
Abstract
The construction of the first genetic map in autotetraploid blueberry has been made possible by the development of new SNP markers developed using genotyping by sequencing in a mapping population created from a cross between two key highbush blueberry cultivars, Draper × Jewel (Vaccinium corymbosum). The novel SNP markers were supplemented with existing SSR markers to enable the alignment of parental maps. In total, 1794 single nucleotide polymorphic (SNP) markers and 233 simple sequence repeat (SSR) markers exhibited segregation patterns consistent with a random chromosomal segregation model for meiosis in an autotetraploid. Of these, 700 SNPs and 85 SSRs were utilized for construction of the ‘Draper’ genetic map, and 450 SNPs and 86 SSRs for the ‘Jewel’ map. The ‘Draper’ map comprises 12 linkage groups (LG), associated with the haploid chromosome number for blueberry, and totals 1621 cM while the ‘Jewel’ map comprises 20 linkage groups totalling 1610 cM. Tentative alignments of the two parental maps have been made on the basis of shared SSR alleles and linkages to double-simplex markers segregating in both parents. Tentative alignments of the two parental maps have been made on the basis of shared SSR alleles and linkages to double-simplex markers segregating in both parents.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Consumer demand for blueberries is at an all-time high due, in part, to their many recognized health benefits in addition to increased worldwide production. Blueberry fruit is rich in phenolic compounds, which have been linked to improved night vision, prevention of macular degeneration, anticancer activity, inhibition of pro-inflammatory molecules and reduced risk of heart disease (Johnson and Arjmandi 2013; Kalt et al. 2007). Blueberry polyphenols have also been shown to increase endothelium-dependent vasodilation as assessed by flow-mediated dilation, leading to an improvement in overall vascular function (Rodriguez-Mateos et al. 2013). Resveratrol content, has been linked to reduced risk of heart disease and cancer, while pterostilbene, the primary antioxidant component of blueberries, has been shown to have both preventive and therapeutic effects on neurological, cardiovascular, metabolic and hematological disorders (McCormack and McFadden 2013; Hangun-Balkir and McKenney 2012; Rimando et al. 2004). In the UK, the high consumer demand for blueberries, combined with the lack of appropriate high-quality cultivars suitable for UK climatic conditions, has resulted in a real need by the industry for the rapid development of new blueberry cultivars with high fruit and nutritional quality and expanded fruiting season to meet the demand for home grown soft fruit. UK blueberries supply only 5 % of demand, and projections have indicated that a rise of 50–100 % in blueberry production up to 10 % of demand is feasible, given appropriate cultivars and management practices (Brazelton 2013).
Highbush blueberry cultivars (Vaccinium corymbosum L.) occur at three ploidy levels, 2n = 2x = 24, 4x = 48 and 6x = 72 (Camp 1945; Megalos and Ballington 1988). Many of the major blueberry varieties cultivated around the world are autotetraploid (Qu et al. 1998; Boches et al. 2005). Autopolyploids (polysomic polyploids) which also include sugarcane (Saccharum spp.) and potato (Solanum spp.) contain more than two genetically similar (homologous) genomes, while allopolyploids (disomic polyploids) consist of two or more distinct (non-homologous) genomes such as wheat (Triticum spp.), strawberry (Fragaria × ananassa) and rapeseed (Brassica spp.). In allopolyploids, the association of two differentiated genomes by means of interspecific hybridization results in the observed chromosome doubling. Autopolyploids, on the other hand, are thought to derive from chromosome doubling of the same genome within a single parental species (Gallais 2003). Pairing during meiosis can occur in autopolyploids between randomly chosen pairs of homologous chromosomes (bivalents) or between more than two homologous chromosomes (multivalents). Each chromosome has the potential to pair randomly with any of its homologues leading to tetrasomic inheritance where all allelic combinations (A1A2A3A4) may be produced in equal frequencies (Soltis and Soltis 1993). A recent genetic map has been published for interspecific diploid blueberry comprising a total of 265 markers across 12 linkage groups and has led to the identification of quantitative trait loci for cold hardiness and chilling requirements (Rowland et al. 2012). The number of markers has been recently increased to 318 (Reid et al. unpublished). However, the tetraploid nature of commercial highbush blueberry makes developing a genetic understanding of traits through linkage mapping significantly more difficult than in the disomic blueberry. One of the issues is the larger number of markers required relative to a diploid map and this has been addressed in our study by the use of SNP markers derived from genotyping by sequencing (GBS) of the mapping population.
While statistical methods for linkage analysis and QTL mapping in diploid species are well developed, polysomic analyses have advanced more slowly (Luo et al. 2001; Hackett et al. 2001). A theoretical model of linkage analysis of dominant markers in an autotetraploid species was developed by Hackett et al. (1998), and this approach was later adopted by Meyer et al. (1998) in the development of a linkage map in potato. Luo et al. (2000) developed a model for the prediction of marker genotypes for autotetraploid parents by analyzing marker phenotypes from segregating data obtained from parents and progeny, and this methodology is utilized in the software TetraploidMap (Hackett and Luo 2003; Hackett et al. 2007). TetraploidMap has been used to develop linkage maps in many autotetraploid crops including blackberry (Castro et al. 2013), rose (Gar et al. 2011), alfalfa (Julier et al. 2003) and potato (Bradshaw et al. 2008). A full range of marker types including fully versus partially informative as well as dominant versus codominant markers often can be found segregating simultaneously in an autotetraploid population (Wu et al. 2001). The genotypes of diploid parents can be predicted by the segregation pattern of the progeny, but this is not always the case with autotetraploids due to the possibility of multiple allele dosage and double reduction (Wu et al. 2001) and they must therefore be deduced by parental and offspring phenotypes combined (Hackett and Luo 2003). Both the occurrence and frequency of double reduction in autopolyploids would be expected to affect the pattern of allelic segregation. The frequency of double reduction will itself be affected by the position of each locus on a chromosome with greater values found toward the distal–proterminal regions and almost null at loci located near centromeres (Welch 1962; Butruille and Boiteux 2000). The estimation of recombination frequencies using microsatellite markers (SSRs) is up to four times more informative than with dominant markers (Luo et al. 2001). Genetic markers and marker pairs differ in their informativeness about recombination, measured by the Fisher information. This is discussed in the context of autotetraploid analysis by Hackett et al. (1998), and variances of different configurations, equal to the inverse of the Fisher information, are given there. For linkage map construction in an autotetraploid species, the most informative types of dominant markers are simplex (AOOO × OOOO), duplex (AAOO × OOOO) and double-simplex (AOOO × AOOO) in the parents. These markers segregate in the progeny in expected presence/absence ratios of 1:1, 5:1 and 3:1, respectively, assuming tetrasomic inheritance (random chromosomal segregation). Other configurations, such as AAOO × AOOO or AAOO × AAOO, are present in higher proportions (11:1 and 35:1) and carry little information about recombination. The precision with which the recombination frequency between a pair of marker can be estimated depends on the type and also the phase (coupling or repulsion). For dominant markers, recombination between simplex markers linked in coupling phase has the highest precision, while simplex markers linked in repulsion have a lower precision. Double-simplex markers also have a higher precision in coupling phase. Duplex-to-simplex linkages, however, have equal precision in coupling and repulsion phase, and so are particularly useful for identifying sets of homologous chromosomes.
Single nucleotide polymorphisms (SNPs) are by far the most abundant mutations identified between related DNA molecules. As a result of the advent of rapid and affordable second generation sequencing technologies, SNPs have become increasingly important as markers in plant research on both a fundamental and applied basis (Ward et al. 2013). Genotyping by sequencing (GBS) has enabled the discovery of large numbers of SNPs and the subsequent creation of high density linkage maps in a range of diploid fruit species, including raspberry, grape, blackcurrant, apple, sweet cherry, and peach (Ward et al. 2013; Barba et al. 2014; Russell et al. 2014; Gardner et al. 2014; Guajardo et al. 2015; Bielenberg et al. 2015). SNP markers can be identified from short reads generated by next generation sequencing (NGS) either by aligning to a reference genome or by de novo assembly (Nielsen et al. 2011). Although there is a draft whole genome assembly for blueberry under preparation (Reid et al. unpublished), this was not available at the time of this study, making de novo assembly necessary.
To date, there is no publically available genetic linkage map for autotetraploid blueberry for QTL mapping of economically important traits such as fruit and nutritional quality. The generation of such a genetic linkage map is a critical step toward providing the framework for blueberry crop improvement through marker-assisted breeding, and we describe here the development of the first linkage map in the autotetraploid blueberry V. Corymbosum.
Methods
Plant material
The mapping population is an F1 population generated from a cross between two tetraploid cultivars: the northern highbush ‘Draper’ released by Michigan State University in 2002 and the southern highbush ‘Jewel’ which was released by the University of Florida in 1999.
The cross was made at Michigan Blueberry Growers Marketing and seedlings were propagated at Michigan State University. ‘Draper’ is an early to mid-season cultivar producing premium quality, firm and sweet fruit with superior shelf life. ‘Jewel’ is an early to mid-season cultivar from a high-yielding plant producing very large, slightly tart fruit. The mapping population segregates for a number of key phenotypic traits and is comprised of 99 individuals.
DNA isolation
Young actively growing blueberry leaves were collected from the two parental cultivars, Draper and Jewel, and the 99 available progeny. Between 30 and 50 mg of leaf tissue was placed in a cluster tube (Corning Life Sciences, Tewksbury, MA, USA) containing a 4-mm stainless steel bead (McGuire Bearing Company, Salem, OR, USA) and samples were frozen in liquid nitrogen prior to storage at −80 °C until extraction. Frozen tissue was homogenized using a Retsch® MM301 Mixer Mill (Retsch Inc. Hann, Germany) at a frequency of 30 cycles/s and three 30 s bursts. Two different DNA extraction methods were used, depending on how the DNA was to be utilized. For microsatellite analysis, the DNA was isolated using commercially available cell lysis and protein precipitation solutions (Qiagen, Hilden, Germany) detailed in Gilmore et al. (2011) following the Qiagen DNeasy mini plant kit protocol. DNA was quantified based on absorbance at 260 nm with the Wallac Victoc 3V, 1420 Multilabel Counter (Perkin Elmer, Massachusetts, USA). For construction of the GBS library, DNA was isolated with the E-Z 96® Plant DNA extraction kit (Omega Bio-Tek, Norcross, GA, USA) as previously described (Gilmore et al. 2011) and concentrations determined with a Quant-iT™ Picogreen® dsDNA Assay kit (Invitrogen, Eugene, OR, USA).
Genotyping by sequencing (GBS) library construction and sequencing
GBS libraries from the two parents and progeny were constructed as described by Ward et al. (2013) and Elshire et al. (2011). Briefly, a set of 96 barcoded adapters were generated from complementary oligonucleotides with a ApeK1 overhang sequence and unique barcodes terminate with 4–8 bp on the 3′ end of its top strand and a 3-bp overhang on the 5′ end of its bottom strand complementary to the ‘sticky’ end generated by ApeK1. The common adapter has only an ApeK1 compatible sticky end. Oligonucleotides comprising the top and bottom strands of each barcode adapter and a common adapter were diluted (separately) in TE (50 mM each) and annealed in a thermocycler (95 °C, 2 min; ramp down to 25 °C by 0.1 °C/s; 25 °C, 30 min; 4 °C hold). DNA samples (100 ng in a volume of 10 µl) were added to individual wells containing barcoded adaptors, dried and then digested for 2 h at 75 °C with ApeKI (New England Biolabs, Ipswitch, MA) in 20 µl volumes containing 16 NEB Buffer 3 and 3.6 U ApeKI. Adapters were then ligated to sticky ends by adding 30 µl of a solution containing 1.66 ligase buffer with ATP and T4 ligase (640 cohesive end units) (New England Biolabs) with incubation at 22 °C for 1 h followed by heating to 65 °C for 30 min to inactivate the T4 ligase. Sets of 48 or 96 digested DNA samples, each with a different barcode adapter, were combined (5 µl each) and purified using a commercial kit (QIAquick PCR Purification Kit; Qiagen, Valencia, CA) according to the manufacturer’s instructions. DNA samples were eluted in a final volume of 50 µl Restriction fragments from each library were then amplified in 50 µl volumes containing 2 µl pooled DNA fragments, 16Taq Master Mix (New England Biolabs), and 25 pmol, each, of the following primers: 5’-AATGATACG GCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT and 5’-CA AGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCG ATCT. Temperature cycling consisted of 72 °C for 5 min, 98 °C for 30 s followed by 18 cycles of 98 °C for 30 s, 65 °C for 30 s, 72 °C for 30 s with a final Taq extension step at 72 °C for 5 min (Elshire et al. 2011). These amplified sample pools constitute a sequencing ‘library.’ Annealed and quantitated unique barcode and common adapters were provided by the Buckler Lab for Maize Genetics and Diversity, Cornell University (Ithaca, NY, USA). The GBS libraries were submitted to the Oregon State University Center for Genome Research and Biocomputing (CGRB) core facilities (Corvallis, OR, USA) for quantitation using a Qubit® fluorometer (Invitrogen, Carlsbad, CA, USA). The size distribution of the libraries was confirmed to range between 180 and 250 bp with the Bioanalyzer 2100 HS-DNA chip (Agilent Technologies, Santa Clara, CA, USA). Libraries were diluted to 10 nM based on Qubit® readings, and quantitative PCR (qPCR) used to quantify the diluted libraries. From each pooled library, 15.5 pM was loaded onto a single lane for single-end Illumina® sequencing of 101 cycles with the HiSeq™ 2000 and analyzed using the version 3 cluster generation and sequencing kits.
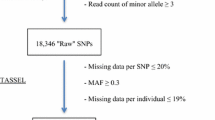
Putative SNP markers were identified using the Universal Network Enabled Analysis Kit (UNEAK) pipeline implemented in TASSEL, as a reference genome sequence was not available to map reads for SNP analysis (Lu et al. 2013). Sequence reads were trimmed to 64 bp "tags" and SNPs were represented by a minimum of five sequences, with SNP loci having a minimum minor allele frequency ≥5 %. Stringent filtering was applied to select markers with less than 10 % missing data.
Development of SSR markers
SSR markers were derived from a number of sources including those previously reported from two expressed sequence tag (EST) libraries developed from cold-acclimated (CA) and non-acclimated (NA) floral buds (Rowland et al. 2003; 2008; Dhanaraj et al. 2004; Boches et al. 2005) of ‘Bluecrop,’ GenBank EST sequences (GVC for GenBank-derived V. corymbosum); 454 sequenced ESTs (VCB); Sanger-sequenced ESTs developed by the New Zealand Institute for Plant & Food Research Ltd (Pr prefix); and genomic sequences from the largest NGS scaffolds of the W85-20 genome assembly (KAN prefix for Kannapolis where the genome is being assembled). Sources and sequences of the majority of SSR markers mapped are described in more detail in Rowland et al. (2014) with any additional marker sequence information and GenBank accession numbers detailed in the supplementary Table (S1). SSR markers were first screened over parents and a subset of progeny to identify alleles that were either present in one parent only and segregating 1:1 or 5:1, or present in both parents and segregating 3:1, respectively. Multi-allelic SSRs which were polymorphic between the parents also were included for further analysis. The whole population was then genotyped with these markers and any SSRs with ≥25 % missing data excluded.
PCR conditions and SSR genotyping
PCRs and capillary electrophoresis platforms varied slightly according to which of the three laboratories (James Hutton Institute, USDA-ARS-NCGR and University of Florida,) genotyped the population. At the James Hutton Institute, a typical 25-μl reaction contained 25 ng template DNA, 1.0 μM forward and reverse primer, 0.2 mM dNTPs, 1 × PCR buffer (containing 10 mm Tris–HCl, 1.5 mm MgCl2, 50 mm KCl, pH 8.3) and 0.1 units Taq polymerase (Roche, Basel, Switzerland). PCR was performed using a Perkin Elmer 9700 Thermal Cycler as follows: 5 min at 95 °C, then 30 s at 95 °C, 30 s at 57 °C and 45 s at 72 °C for 40 cycles followed by 10 min at 72 °C. PCR products were resolved and visualized on a 1.5 % agarose gel to assess success of amplification, then analyzed on an ABI 3730 DNA sequence analyzer (Applied Biosystems, Foster City, USA) using Rox 500 (Applied Biosystems) as an internal size standard. Allele sizes were visualized using GeneMapper software version 3.7 (Applied Biosystems).
At the USDA-ARS-NCGR, PCRs contained 1 × reaction buffer, 2 mM MgCl2, 0.2 mM dNTPs, 0.50 µM fluorescent M13 primer (Shuelke 2000), 0.12 µM forward primer, 0.50 µM reverse primer, 0.075 units of GoTaq® DNA Polymerase (Promega Corporation, Madison, WI, USA), and 4.5 ng genomic DNA in a total volume of 15 µL (Rowland et al. 2014). At the University of Florida, a 10-μl PCR contained 20 ng template DNA, 0.75 μM forward primer, 0.3 µM reverse primer, 0.3 µM M13 fluorescent tag primer (Shuelke 2000), 0.2 mM dNTPs, 2 mM MgCl2, 1 × GoTaq PCR buffer and 0.025 units GoTaq DNA polymerase (Promega Corp, Madison, WI). In all three laboratories, band polymorphisms were scored in individuals as present (1), absent (0) or missing data (9), where markers failed to amplify in an individual.
Linkage map construction
Our mapping strategy involved constructing maps for the two parents separately using simplex and duplex markers, and then aligning these approximately using simplex to double-simplex coupling linkages. Details of the theory for dominant markers can be found in Hackett et al. (1998) and Meyer et al. (1998).
In an allotetraploid species, simplex and double-simplex show the same expected segregation ratios as above, however duplex markers either do not segregate, or segregate in a 3:1 presence/absence ratio depending on the chromosomal pairing. This could form the basis of a test for the type of ploidy, especially in a large population. However, an alternative is to compare the proportion of simplex coupling and repulsion linkages: In an allopolyploid, these should be detected in equal frequencies, while in an autotetraploid few repulsion linkages will be detected (Wu et al. 1992; Al-Janabi et al. 1992).
TetraploidMap was used to explore the joint allele frequencies for each SSR separately, in order to assess the most likely parental genotypes and the goodness of fit between the observed and expected joint allele frequencies. The SSRs were also tested to see whether there was evidence of offspring genotypes that could only be obtained from double reduction (e.g., if the parental genotypes are inferred to be ABCC × DDDD, then an offspring with phenotype BD can only arise from double reduction in the first parent), however none were found. If no parental genotype model gave a satisfactory fit to the observed joint frequencies, then the alleles were analyzed separately, as dominant markers, but labeled as a, b, etc., to show the origin.
Linkage maps were constructed separately for markers segregating in ‘Draper’ followed by those segregating in ‘Jewel.’ The marker sets in ‘Draper’ were assembled by combining SSR markers polymorphic in ‘Draper’ with GBS markers suggested by a preliminary filter to be heterozygous in ‘Draper,’ homozygous in ‘Jewel’ and segregating in a 1:1 or 5:1 ratio in the progeny, based on a Chi-square test of goodness of fit. Markers for which there was a significant lack of fit (p < 0.001) were excluded. This was then repeated to identify markers heterozygous in ‘Jewel’ and homozygous in ‘Draper’ for ‘Jewel’ map construction.
Each set of markers was clustered into linkage groups using group average clustering with a distance measure based on the significance level of a Chi-square test for independent segregation as described by Luo et al. (2001) in the context of a tetraploid potato analysis. Any co-segregating markers were excluded. The TetraploidMap software (Hackett et al. 2007) was used to calculate recombination frequencies and LOD scores between all pairs of markers, for each cluster separately. As the resultant linkage groups, were too large to be ordered within TetraploidMap the pairwise recombination frequencies and LOD scores for independence for each linkage group were analyzed with JoinMap 4 (Van Ooijen 2006). The linkage groups were checked one group at a time within JoinMap to confirm linkage and eliminate any markers with LOD scores less than four to any of the others in that linkage group, and the remaining markers were ordered using its regression mapping algorithm, with default parameters. The software MapChart (Voorrips 2002) was then used to display the linkage maps which were numbered randomly.
Double-simplex markers from the GBS analysis were not used in the cluster analysis, as they have low Fisher information unless they are in coupling phase (Meyer et al. 1998). However, they were tested for coupling associations with the simplex markers from both ‘Draper’ and ‘Jewel’ using a Chi-square test for independent segregation and inferring association if the test for independent segregation was rejected with significance p < 0.001. Linkage groups from ‘Draper’ and ‘Jewel’ were then aligned on the basis of at least one double-simplex marker showing an association with simplex SNPs in both the ‘Draper’ and the ‘Jewel’ groups.
Results
Genotyping by sequencing libraries
The GBS library constructed from the ‘Draper’ × ‘Jewel’ mapping population generated 153M single-end reads. The number of reads ranged from a minimum of 259,300 in progeny CF89 to 2.7 million in CF9 and averaged 1.5 million reads per sample (Fig. S1). Unique tag counts ranged from a low of 145,038 tags in CF89 to 596,130 tags per individual sequenced in this lane and were 422,463 on average. The proportion of unique tags per read was 30 % on average across all the individuals sequenced and was highest in individuals with the lowest number of reads: CF89, CF90 and CF95 (Fig. S1). Using the UNEAK pipeline, 109,044 putative SNPs were identified. CF89, CF90 and CF95 had the highest percent of missing data at 49, 40 and 27 % of the SNPs identified. Filtering to exclude any SNPs with ≥10 % missing values resulted in 17,846 SNPs. Heterozygous SNPs (5224 in ‘Draper’ and 6533 in ‘Jewel’) were further screened to select those which exhibited only one allele segregating in a 1:1 or 5:1 ratio (1101 in ‘Draper’ and 673 in ‘Jewel’) based on a chi-square goodness of fit test, excluding those with significant distortion (p < 0.001). SNP counts were only considered missing where 0/0 read depths were recorded.
SSR marker analysis
A total of 295 SSR primer pairs were tested for polymorphism between the parents of the mapping population. Nine primer pairs failed to amplify and products of a further 53 primers were homozygous in both parents. From the 233 primer pairs successfully scored, a total of 402 markers were mapped. Of these, 207 were codominant SSR markers, with two or more alleles segregating, while the remaining 195 were individual bands for products from an SSR primer pair that were scored as dominant markers. With codominant markers, such as SSRs, it is easy to detect the presence of double reduction (two sister chromatids recovered from a single gamete). However, there were no offspring genotypes identified in this dataset that could be obtained only through double reduction.
Comparison of the frequency of simplex coupling and repulsion linkages
In a cross between allopolyploids, an analysis of simplex markers assuming disomic inheritance should be equally likely to detect coupling and repulsion linkages. However, for ‘Draper’ (1143 simplex markers), there were 2214 pairs with coupling recombination frequency less than 0.1, however none with a repulsion recombination frequency less than 0.1. For ‘Jewel’ (681 simplex markers), there were 810 pairs with coupling recombination frequency less than 0.1, but again none with a repulsion recombination frequency less than 0.1. Similar comparisons at recombination frequencies of 0.2 and 0.25 showed very small number of repulsion linkages in comparison with the coupling linkages. We were therefore confident in our analysis of this cross using an autotetraploid rather than an allopolyploid model.
Linkage map construction
The initial clustering analysis of 1260 markers identified 12 linkage groups in ‘Draper.’ After calculation of recombination frequencies and LOD scores with TetraploidMap, these markers were used to construct linkage maps at a minimum LOD score of 4 in JoinMap. The resulting map had a total length of 1621 cM and comprised 785 mapped markers, among which 33 were co-dominant SSRs, 52 were dominant SSRs and 700 were SNPs (Table 1). Linkage group coverage ranged from 38 to 104 markers with the average number of markers per LG being 65 and the number of markers mapped across all 12 linkage groups totaling 785. The largest gap between markers was 10.9 cM (LG 7). Clusters formed in JoinMap were separated based on a minimum LOD score of 4, and this resulted in an average loss of markers in ‘Draper’ of 15.5 per linkage group; in ‘Jewel,’ an average of 3 markers per group was discarded.
Fewer markers (856) were available for the initial cluster analysis for ‘Jewel’ map, than for ‘Draper’. This resulted in the ‘Jewel’ groups being more fragmented than for ‘Draper’ and the clustering analysis in TetraploidMap produced 32 large groupings with a further 12 smaller groups of between 2 and 7 markers. The larger groups detected and groups containing markers linking ‘Jewel’ to ‘Draper’ were then ordered into linkage maps.
Associations between the ‘Draper’ and ‘Jewel’ maps were identified utilizing double-simplex SNP markers (heterozygous in both parents). Where possible these associations were confirmed by SSRs mapped to linkage groups in both parents. Twenty linkage groups from ‘Jewel’ were aligned with ‘Draper’ maps by this approach. These 20 groups consisted of 536 markers, comprising 59 codominant SSR markers and 27 dominant markers and 450 SNP markers from the GBS dataset. Linkage group coverage ranged from 7 to 62 markers with the average number of markers per LG being 27. The total number of markers mapped in ‘Jewel’ was 536 covering 1610 cM, with the largest gap between markers being 27.2 cM (LG 11b). Table 2 gives details of the markers mapped to each linkage group, and Table 3 shows the double-simplex markers and SSRs on which the alignments are based, and the closest simplex marker from each of ‘Draper’ and ‘Jewel’ to the double-simplex marker. The linkage maps are shown in Fig. 1.












Genetic linkage map constructed in the tetraploid blueberry population, ‘Draper’ × ‘Jewel.’ Underlined and bold markers show the closest linked double-simplex markers for each of the parents, while SSR markers linking parental maps are shown with a line between the maps (Table 3)
Discussion
We report the first application of GBS in blueberry. We have generated thousands of SNPs located across the V. corymbosum genome and utilized a proportion of them to construct the first genetic map in tetraploid blueberry, indeed in any polyploid woody perennial fruit crop. Genotyping of an F1 population enabled the genetic mapping of 785 SNP markers at LOD 4 or above to the 12 linkage groups of ‘Draper,’ and 536 to 20 linkage groups in ‘Jewel.’ Mapping of homologous chromosomes awaits future enlargement of the marker dataset and we are confident that GBS will provide such resources in a future study. However the GBS data obtained was not sufficient in terms of read depth coverage to call allele dosage and so was used to determine presence or absence of alleles only.
We found that excluding SNP markers with >10 % missing data led to a decrease in the number of SNPs available for linkage map construction and this is consistent with previous reports in grape (Barba et al. 2014), sweet cherry (Guajardo et al. 2015) and black raspberry (Bushakra et al. 2015). In blueberry, the number of SNPs (109,044) identified by the non-reference guided UNEAK pipeline was much larger than that identified by a reference-guided pipeline in black raspberry (57,238) constructed using the same protocol and sequenced in the same Illumina® run. Number of SNPs identified in the separate studies on sweet cherry and grape were lower than those in blueberry, at 11,854 and 42,172 respectively. Excluding SNP markers with >10 % missing data in blueberry led to a 6-fold reduction in the number of SNPs (17,846 SNPs) and 7.2-fold reduction in black raspberry (7911 SNPs). Using this same arbitrary filtering parameter of excluding SNP markers with >10 % missing data, 1.4-fold and 2.5-fold reductions in the number of SNPs were observed in the diploid grape (16,833 SNPs) and sweet cherry (8476 SNPs) respectively, though other differences between the studies may have come into play here. The relatively large number of SNPs identified in blueberry compared with black raspberry, constructed and sequenced simultaneously, along with the higher rate of SNP reduction in SNP markers with >10 % missing data is noteworthy and might have resulted from the autopolyploidy of the blueberry, sub-optimal DNA quality, and/or use of a non-reference-guided SNP detection pipeline necessitated by the absence of a reference genome. The average proportion of unique tags to reads per sample in blueberry (30 %) was almost twofold higher than that observed in black raspberry (17 %) (Bushakra et al. unpublished). We believe this is due to the presence of two genomes per locus in blueberry when compared to the single diploid genome of black raspberry. Since autopolyploidy caused higher amount of variation compared with the diploid, a corresponding higher coverage is needed to obtain well-supported SNP calls with >5 supporting reads.
Of the 17,846 blueberry SNPs evaluated for mapping in this study, 785 (4.4 %) were mapped in ‘Draper’ and 450 SNPs (2.5 %) were mapped in ‘Jewel.’ Although a large number of markers have been excluded in this study, such loses are in line with previous studies and we decided that priority should be given to the quality of markers used over the quantity. In sweet cherry, of 8476 SNPs selected for mapping, 443 SNPs (5 %) were mapped in the maternal parent ‘Rainier,’ and 474 SNPs (5.6 %) in the paternal parent, ‘Riverdel’ (Guajardo et al. 2015). In grape, of 16,833 SNPs, linkage maps were constructed with 1146 SNPs (6.8 %) in V. rupestris B38 and 1215 SNPs (7.2 %) in ‘Chardonnay,’ while in black raspberry 399 (5 %) of the 7911 SNPs were mapped in the parents (Bushakra et al. 2015).
All four parental homologues were identified across four linkage groups in ‘Draper’ while a further five linkage groups recovered three homologues (Table S2). In ‘Jewel’ however, only three homologues could be resolved in four linkage groups with eight more recovering only two (Table S2). We are not aware of any group calling allele dosage in a tetraploid crop using genotyping by sequencing. A recent study by Rocher et al. (2015) in tetraploid alfalfa (Medicago sativa) found only 23 % of SNP loci were deemed as having reliable genotype calls in 50 % of plant samples but sequencing depth was not sufficient for tetraploid allele dosage. A sequencing depth of between 60 and 80× has been estimated as appropriate in heterogeneous cultivars of tetraploid potato in order to establish reliable allele dosage (Uitdewilligen et al. 2013).
Due to the small population size, only tentative linkages were assigned in this study and best alignments reported between parents were based on this. It will be necessary to repeat this study in a larger population of at least 275 seedlings to obtain greater confidence in identifying markers which are truly duplex, which would give an increased power to detect linkages. Another area for improvement is the number of markers required to cover all 96 chromosomes in blueberry. With one simplex marker per 5 cM and assuming an average chromosome length of ~100 cM, would require a total of 1920 simplex markers. Then, in order to align homologous chromosomes together, a further 2 × 12 sets of duplex markers (480 in total) would give an overall requirement of ~2500 markers. Only next generation sequencing technologies could supply these and we have demonstrated their potential in this study.
The linkage map developed for ‘Draper’ and ‘Jewel’ required the power of both TetraploidMap and JoinMap 4 to order markers correctly. There were too many markers in this dataset to analyze using TetraploidMap in isolation as in its current format TetraploidMap cannot handle large enough datasets for all analyses required (Hackett et al. 2007).
Traditional blueberry breeding has been complicated by a relatively long juvenile period (approximately 3 years) and polyploid nature of the plant as well as by severe inbreeding depression preventing the development of inbred lines (Die and Rowland 2013). Phenotypic selection of blueberry is often complicated by incomplete expression of traits through a combination of environmental factors including: climate, soil type and abiotic and biotic pressures (Collard et al. 2005; Winter and Kahl 1995). Blueberry breeding programmes would benefit from the further development of genomic and genetic resources such as the map developed in this study, to enable identification of genetic markers for key breeding traits. Such markers will then be available for implementation of marker-assisted breeding (MAB), as is currently being applied by breeders of other woody perennial fruit crops. MAB is particularly important to improve the effectiveness of selection of parental plants for crosses and for early generational seedling selection, to eliminate offspring with undesirable traits before they are field planted. A key advantage of MAB is that selection based on molecular markers is not affected by the environment or by the developmental stage of plants.
Generation of a genetic linkage map is a critical step toward providing the framework for knowledge-based crop improvement. Traits that are difficult to phenotype, such as the chilling requirement and cold tolerance of individual progeny could be more accurately predicted with the allocation of appropriate markers. Marker-assisted breeding is a powerful aid to in genetic improvement particularly when combining traits like such as adaptation and season extension with other significant traits including fruit quality and pest and disease resistance. Woody perennials, like blueberry, are especially suitable for improvement via marker-assisted breeding because of their long generation times, high heterozygosity, self-incompatibility, inbreeding depression, and polyploidy of commercial types (Russell et al. 2014; Barba et al. 2014; Guajardo et al. 2015) and this study is a step towards achieving this goal in blueberry.
References
Al-Janabi SM, Honeycutt RJ, McClelland M, Sobral BWS (1992) A genetic linkage map of Saccharum spontaneum L. ‘SES 208’. Genetics 134:1249–1260
Barba P, Cadle-Davidson L, Harriman J, Glaubitz JC, Brooks S, Hyma K, Reisch B (2014) Grapevine powdery mildew resistance and susceptibility loci identified on a high-resolution SNP map. Theor Appl Genet 127:73–84
Bielenberg DG, Rau B, Fan S, Gasic K, Abbott A.G, Reighard GL, Okie WR, Wells CE (2015) Genotyping by sequencing for SNP-based linkage map construction and QTL analysis of chilling requirement and bloom date in peach [Prunus persica (L.) Batsch]. PLoS ONE 10(10):e0139406
Boches PS, Bassil NV, Rowland LJ (2005) Microsatellite markers for Vaccinium from EST and genomic libraries. Mol Ecol Notes 5:657–660
Bradshaw JE, Hackett CA, Pande B, Waugh R, Bryan GJ (2008) QTL mapping of yield, agronomic and quality traits in tetraploid potato (Solanum tuberosum subsp. tuberosum). Theor Appl Genet 116:193–211
Brazelton C (2013) World Acreage and Production. North American Blueberry Council. http://www.chilealimentos.com/2013/phocadownload/Aprocesados_congelados/nabc_2012-world-blueberry-acreage-production.pdf. Accessed 9 Feb 2015
Bushakra JM, Bryant DW, Dossett M, Vining KJ, Van Buren R, Gilmore BS, Lee J, Mockler TC, Finn CE, Bassil NV (2015) A genetic linkage map of black raspberry (Rubus occidentalis) and the mapping of Ag4 conferring resistance to the aphid Amphorophora agathonica. Theor Appl Genet 128(8):1631–1646
Butruille DV, Boiteux LS (2000) Selection–mutation balance in polysomic tetraploids: impact of double reduction and gametophytic selection on the frequency of subchromosomal localization of deleterious mutations. Proc Natl Acad Sci USA 97:6608–6613
Camp WH (1945) The North American blueberries with notes on other groups of Vacciniaceae. Brittonia 5:203–275
Castro P, Stafne ET, Clark JR, Lewers KS (2013) Genetic map of the primocane-fruiting and thornless traits of tetraploid blackberry. Theor Appl Genet 126(10):2521–2532
Collard BCY, Jahufer MZZ, Brouwer JB, Pang ECK (2005) An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: the basic concepts. Euphytica 142:169–196
Dhanaraj AL, Slovin JP, Rowland LJ (2004) Analysis of gene expression associated with cold acclimation in blueberry floral buds using expressed sequence tags. Plant Sci 166:863–872
Die JV, Rowland LJ (2013) Superior cross-species reference genes: a blueberry case study. PLoS ONE 8(9):e73354
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6(5):e19379
Gallais A (2003) Quantitative genetics and breeding methods in autopolyploid plants. INRA Editions, Paris
Gar O, Sargent DJ, Tsai CJ, Pleban T, Shalev G, Byrne DH, Zamir D (2011) An Autotetraploid Linkage Map of Rose (Rosa hybrida) Validated Using the Strawberry (Fragaria vesca) Genome Sequence. PloS ONE 6(5):e20463
Gardner KM, Brown P, Cooke TF, Cann S, Costa F, Bustamante C, Velasco R, Troggio M, Myles S (2014) Fast and cost-effective genetic mapping in apple using next-generation sequencing. G3: Genes|Genomes|Genet 4(9):1681–1687
Gilmore BS, Bassil NV, Hummer KE (2011) DNA extraction protocols from dormant buds of twelve woody plant genera. J Am Pomol Soc 65:201–207
Guajardo V, Solis S, Sagredo B, Gainza F, Munoz C, Gasic K, Hinrichsen P (2015) Construction of high density sweet cherry (Prunus avium L.) linkage maps using microsatellite markers and SNPs detected by genotyping-by-sequencing (GBS). PLoS ONE 10(5):e0127750
Hackett CA, Luo ZW (2003) TetraploidMap: construction of a linkage map in autotetraploid species. J Hered 94:358–359
Hackett CA, Bradshaw JE, Meyer RC, McNicol JW, Milbourne D, Waugh R (1998) Linkage analysis in autotetraploid species: a simulation study. Genet Res Camp 71:143–154
Hackett CA, Bradshaw JE, McNicol JW (2001) Interval mapping of QTLs in autotetraploid species. Genetics 159:1819–1832
Hackett CA, Milne I, Bradshaw JE, Luo ZW (2007) TetraploidMap for Windows: linkage map construction and QTL mapping in autotetraploid species. J Hered 98:727–729
Hangun-Balkir Y, McKenney L (2012) Determination of antioxidant activities of berries and resveratrol. Green Chem Lett Rev 5(2):147–153
Johnson SA, Arjmandi BH (2013) Evidence for anti-cancer properties of blueberries: a mini review. Anticancer Agents Med Chem 13(8):1142–1148
Julier B, Flajoulot S, Barre P, Cardinet G, Santoni S, Huguet T, Huyghe C (2003) Construction of two genetic linkage maps in cultivated tetraploidalfalfa (Medicago sativa) using microsatellite and AFLP markers. BMC Plant Biol 3:9
Kalt W, Joseph JA, Shukitt-Hale B (2007) Blueberries and human health. A review of the current research. J Am Pomol Soc 61:151–160
Lu F, Lipka AE, Glaubitz J, Elshire R, Cherney JH, Casler MD, Buckler ES, Costich DE (2013) Switchgrass genomic diversity, ploidy, and evolution: novel insights from a network-based SNP discovery protocol. PLoS Genet 9(1):e1003215
Luo ZW, Hackett CA, Bradshaw JE, McNicol JW, Milbourne D (2000) Predicting parental genotypes and gene segregation for tetrasomic inheritance. Theor Appl Genet 100:1067–1073
Luo ZW, Hackett CA, Bradshaw JE, McNicol JW, Milbourne D (2001) Construction of a genetic linkage map in tetraploid species using molecular markers. Genetics 157:1369–1385
McCormack D, McFadden D (2013) A review of Pterostilbene antioxidant activity and disease modification. Oxid med Cell Longev 2013:575482
Megalos BS, Ballington JR (1988) Unreduced pollen frequencies versus hybrid production in diploid- tetraploid Vaccinium crosses. Euphytica 39:271–278
Meyer RC, Milbourne D, Hackett CA, Bradshaw JE, McNicol JW, Waugh R (1998) Linkage analysis in tetraploid potato and association of markers with quantitative resistance to late blight (Phytophthora infestans). Mol Gen Genet 259:150–160
Nielsen R, Paul JS, Alberechtsen A, Song YS (2011) Genotype and SNP calling from next generation sequencing data. Nat Rev Genet 12:443–451
Qu L, Hancock JF, Whallon JH (1998) Evolution in an autopolyploid group displaying predominantly bivalent pairing at meiosis: genomic similarity of diploid Vaccinium darrowii and autotetraploid Vaccinium corymbosum (Ericaceae). Am J Bot 85:698–703
Rimando AM, Kalt W, Magee JB, Dewey J, Ballington JR (2004) Resveratrol, pterostilbene, and piceatannol in Vaccinium berries. J Agric Food Chem 52(15):4713–4719
Rocher S, Jean M, Castonguay Belzile F (2015) Validation of genotyping by sequencing analysis in populations of tetraploid Alfalfa by 454 sequencing. PLoS ONE 10(6):e0131918
Rodriguez-Mateos A, Rendeiro C, Bergillos-Meca T, Tabatabaee S, George TW, Heiss C, Spencer JPE (2013) Intake and time dependence of blueberry flavonoid-induced improvements in vascular function: a randomized, controlled, double-blind, crossover intervention study with mechanistic insights into biological activity. Am J Clin Nutr 98:1179–1191
Rowland LJ, Mehra S, Dhanaraj AL, Ogden EL, Arora A (2003) Identification of molecular markers associated with cold tolerance in blueberry. Acta Hort 625:59–69
Rowland LJ, Dhanaraj AL, Naik D, Alkharouf N, Matthews B, Arora R (2008) Study of cold tolerance in blueberry using EST libraries, cDNA microarrays, and subtractive hybridization. HortSci 43:1975–1981
Rowland LJ, Bell D, Alkharouf N, Bassil N, Drummond F, Beers L, Buck E, Finn C, Graham J, McCallum S, Hancock J, Polashock J, Olmstead J, Main D (2012) Generating genomic tools for blueberry improvement. J Fruit Sci 12:276–287
Rowland LJ, Ogden EL, Bassil N, Buck EJ, McCallum S, Graham J, Brown A, Wiedow C, Campbell AM, Haynes KG, Vinyard BT (2014) Construction of a genetic linkage map of an interspecific diploid blueberry population and identification of QTL for chilling requirement and cold hardiness. Mol Breed 34:2033–2048
Russell J, Hackett C, Hedley P, Liu H, Milne L, Bayer M, Marshall D, Jorgensen L, Gordon S, Brennan R (2014) The use of genotyping by sequencing in blackcurrant (Ribes nigrum): developing high-resolution linkage maps in species without reference genome sequences. Mol Breed 33:835–849
Shuelke M (2000) An economic method for the fluorescent labelling of PCR fragments. Nat Am Inc 18:233–234
Soltis DE, Soltis PS (1993) Molecular data and the dynamic nature of polyploidy. Crit Rev Plant Sci 12:243–273
Uitdewilligen J, Wolters AM, D’Hoop B, Borm T, Visser R, Eck H (2013) A next generation sequencing method for genotyping-by-sequencing of highly heterozygous autotetraploid potato. PLoS ONE 8(5):e62355
Van Ooijen JW (2006) JoinMap ® 4; Software for the calculation of genetic linkage maps in experimental populations. Kyazma BV, Wageningen
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Ward JA, Bhangoo J, Fernández-Fernández F, Moore P, Swanson JD, Viola R, Velasco R, Bassil N, Weber CA, Sargent DJ (2013) Saturated linkage map construction in Rubus idaeus using genotyping by sequencing and genome-independent imputation. BMC Genom 14:2
Welch JE (1962) Linkage in autotetraploid maize. Genetics 47:367–396
Winter P, Kahl G (1995) Molecular marker technologies for plant improvement. World J Microbiol Biotechnol 11:438–448
Wu KK, Burnquist W, Sorrells ME, Tew TL, Moore PH, Tanskley SD (1992) The detection and estimation of linkage in polyploids using single-dose restriction fragments. Theor Appl Genet 83:294–300
Wu R, Gallo-Meager M, Littell RC, Zeng ZB (2001) A general polyploidy model for analyzing gene segregation in outcrossing tetraploid species. Genetics 159:869–882
Acknowledgments
This work was funded through Horticulture Link (HL0190), and all contributing partners are gratefully acknowledged. Project collaboration with the Specialty Crop Research Initiative-funded project (Grant 2008-51180-04861 entitled ‘Generating genomic tools for blueberry improvement’) has provided valuable access to plant material, genetic resources and advice. Support for this work from the Scottish Government’s Rural and Environment Science and Analytical Services Division (RESAS) is gratefully acknowledged. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the James Hutton Institute or any of the other agencies involved in this research. We thank all reviewers for their comments and Sue Gardiner for assisting in preparation of this ms.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
McCallum, S., Graham, J., Jorgensen, L. et al. Construction of a SNP and SSR linkage map in autotetraploid blueberry using genotyping by sequencing. Mol Breeding 36, 41 (2016). https://doi.org/10.1007/s11032-016-0443-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-016-0443-5