings which did not contain the P. canescens CLSR_G4 R haplotype were all susceptible to CLS. There were, however, three individuals which had the P. canescens CLSR_G4 haplotype, but were also susceptible to CLS (Fig. 3). This, as with the case of sweet cherry, indicates that the presence of the P. canescens-derived CLSR_G4 R haplotype does not guarantee that the tree will be resistant, but without it, trees are susceptible.

Polyacrylamide gel images of PCR products for four SSR markers (Supplementary Table 4) on sour cherry (Supplementary Table 2). SSR markers were designed and used to validate the presence of the P. canescens-derived CLSR_G4 R haplotype in sour cherry. a CLS026, b CLS028, c CLS004, and d CLS005. Arrows denote the location and fragment size of the P. canescens allele. Individuals with an asterisk are resistant to cherry leaf spot
Discussion
One major QTL controlling CLS resistance, named CLSR_G4, was identified on G4. Individuals must possess the P. canescens-derived R haplotype for this QTL in order to be disease resistant. This is in agreement with the phenotypic data in sweet cherry which suggested that resistance to CLS was associated with the presence of a major gene from P. canescens. However, a two gene model, where both parents are heterozygous for a second gene, is needed to explain the genetic control of CLS resistance (Fig. 4a, b). This two gene model is consistent with the finding in sweet cherry where five of the 15 individuals with the P. canescens-derived R haplotype for CLSR_G4 were CLS susceptible. In Fig. 4a, these five susceptible individuals are represented with the genotype Aabb and the 10 resistant individuals are represented by the genotypes AaBB, AaBb, and AaBb.

Proposed two gene model for disease resistance in a sweet cherry and b sour cherry. Individuals are resistant when dominant alleles are present at two unlinked loci, the P. canescens-derived R haplotype for CLSR_G4 is represented as locus ‘A,’ and a proposed second locus, ‘B.’ Disease-resistant parents F5-18-167 (sweet) and 23-23-13 (sour) are shown to be heterozygous at both loci (AaBb and A1a1a2a2B1b1b2b2 for sweet and sour cherry, respectively) where parent 2 (sweet), ‘Montmorency’ (sour) and ‘UF’ (sour) are shown to be homozygous recessive for the ‘A’ locus and heterozygous for the proposed second locus needed to confer resistance. Squares highlighted in gray identify those genotypes proposed to confer CLS resistance
A two gene model can also explain the genetic control of CLS resistance in sour cherry. If a sour cherry individual has the P. canescens-derived R haplotype for CLSR_G4, represented as A 1 , it must also have B 1 to be resistant to CLS (Fig. 4b). Two-sevenths and one-fifth of the progeny with the P. canescens-derived R haplotype for CLSR_G4 were found to be susceptible in the populations ‘Montmorency’ × 23-23-13 and ‘UF’ × 23-23-13, respectively. The observed percentages of susceptible individuals were close to the expected one-fourth of those predicted for individuals carrying the P. canescens-derived R-haplotype.
It is possible that sweet cherry and sour cherry have genes that contribute to CLS resistance as ranges of CLS resistance and susceptibility exist in both these species. For example, partial resistance to CLS in cherry was identified and characterized based on infection severity and subsequent defoliation (Sjulin et al. 1989). Our inability to identify the hypothesized second QTL needed to confer resistance is not likely due to the lack of marker coverage as the largest genome wide gap among the SNP markers was 12.1 cM on G7 followed by 8 cM on G2 (Supplementary Table S3). Instead, the hypothesized second gene may actually be two or more genes and the population sizes may have been too small to detect more minor gene effects that are characteristic of horizontal resistance. Larger population sizes in sour cherry are being generated to investigate this possibility.
The SSR markers designed in this study allowed the determination of whether an individual had the P. canescens CLSR_G4 R haplotype. These markers were especially useful in sour cherry where the SNP markers alone could not be used to determine the presence or absence of the R haplotype. In sour cherry, some individuals can have chromosomes that are represented by more than the four copies expected for a tetraploid. This is due to the segmental allo-polyploid nature of sour cherry (Beaver and Iezzoni 1993; Beaver et al. 1995). Cytological studies on sour cherry have revealed that multivalent and univalent formations at meiosis are common (Schuster 2000; Schuster and Wolfram 2005). If a seeding has five copies of a chromosome, for example, it is difficult to determine the seedling’s genotype for that region using SNP data alone.
The SSR markers for the P. canescens CLSR_G4 region enable the use of marker-assisted selection to increase the efficiency of breeding CLS-resistant cherry cultivars. Those seedling populations predicted to be segregating for P. canescens-derived CLS resistance can be screened in the greenhouse for the presence of the SSR alleles that tag the R haplotype. Those seedlings that do not have the R haplotype can be discarded while those seedlings with the R haplotype can be maintained and planted in the breeding field. Based on the two allele model for the genetic control of CLS resistance, three-fourths of the seedling planted in the field would be predicted to be CLS resistant as opposed to only three-eighths of the seedlings if the DNA test had not been performed. Discarding susceptible seedlings prior to field planting results in reduced field maintenance costs and allow for resources to be targeted to those seedlings more likely to be CLS resistant.
References
Beaver JA, Iezzoni AF (1993) Allozyme inheritance in tetraploid sour cherry (Prunus cerasus L.). J Am Soc Hortic Sci 118:873–877
Beaver JA, Iezzoni AF, Ramn C (1995) Isozyme diversity in sour, sweet and ground cherry. Theor Appl Genet 90:847–852
Bink MCAM, Uimari P, Sillanpaa MJ, Janss LLG, Jansen RC (2002) Multiple QTL mapping in related plant populations via a pedigree-analysis approach. Theor Appl Genet 104:751–762
Bink MCAM, Boer MP, ter Braak CJF, Jansen J, Voorrips RE, van de Weg WE (2008) Bayesian analysis of complex traits in pedigreed plant populations. Euphytica 161:85–96
Budan S, Mutafa I, Stoian I, Popescu I (2005) Field evaluation of cultivar susceptibility to leaf spot at Romania’s sour cherry genebank. Acta Hortic 667:153–158
Howell GS, Stackhouse SS (1973) The effect of defoliation time on acclimation and dehardening in tart cherry (Prunus cerasus L.). J Am Soc Hortic Sci 98:132–136
Illumina (2010) GenomeStudio genotyping module v1.0, user guide. Illumina, Towne Centre Drive
Keitt GW, Blodgett EC, Wilson EE, Magie RO (1937) The epidemiology and control of cherry leaf spot. Univ Wisc Agric Exp Stn Res Bull 132:1–117
Omstead JW, Sebolt AM, Cabrera A, Sooriyapathirana SS, Hammar S, Iriarte G, Wang D, Chen CY, van der Knaap E, Iezzoni AF (2008) Construction of an intra-specific sweet cherry (Prunus avium L.) genetic linkage map and synteny analysis with the Prunus reference map. Tree Genet Genomes 4:897–910
Peace C, Bassil N, Main D, Ficklin S, Rosyara UR, Stegmeir T, Sebolt A, Gilmore B, Lawley C, Mockler TC, Bryant DW, Wilhelm L, Iezzoni A (2012) Development and evaluation of a genome-wide 6K SNP array for diploid sweet cherry and tetraploid sour cherry. PLoS One 7(12):e48305
Proffer TJ, Berardi R, Ma Z, Nugent JE, Ehret GR, McManus PS, Jones AL, Sundin GW (2006) Occurrence, distribution, and polymerase chain reaction-based detection of resistance to sterol demethylation inhibitor fungicides in populations of Blumeriella jaapii in Michigan. Phytopathology 96:709–717
Richards D (1986) Tree growth and productivity—the role of roots. Acta Hortic 175:27–36
Rosyara UR, Bink MCAM, van de Weg E, Zhang G, Wang D, Sebolt A, Dirlewanger E, Quero-García J, Schuster M, Iezzoni AF (2013) Fruit size QTL identification and the prediction of parental QTL genotypes and breeding values in multiple pedigreed populations in sweet cherry. Mol Breed 32:875–887
Schuster M (2000) Genome investigation of sour cherry, Prunus cerasus L. Acta Hortic 538:375–379
Schuster M, Tobutt KR (2004) Screening of cherries for resistance to Leaf Spot, Blumeriella jaapii. Acta Hortic 663:239–244
Schuster M, Wolfram B (2005) Sour cherry breeding at Dresden-Pillnitz. Acta Hortic 667:127–130
Schuster M, Grafe C, Hoberg E, Schütze W (2013) Interspecific hybridization in sweet and sour cherry breeding. Acta Hortic 976:79–86
Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K (2001) dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29(1):308–311
Sjulin TM, Jones AL, Andersen RL (1989) Expression of partial resistance to cherry leaf spot in cultivars of sweet, sour, duke, and European ground cherry. Plant Dis 73:56–61
Sonneveld T, Robbins TP, Tobutt KR (2006) Improved discrimination of self-incompatibility S-RNase alleles in cherry and high throughput genotyping by automated sizing of first intron polymerase chain reaction products. Plant Breed 125:305–307
Verde I, Abbott AG, Scalabrin S, Jung S, Shu S, Marroni F, Zhebentyayeva T, Dettori MT, Grimwood J, Cattonaro F, Zuccolo A, Rossini L, Jenkins J, Vendramin E, Meisel LA, Decroocq V, Sosinski B, Prochnik S, Mitros T, Policriti A, Cipriani G, Dondini L, Ficklin S, Goodstein DM, Xuan P, Del Fabbro C, Aramini V, Copetti D, Gonzalez S, Horner DS, Ralchi R, Lucas S, Mica E, Maldonado J, Lazzari B, Bielenberg D, Pirona R, Miculan M, Barakat A, Testolin R, Stella A, Tartarini S, Tonutti P, Arús P, Orellana A, Wells C, Main D, Vizzotto G, Silva H, Salamini F, Schmutz J, Morgante M, Rokhsar DS (2013) The high-quality draft of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat Genet 45:487–494
Wharton P, Iezzoni A (2005) Development of a protocol for screening cherry germplasm for resistance to cherry leaf spot. Acta Hortic 667:509–514
Wharton PS, Iezzoni A, Jones AL (2003) Screening cherry germplasm for resistance to cherry leaf spot. Plant Dis 87:471–477
Acknowledgments
This project was supported by the USDA National Institute of Food and Agriculture (NIFA) hatch project number MICL02032 and the USDA-NIFA-Specialty Crop Research Initiative project, RosBREED: Enabling marker-assisted breeding in Rosaceae (2009-51181-05808).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Stegmeir, T., Schuster, M., Sebolt, A. et al. Cherry leaf spot resistance in cherry (Prunus) is associated with a quantitative trait locus on linkage group 4 inherited from P. canescens . Mol Breeding 34, 927–935 (2014). https://doi.org/10.1007/s11032-014-0086-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11032-014-0086-3