
Abstract
An F2 population was developed from a cross between a mur-cytoplasmic male sterile broccoli line and a restorer Chinese kale line. Phenotypic analysis of F2 plants indicated that the pollen fertility is controlled by two genes and segregated in a duplicate gene interaction mode with a ratio of 15:1. A total of 236 single nucleotide polymorphism (SNP) markers were developed utilizing 1,448 primers designed for production of expressed sequence tag (EST)-SNP markers of Raphanus sativus and analyzed by the dot-blot technique in 205 F2 individuals. A linkage map was constructed with a total of 142 markers and these markers were assigned to nine linkage groups together with simple sequence repeat markers mapped previously on the published linkage maps of Brassica oleracea. The linkage map spanned 909 cM with an average marker distance of 6.4 cM. A fertility restorer locus (Rfm1) was mapped on LG1, corresponding to chromosome 3, along with a flower color locus at a distance of 25 cM. SNP markers flanking the Rfm1 locus were BoCL2642s at a distance of 2.5 cM on one side and BoCL2901s at a distance of 7.5 cM on the other side. All the SNP markers showed homology with Arabidopsis thaliana and Brassica rapa genome sequences. Three pentatricopeptide repeat genes of the P-subfamily, particularly expressed in buds of the restorer line, were identified and these genes could be potential candidate fertility restorer genes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The family Brassicaceae is an economically important source of vegetables, oilseeds and forages. Brassica oleracea is one of the major species in this family, which includes broccoli, cauliflower, cabbage and kale. Commercial hybrid seed production of B. oleracea vegetables is performed using self-incompatibility, but sometimes its instability results in contamination of selfed seeds in hybrid seeds. Cytoplasmic male sterility (CMS) together with fertility restoration is a reliable hybrid seed production tool in crop species and also an excellent model for studying nuclear–cytoplasmic gene interaction. CMS is the maternally inherited inability of plants to produce functional pollen and is associated with the expression of novel chimeric open reading frames (ORFs) encoded by the mitochondrial genome. The chimeric ORFs differ among the CMS systems, but often carry a recognizable segment of coding or flanking sequences of essential mitochondrial genes. The nuclear genes that suppress or compensate for mitochondrial dysfunction and restore fertility to CMS plants are designated fertility restorer (Rf) genes. The CMS-Rf system is commercially used for hybrid seed production of Brassica napus, Brassica juncea, onion, etc.
In Brassicaceae, several different cytoplasmic male sterile lines have been identified and these can be grouped under two categories, alloplasmic and autoplasmic. CMS lines have been developed in B. napus, B. rapa and B. juncea, and four sources of Brassica CMS, viz., ogu/kosena, pol, nap and tour, have been well characterized at the molecular level (Schnable and Wise 1998). In each case, a novel open reading frame has been identified in transcripts of normal mitochondrial genes. Identification of a new CMS line and a restorer gene is always beneficial as a new source material for hybrid breeding. Each restorer gene is unique in nature and molecular study helps to elucidate the mechanism of gene action. A Diplotaxis muralis-based CMS line of B. oleracea (termed here mur CMS B. oleracea) has been developed previously and a novel open reading frame, orf72, associated with male sterility has been identified (Shinada et al. 2006).
For most alloplasmic CMS systems, fertility restorer genes are required to be introduced from cytoplasm donor species. Restoration is mostly governed by a single restorer gene in all the known Brassica CMS-Rf systems except tour CMS B. napus, in which two dominant genes, Rft1 and Rft2, have been reported for fertility restoration (Janeja et al. 2003). Even a single restorer gene has been found to restore pollen fertility in two different CMS Brassica lines developed from different cytoplasms (Bhat et al. 2005). A number of restorer genes have been cloned in maize, petunia, radish, rice and sorghum, and all except rf2 in maize (Cui et al. 1996), Rf17 of CW-CMS rice (Fujii and Toriyama 2005) and Rf2 in Lead rice CMS (Itabashi et al. 2011) encode pentatricopeptide repeat (PPR) proteins targeted at mitochondria (Bentolila et al. 2002; Brown et al. 2003; Desloire et al. 2003; Koizuka et al. 2003; Kazama and Toriyama 2003; Klein et al. 2005). CMS restorer genes belong to the P-subfamily of PPR genes, which are categorized based on their C-terminal domain structure (Small and Peeters 2000; Lurin et al. 2004). All restorer genes cloned to date have been identified by a map-based cloning approach, in which the first step is to find DNA markers linked with a fertility restorer locus followed by fine mapping for positional cloning of the gene.
The objective of the present study was molecular mapping of a male fertility restorer locus of mur CMS B. oleracea and analysis of an Arabidopsis thaliana homologous region for identification of genes encoding PPR proteins. A linkage map was constructed using expressed sequence tag (EST)-based single nucleotide polymorphism (SNP) markers based on an F2 population derived from a CMS line and a restorer line of B. oleracea. A total of 142 loci were assigned to nine linkage groups (LGs) with a total coverage of 909 cM. A fertility restorer locus (Rfm1) was mapped on LG1, corresponding to chromosome 3 of B. oleracea, along with a flower color gene. Flanking EST-SNP markers for Rfm1 were identified covering a region of 10 cM. Based on sequence homology of a region between the markers flanking the Rfm1 locus with the genome sequences of Arabidopsis thaliana and Brassica rapa, three genes encoding PPR proteins could be identified. These genes are expressed only in buds of the fertility restorer line and are potential candidate genes for male fertility restoration.
Materials and methods
Plant materials
A CMS line of alloplasmic origin in Brassica rapa has been developed by substituting the nucleus of the wild species Diplotaxis muralis with the nucleus of B. rapa var. chinensis through repeated backcrossings of an intergeneric hybrid of D. muralis × B. rapa with B. rapa as a pollinator for eight generations (Hinata and Konno 1979). Similarly a CMS line in broccoli (B. oleracea var. italica) harboring D. muralis cytoplasm has also been developed by N. Konno and K. Hinata (unpublished), named mur CMS B. oleracea. A male fertility restorer line has been developed in Chinese kale (B. olearcea var. alboglabra) by backcrossing with another male semi-sterile alloplasmic line of B. oleracea having D. muralis cytoplasm (Shinada et al. 2006). A segregating F2 population generated from a cross between CMS broccoli and restorer Chinese kale was used for genotyping.
Phenotyping and genomic DNA extraction
A segregating F2 population was grown in a greenhouse from July 2009 to March 2010 in Sendai, Japan. Pollen fertility and sterility of 205 plants were investigated at the flowering stage through visual examination of pollen grain dust and stainability with 2 % acetocarmine. Seed setting ability was also examined after self-pollination in some plants. Based on pollen grain fertility, plants were categorized as male fertile (male fully fertile and male semi-fertile) or male sterile, and analyzed in order to decipher the genetics of fertility restoration. The F2 population also showed segregation for white and yellow flower color, and phenotypic data were obtained for molecular mapping of a flower color gene. Genomic DNA was extracted from young leaves of all the individuals using the modified CTAB protocol (Doyle and Doyle 1990). The DNA was quantified by ethidium bromide staining after electrophoresis on agarose gel and used as a PCR template in genotyping of the F2 population with EST-SNP and simple sequence repeat (SSR) markers.
Development of EST-based SNP markers and detection of polymorphism
The EST sequences of radish published on the radish sequence database (http://radish.plantbiology.msu.edu) have been explored to design primer pairs for specific amplification of genes and to identify SNPs for production of EST-based SNP markers in Raphanus sativus (Li et al. 2011). Since R. sativus and B. oleracea belong to the same tribe Brassiceae, these radish primer sequences were utilized for production of EST-based SNP markers in B. oleracea. These primers were used in PCR amplification from genomic DNA of the parental lines, i.e., CMS broccoli and restorer Chinese kale. A 20-μl reaction mixture contained 10 ng of plant genomic DNA, 10 pmol of each primer, 1× ExTaq buffer, 2 nmol of each dNTP, and 0.5 U of Taq DNA polymerase (ExTaq, Takara Biomedicals, Japan). PCR was performed in a thermal cycler (Eppendorf) with the following cycling conditions: initial denaturation at 94 °C for 5 min, 40 cycles of 94 °C for 30 s, 56 °C for 30 s and 72 °C for 1 min. Five microliters of the PCR product were electrophoresed on 1.2 % agarose gel, and amplified fragment sizes ranged from 300 to 600 bp. Single fragment PCR products were selected and sequenced by the Sanger method. High quality sequences were aligned and analyzed to identify SNPs using SEQUENCHER version 4.7 software (Gene Codes Corporation, MI, USA).
Sequences having SNPs between CMS broccoli and restorer Chinese kale were used for designing probes for dot-blot-SNP analysis, which is a cost-effective and highly efficient SNP analysis method (Shiokai et al. 2010a). For each marker, a set of two oligonucleotide probes of 48 nucleotides, comprising a sequence of 17 nucleotides with an SNP in the middle, a 6-nucleotide spacer and a bridge sequence of 25 nucleotides, were designed. The hybridization conditions for the probes were predicted using DINAMelt web server (http://www.bioinfo.rpi.edu/applications/hybrid/) and slight modifications were made in hybridization temperature or salt concentration to achieve optimum dot-blot results as described by Shiokai et al. (2010b). Genomic regions having SNPs were amplified by multiplex PCR using five or six primer pairs mixed together based on annealing temperature and other parameters (Kaplinski et al. 2005). The PCR reaction was set up in a 10-μl volume consisting of 10 ng template DNA, 10 pM of each primer, 1× KAPATaq buffer (without MgCl2), 1.75 mM MgCl2, 0.25 mM of each dNTP and 0.25 U of KAPATaq DNA polymerase (Kapa Biosystems, Boston, MA, USA). The PCR conditions were initial denaturation at 94 °C for 4 min, 35 cycles of 94 °C for 30 s, 56 °C for 30 s and 72 °C for 1 min, and final extension at 72 °C for 5 min. PCR-amplified products were denatured by mixing an equal volume of denaturation solution containing 0.4 N NaOH and 10 mM EDTA before blotting onto nylon membrane by Multi-pin Blotter (ATTO, Japan). SNP detection was performed using the dot-blot-SNP analysis procedure (Shiokai et al. 2010a), except that the washing and hybridization temperature were changed as per the probe.
SSR marker analysis
SSR markers from published literature (Supplementary Table 1) were screened for polymorphism between parental genotypes to select potential anchor markers for the B. oleracea genetic map. PCR amplification of SSR markers was performed using the reaction conditions given in the reference data of the markers. The amplified products were resolved on 8 % polyacrylamide gel in 1× TBE buffer and visualized under UV after staining with ethidium bromide. Markers polymorphic between the parents were used for genotyping of F2 plants to assign linkage groups to those of the reference linkage map (Lowe et al. 2004; Piquemal et al. 2005; Iniguez-Luy et al. 2008). Two SCAR markers, M3.4 and Nit-2, were also analyzed on 3 % agarose gel (Supplementary Table 1).
Linkage analysis and map construction
Linkage analysis and map construction were performed using JoinMap 4.0 software (Van Ooijen 2006) and linked loci were grouped with independent LOD parameter into nine linkage groups. The marker order was confirmed by a regression mapping algorithm on the basis of a minimum LOD score of 1.0 and a maximum recombination fraction of 0.4. The Kosambi map function was used to estimate genetic distances in cM (Kosambi 1944). The EST-based SNP markers were named <Bo> <EST name> <s> and sequence characterized amplified region (SCAR) markers were designated with <c> at the end, with names following the international nomenclature (De Vincente et al. 2004).
Sequence comparison with Arabidopsis thaliana and Brassica rapa genomes
The sequences of EST-based SNP loci on a linkage map were aligned with the genome sequences of A. thaliana (TAIR; http://www.arabidopsis.org) and B. rapa (Wang et al. 2011) using the BLAST tool of the Brassica database BRAD (http://brassicadb.org/brad) (Feng et al. 2011), and homologous regions were searched. The E value, a statistical significance threshold for reporting matches against database sequences, was set at 0.01 for sequence alignment. The sequences of marker loci were regarded as homologous to the genomes with a threshold value of E < 10−10. Regions having at least three loci with conserved collinearity with A. thaliana and B. rapa were considered to be homologous syntenic regions. A single non-collinear homolog in the syntenic region was ignored.
Identification of PPR genes and expression analysis
EST-SNP markers flanking Rfm1 showed sequence collinearity with the B. rapa and A. thaliana genome sequences. A selected syntenic region of the A. thaliana sequence was searched for PPR genes in the browser (http://www.plantenergy.uwa.edu.au/gb2/gbrowse/atbrowser/) and primers were designed based on PPR-encoding genes of A. thaliana (O’Toole et al. 2008). A homologous B. rapa sequence region was also analyzed by GENSCAN (http://genes.mit.edu/GENSCAN.html/) to predict genes, and deduced amino acid sequences were aligned with known sequences of A. thaliana proteins using the BLASTP program of NCBI. Predicted peptides having high similarity to A. thaliana PPR proteins were identified. Genes encoding PPR proteins were selected to design specific primers for PCR (Supplementary Table 5). Genomic DNAs of CMS broccoli and restorer Chinese kale were used as templates in PCR amplification. The 20-μl reaction mixture contained 10 ng of plant genomic DNA, 10 pmol of each primer, 1× ExTaq buffer, 2.5 nmol of each dNTP and 0.5 U of Taq DNA polymerase (ExTaq, Takara Biomedicals, Japan). PCR was performed in a thermal cycler with the following cycling conditions: initial denaturation at 94 °C for 4 min, 40 cycles of 94 °C for 30 s, annealing temperature (as given in Supplementary Table 5) for 30 s and 72 °C for 45 s. PCR products were resolved on 1.5 % agarose gel and visualized under UV after staining with ethidium bromide.
RNA was extracted from young leaves and buds of CMS broccoli and restorer Chinese kale using SV Total RNA Isolation System (Promega Corp.) according to the manufacturer’s instructions. First strand cDNA was synthesized from 1 μg total RNA using Pd(N)6 primer and reverse transcriptase of the first strand cDNA synthesis kit (GE Healthcare, UK). RT-PCR was performed using PPR gene-specific primers with the same PCR conditions as used in amplification of genomic DNA. The actin gene primer set was used as a positive control of RT-PCR. The amplified products were separated on 1.5 % agarose gel and visualized under UV after staining with ethidium bromide.
Results
Phenotypic analysis of pollen fertility
An F2 population segregating for pollen fertility was used for linkage map construction and gene mapping. Phenotypic differences were clearly distinguishable between fully fertile and sterile flowers, as the sterile flowers had comparatively short filaments with stunted anthers having a small quantity of pollen, whereas fully fertile flowers had long filaments positioned above a stigma and a high quantity of pollen (Fig. 1). The semi-fertile flowers had medium size filaments and a comparatively smaller quantity of pollen than fertile ones. Pollen grain stainability showed a clear difference between semi-fertile and fertile, which was reconfirmed by visual examination. The pollen grain fertility was analyzed in 205 individuals, and 191 plants were classified as fertile (170 full fertile and 21 semi-fertile) and 14 as sterile. The phenotypic segregation fitted well with the 15:1 ratio (χ2 value 0.117), revealing that fertility restoration was controlled by two genes under the duplicate gene interaction mode (Table 1). The genetic mode of fertility restoration was sporophytic and all the plants in F1 progeny were fully fertile. In the F2 population, there were 153 white flower plants and 52 yellow flower plants, suggesting that white petal color (from Chinese kale) was dominant over yellow petal color (broccoli) and is under monogenic control (segregation ratio 3:1). In this segregation analysis, we were unable to detect linkage between the genes for fertility restoration and a flower color gene. Reciprocal crosses made between B. oleracea cultivars of yellow petal color and B. oleracea var. alboglabra of white petal color yielded the identical results, hence confirming that the inheritance of flower color is not influenced by the cytoplasm.

Flowers of Diplotaxis muralis: a male sterile, b semi-fertile and c fertility-restored B. oleracea
SNP analysis and molecular mapping of a restorer gene
A total of 1,448 primer pairs derived from radish EST sequences were used in PCR amplification of DNAs from parental lines and 729 (~50 %) of these primer pairs yielded single band amplification. Out of these, 720 amplified products were sequenced and data showing a sequence quality score >90 % were analyzed for SNP identification. In 633 DNA fragments, sequence data were obtained from both CMS broccoli and restorer Chinese kale and aligned properly, while 87 sequences were obtained either from one of the parental lines or were poorly aligned. The aligned sequence data of 633 fragments covering ~300 kb showed 1,113 nucleotide variations between the parental lines. A total of 236 fragments having SNPs were identified by comparing parental sequences. The frequency of variable bases, i.e., SNPs and indels, was 1/268 bp and the frequency of SNP was 1/325 bp (Table 2). Sequences having SNPs between the parental lines were used for designing probes of SNP markers. SNPs of 204 markers which have been developed for quantitative trait locus analysis using an F2 population obtained from a cross between cabbage and broccoli (Kifuji et al. unpublished) were also screened and 48 SNP markers were found to be polymorphic between CMS broccoli and restorer Chinese kale. Among 122 SSR markers surveyed for polymorphism, 15 single-locus SSR markers covering all the linkage groups showed segregation in the F2 population.
In total, 163 markers segregated in the 205 F2 plants genotyped. By linkage analysis with JoinMap 4.0 software, 142 markers (125 EST-SNPs, 15 SSR, one SCAR and one flower color) could be assigned to nine linkage groups, designated LG1–LG9, while the others remained ungrouped. Primer sequences, probe sequences and hybridization conditions of the mapped SNP markers are given in Supplementary Table 2. One to three SSR markers were assigned to each linkage group and on this basis the linkage groups were designated C1–C9 according to the reference linkage map of B. oleracea (Fig. 2). The linkage map spanned 909 cM with an average distance between markers of 6.4 cM, a minimum distance of 0.2 cM and a maximum distance of 23 cM. The largest linkage group (LG1) comprised 30 markers and had a length of 182.4 cM, while the smallest group (LG9) had eight loci with 44.6 cM. Based on the estimated physical length of 596 Mb in B. olercaea (Johnston et al. 2005), the average physical distance between markers for this map is estimated to be 4.9 Mb (estimated 1 cM = 766 kb).

Genetic linkage map of B. oleracea based on EST-SNP markers with comparative maps of A. thaliana and B. rapa. The linkage groups are labeled as LG1–LG9 in order of length and their correspondence to the C genome chromosome (C1–C9). Marker positions (in cM) are shown on the left-hand side with the corresponding marker names on the right-hand side of each LG. Each locus was tested for homology with A. thaliana and B. rapa and is represented in the horizontal bar within LGs (colored according to Parkin et al. 2005) and verticals bars shown to the right of LGs (colored according to the key at the bottom of the figure) for A. thaliana and B. rapa chromosomes respectively. FC on LG1 represents flower color
Linkage analysis of phenotypic data by JoinMap 4.0 assigned the Rfm1 locus to LG1 (chromosome 3) of B. oleracea. We were unable to locate the exact position of another restorer locus (Rfm2) on the linkage map, but, comparing segregation distortion for markers in sterile and semi-fertile plants, it seemed to be positioned on LG3 between markers BoCL6818s and BoCL7968s. SNPs flanking the Rfm1 locus were BoCL2642s on one side and BoCL2901s on other side at a distance of 2.54 and 7.48 cM, respectively. The flower color locus was also assigned to LG1 with a distance of 25 cM from Rfm1 locus. The closest marker to the flower color locus was BoCL3107s, which was identified at a distance of 5.88 cM.
Syntenic relation with A. thaliana and B. rapa
All the EST-SNP markers used in the present study showed homology with A. thaliana and B. rapa genome sequences in BLAST analysis with the BRAD software. There were 115 loci homologous to A. thaliana at a significance threshold E < 10−10, distributed over all the nine linkage groups. Collinearity was interrupted by the presence of markers showing homology to other regions of the B. oleracea map at a significant E value. Matching nucleotide lengths, E values and chromosome names are given in Supplementary Table 3. Based on criteria of three or more (continuous) collinear markers, we identified 11 regions syntenic with the genome of A. thaliana. LG8 was completely collinear with chromosome 5 of A. thaliana, whereas LG1 and LG3 were homologous to a 55-cM region and a 40-cM region of A. thaliana chromosome 5, respectively. LG7 had segmental homology to all five chromosomes of A. thaliana. LG9 was homologous to chromosome 1 of A. thaliana. Homology of each locus to the B. rapa genome sequence was searched for by the BLAST tool of BRAD for the recently published B. rapa genome sequence (Wang et al. 2011) and a total of 24 syntenic segments were identified by comparison with our linkage map of B. oleracea based on the criteria of three or more (continuous) collinear markers. LG1 had four collinear regions, three of which were collinear with chromosome 3 of B. rapa. LG2 (C6), LG4 (C1), LG5 (C8) and LG8 (C2) showed synteny with B. rapa chromosomes 7, 1, 9 and 2, respectively (Fig. 2).
Identification of PPR genes in the syntenic region and expression analysis
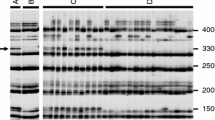
The EST-SNP markers BoCL2901s and BoCL2642s flanking Rfm1 showed sequence homology with regions of 608,752–609,089 and 2,439,089–2,439,370 bp, respectively, on chromosome 3 of A. thaliana and homology with regions of 14,577,244–14,577,510 and 15,565,034–15,565,334 bp, respectively, on chromosome 3 of B. rapa. The homologous segment of A. thaliana (~3 Mb of chromosome 3) contained 18 genes encoding PPR proteins. Nine of them were P-subfamily PPR genes, which encode PPR proteins targeted to mitochondria or plastid (Supplementary Table 4). The region of B. rapa c. 1 Mb in size was analyzed by GENSCAN and a total of 216 genes were predicted. These were searched to detect identity with A. thaliana PPR proteins by the BLASTP program and the deduced amino acid sequences showed identities with 16 PPR proteins. The highest identity (87 %, E value 0) was between the B. rapa predicted peptide and AtPPR_3g06920. Interestingly, AtPPR_3g06920 was one of the 18 PPR genes identified in the region of ~3 Mb on A. thaliana chromosome 3. In most cases, the CMS restorer genes encode PPR proteins of the P-subfamily targeted to mitochondria and these Rf PPR genes are present in clusters together with other non-restorer PPR genes (Brown et al. 2003; Koizuka et al. 2003; Barr and Fishman 2010). We utilized the A. thaliana sequence information for designing eight specific primer pairs for P-subfamily PPR genes. In PCR using a primer pair designed from the sequence of AtPPR_3g02490, two faint bands were amplified in parental genomes with slight variation in size. Amplified products having expected sizes were obtained from genomic DNAs of both CMS broccoli and restorer Chinese kale by the primer pairs of AtPPR_3g07290, AtPPR_3g06430, AtPPR_3g049650 and AtPPR_3g06920, whereas there was no amplification by the primer pairs of AtPPR_3g02650, AtPPR_3g04130 and AtPPR_3g09060 (Supplementary Table 5). PCR products of parental genotypes had no size polymorphism and showed monomorphic bands (Fig. 3).

Analysis of PPR genes. a PCR amplification of PPR genes in genomic DNA of CMS broccoli (St) and restorer Chinese kale (Fr). b, c Expression analysis of PPR genes using RT-PCR in buds and leaves of CMS and restorer plants. SB CMS bud; FB restorer bud; SL CMS leaf; FL restorer leaf; M 100-kb DNA ladder. The actin gene was used as a control
RT-PCR was performed using total RNA from leaves and buds of CMS broccoli and restorer Chinese kale. Using the primer pair of AtPPR_3g02490, two bands (approximately 500 and 700 bp) were obtained in the buds and leaves of the restorer line by RT-PCR and a single band was detected in the leaves of CMS lines, whereas there was no amplification in the CMS buds. The primer pair of AtPPR_3g07290 generated faint amplification of 500 bp in the buds of both the CMS and restorer lines. Using the primer pairs of AtPPR_3g06430 and AtPPR_3g06920, products having sizes of 700 and 350 bp, respectively, were amplified exclusively in the restorer buds with no amplification in the CMS buds nor in the leaves of the CMS and restorer lines. The primer pair of AtPPR_3g09650 generated a product of 350 bp in the restorer buds and faint amplification in the leaves of the CMS and restorer lines. RT-PCR was replicated and consistency of results was confirmed (Fig. 3).
Discussion
In most Brassica CMS systems, a single gene restores pollen fertility, e.g., pol CMS of B. napus (Fang and McVetty 1989) and mori CMS of B. juncea (Prakash et al. 1998), whereas fertility restoration in mur CMS B. oleracea was found to be under the control of two loci functioning in a duplicated mode of gene interaction. Janeja et al. (2003) have also reported two dominant genes, Rft1 and Rft2, required for fertility restoration in tour CMS B. napus, in which Rft1 alone can restore pollen fertility completely.
Developing a linkage map of DNA markers is a prerequisite for molecular mapping of important agronomic traits and facilitating marker-assisted breeding. Most of the restorer genes cloned so far have been identified by a map-based cloning approach, in which the first step is to find DNA markers linked with an Rf locus followed by fine mapping and synteny analysis with known genomes. Although the Rf2 gene of CMS T-maize encoding a mitochondrial aldehyde dehydogenase has been identified by a transposon tagging strategy (Cui et al. 1996), molecular tagging and fine mapping of restorer loci in other species, e.g., petunia, radish and rice, have been performed for the cloning of genes. We constructed a linkage map of B. oleracea using EST-based SNP markers with a segregating F2 population of CMS and restorer parents. In the primers designed from R. sativus EST sequences, 50 % primers yielded single band amplification in the parental lines, and 236 ESTs having SNPs (37 %) were identified. This result proved the transferability of EST information to other Brassicaceae species and the utility of resources in marker development. Rfm1 of mur CMS B. oleracea was mapped on LG1 (B. oleracea chromosome 3) with flanking markers at a distance 2.54 cM on one side and 7.48 cM on the other side. For the tour Brassica restorer gene, Trendelkamp et al. (1999) have identified 11 amplified fragment length polymorphism (AFLP) markers linked with the restorer gene, whereas Janeja et al. (2003) have found two AFLP markers by using near isogenic lines. Two AFLP markers and one close (0.6 cM) SCAR marker linked to the fertility restorer gene have been developed in mori CMS B. juncea (Ashutosh et al. 2007). Since AFLP markers could not be used directly for map-based cloning of genes, they must be converted to sequence-tagged markers, such as SCAR, cleaved amplified polymorphic sequence or SNP markers. Furthermore, AFLP markers frequently reside in intergenic regions, which is not suitable for synteny analysis. The present EST-SNP linkage map would be informative for identification and positional cloning of agronomically important genes in B. olercaea.
Chromosome 5 of A. thaliana showed synteny to the complete LG8 (B. oleracea chromosome 2), a 55-cM LG1 segment (B. oleracea chromosome 3) and a 40-cM LG3 segment (B. oleracea chromosome 9). Repeated long syntenic regions have been reported for B. oleracea corresponding to chromosomes 2, 3 and 5 of A. thaliana genome by comparing restriction fragment length polymorphism and EST sequences (Lan et al. 2000; Babula et al. 2003; Parkin et al. 2005) but not for the whole linkage group. We also found repeated long regions in B. oleracea chromosomes syntenic to A. thaliana chromosomes 2, 3 and 5, but a single syntenic region for chromosomes 1 and 4 of A. thaliana. Brassica species has 87 % sequence identity in coding regions with A. thaliana (Cavell et al. 1998) and some genes have counterparts. Other genes have no apparent counterparts, but synteny is preserved between Brassica and A. thaliana. Transcriptome mapping followed by homology analysis between B. oleracea and A. thaliana have revealed extensive collinearity of the genomes and duplication, mostly of chromosomes 1 and 5 of A. thaliana (Li et al. 2003). Furthermore, in the present study, sequence homology analysis identified 24 syntenic segments in the recently published B. rapa genome. LG1 had four collinear regions, three of which showed synteny with chromosome 3 of B. rapa. These results indicate the suitability of sequence-based markers for comparative genomic studies. Radish EST sequences have been utilized extensively to generate an SNP linkage map of R. sativus and comparative studies have revealed high homeology among Brassica species (Li et al. 2011).
A. thaliana-derived markers have been utilized in high-density mapping of the Rfp restorer locus of B. napus and have supported the extended collinearity between the B. napus Rfp region and an orthologous segment of A. thaliana genome, with a single exception (Formanova et al. 2010). Cloning of the restorer genes of the radish CMS systems Ogura and Kosena has been performed utilizing the close synteny between radish and A. thaliana genomes following a map-based approach, and Rf-encoded PPR genes have been revealed to be present in clusters together with other non-restorer PPR genes (Brown et al. 2003; Desloire et al. 2003; Koizuka et al. 2003). The flanked markers of the Rfm1 gene showed homology with the A. thaliana region containing 18 genes encoding PPR proteins and nine of these belong to the P-subfamily of the PPR genes. One of the PPR genes, AtPPR_3g06920, was also found in a syntenic B. rapa region around the Rfm1 locus. Analyzing syntenic genomic regions from A. thaliana and B. rapa, Geddy and Brown (2007) have shown that the location and direction of PPR genes are less conserved in collinear regions and often appear in different chromosomal contexts. PPR regions were aligned for a small region, but in the present studies we considered a large region of c. 3 Mb for analysis and found conserved PPR genes. Kato et al. (2007) have reported that the Rf-1 locus of rice contains several duplicated copies of the restorer gene. Furthermore, the gene order between clusters from different species is conserved, suggesting that the Rf1 locus may have been generated by homologous recombination. The PPR protein-encoding genes in B. oleracea, similar to AtPPR_3g06430, AtPPR_3g09650 and AtPPR_3g06920, which were deduced from the syntenic region of A. thaliana, showed gene expression exclusively in fertile buds. All three PPR genes belong to the P-subfamily category in A. thaliana. A target organelle of protein encoded by AtPPR_3g06920 is unknown, whereas AtPPR_3g06430 and AtPPR_3g09650 encode proteins targeted to plastids as per Predotar prediction (Lurin et al. 2004). Based on the findings of the present study, we consider these genes to be potential candidate restorer genes, but additional molecular analysis is required to confirm their relevance and functional role in fertility restoration.
References
Ashutosh, Sharma PC, Prakash S, Bhat SR (2007) Identification of AFLP markers linked to the male fertility restorer gene of CMS (Moricandia-arvensis) Brassica juncea and conversion to SCAR marker. Theor Appl Genet 114:385–392
Babula D, Kaczmarek M, Barakat A, Delseny M, Quiros CF, Sadowski J (2003) Chromosomal mapping of Brassica oleracea based on ESTs from Arabidopsis thaliana: complexity of the comparative map. Mol Genet Genomics 268:656–665
Barr CM, Fishman L (2010) The nuclear component of a cytonuclear hybrid incompatibility in Mimulus maps to a cluster of pentatricopeptide repeat genes. Genetics 184:455–465
Bentolila S, Alfonso AA, Hanson MR (2002) A pentatricopeptide repeat-containing gene restores fertility to cytoplasmic male-sterile plants. Proc Natl Acad Sci USA 99:10887–10892
Bhat SR, Prakash S, Kirti PB, Dinesh Kumar V, Chopra VL (2005) A unique introgression from Moricandia arvensis confers male fertility upon two different cytoplasmic male-sterile lines of Brassica juncea. Plant Breed 124:117–120
Brown GG, Formanova N, Jin H, Wargachuk R, Dendy C, Patil P, Laforest M, Zhang J, Cheung WY, Landry BS (2003) The radish Rfo restorer gene of Ogura cytoplasmic male sterility encodes a protein with multiple pentatricopeptide repeats. Plant J 35:262–272
Cavell AC, Lydiate DJ, Parkin IA, Dean C, Trick M (1998) Collinearity between a 30-centimorgan segment of Arabidopsis thaliana chromosome 4 and duplicated regions within the Brassica napus genome. Genome 41:62–69
Cui X, Wise RP, Schnable PS (1996) The rf2 nuclear restorer gene of male-sterile T-cytoplasm maize. Science 272:1334–1336
De Vincente C, Thomas M, Adriana A (2004) Descriptors for genetic markers technologies. IPGRI report, Rome
Desloire S, Gherbil W, Laloui S, Marhadour V, Clouet V, Cattolico L, Falentin C, Giancola S, Renard M, Budar F, Small I, Caboche M, Delourme R, Bendahmane A (2003) Identification of the fertility restoration locus Rfo in radish as a member of the pentatricopeptide-repeat protein family. EMBO Rep 4:588–594
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Fang GH, McVetty PBE (1989) Inheritance of male-fertility restoration and allelism of the restorer gene for the polima CMS system in oilseed rape. Genome 32:1044–1047
Feng C, Shengyi L, Jian W, Lu F, Silong S, Bo L, Pingxia L, Wei H, Wa Xiaowu (2011) BRAD, the genetics and genomics database for Brassica plants. BMC Plant Biol 11:136
Formanova N, Stollar R, Geddy R, Mahe L, Laforest M, Landry BS, Brown GG (2010) High resolution mapping of the Brassica napus Rfp restorer locus using Arabidopsis-derived molecular markers. Theor Appl Genet 120:843–851
Fujii S, Toriyama K (2005) Molecular mapping of the fertility restorer gene for ms-CW-type cytoplasmic male sterility of rice. Theor Appl Genet 111:696–701
Geddy R, Brown GG (2007) Genes encoding pentatricopeptide repeat (PPR) proteins are not conserved in location in plant genomes and may be subject to diversifying selection. BMC Genomics 8:130
Hinata K, Konno N (1979) Studies on a male sterile strain having the Brassica campestris nucleus and the Diplotaxis muralis cytoplasm. I. On the breeding procedure and some characteristics of the male sterile strain. Jpn J Breed 29:305–311
Iniguez-Luy FL, Voort AV, Osborn TC (2008) Development of a set of public SSR markers derived from genomic sequence of a rapid cycling Brassica oleracea L. genotype. Theor Appl Genet 117:977–985
Itabashi E, Iwata N, Fujii S, Kazama T, Toriyama K (2011) The fertility restorer gene, Rf2, for lead rice-type cytoplasmic male sterility of rice encodes a mitochondrial glycine-rich protein. Plant J 65:359–367
Janeja HS, Banga SS, Lakshmikumaran M (2003) Identification of AFLP markers linked to fertility restorer genes for tournefortii cytoplasmic male-sterility system in Brassica napus. Theor Appl Genet 107:148–154
Johnston JS, Pepper AE, Hall AE, Chen ZJ, Hodnett G, Drabek J, Lopez R, Price HJ (2005) Evolution of genome size in Brassicaceae. Ann Bot 95:229–235
Kaplinski L, Andreson R, Puurand T, Remm M (2005) MultiPLX: automatic grouping and evaluation of PCR primers. Bioinformatics 21:1701–1702
Kato H, Tezuka K, Feng YY, Kawamoto T, Takahashi H, Mori K, Akagi H (2007) Structural diversity and evolution of the Rf-1 locus in the genus Oryza. Heredity 99:516–524
Kazama T, Toriyama K (2003) A pentatricopeptide repeat-containing gene that promotes the processing of aberrant atp6 RNA of cytoplasmic male-sterile rice. FEBS Lett 544:99–102
Klein RR, Klein PE, Mullet JE, Minx P, Rooney WL, Schertz KF (2005) Fertility restorer locus Rf1 of sorghum (Sorghum bicolor L.) encodes a pentatricopeptide repeat protein not present in the collinear region of rice chromosome 12. Theor Appl Genet 111:994–1012
Koizuka N, Imai R, Fujimoto H, Hayakawa T, Kimura Y, Kohno-Murase J, Sakai T, Kawasaki S, Imamura J (2003) Genetic characterization of a pentatricopeptide repeat protein gene, orf687 that restores fertility in the cytoplasmic male sterile Kosena radish. Plant J 34:407–415
Kosambi DD (1944) The estimation of map distance from recombination value. Ann Eugen 12:172–175
Lan TH, DelMonte TA, Reischmann KP, Hyman J, Kowalski SP, McFerson J, Kresovich S, Paterson AH (2000) An EST-enriched comparative map of Brassica oleracea and Arabidopsis thaliana. Genome Res 10(6):776–788
Li G, Gao M, Yang B, Quiros CF (2003) Gene for gene alignment between the Brassica and Arabidopsis genomes by direct transcriptome mapping. Theor Appl Genet 107:168–180
Li F, Hasegawa Y, Saito M, Shirasawa S, Fukushima A, Ito T, Fujii H, Kishitani S, Kitashiba H, Nishio T (2011) Extensive chromosome homoeology among Brassiceae species were revealed by comparative genetic mapping with high-density EST-based SNP markers in radish (Raphanus sativus L.). DNA Res 18:401–411
Lowe AJ, Moule C, Trick M, Edwards KJ (2004) Efficient large scale development of microsatellites for marker and mapping applications in Brassica crop species. Theor Appl Genet 108:1103–1112
Lurin C, Andres C, Aubourg S, Bellaoui M, Bitton F, Bruyere C, Caboche M, Debast C, Gualberto J, Hoffmann B, Lecharny A, Le Ret M, Matrin-Magnietee M-L, Mireau H, Peeters N, Renou J-P, Szurek B, Taconnat L, Small I (2004) Genome-wide analysis of Arabidopsis pentatricopeptide repeat proteins reveals their essential role in organelle biogenesis. Plant Cell 16:2089–2103
O’Toole N, Hattori M, Andres C, Iida K, Lurin C et al (2008) On the expansion of the pentatricopeptide repeat gene family in plants. Mol Biol Evol 25:1120–1128
Parkin IA, Gulden SM, Sharpe AG, Lukens L, Trick M et al (2005) Segmental structure of the Brassica napus genome based on comparative analysis with Arabidopsis thaliana. Genetics 171:765–781
Piquemal J, Cinquin E, Couton F, Rondeau C, Seignoret E et al (2005) Construction of an oilseed rape (Brassica napus L.) genetic map with SSR markers. Theor Appl Genet 111:1514–1523
Prakash S, Kirti PB, Bhat SR, Gaikwad K, Kumar VD, Chopra VL (1998) A Moricandia arvensis based cytoplasmic male sterility and fertility restoration systems in Brassica juncea. Theor Appl Genet 97:488–492
Schnable PS, Wise RP (1998) The molecular basis of cytoplasmic male sterility and fertility restoration. Trends Plant Sci 3:175–180
Shinada T, Kikuchi Y, Fujimoto R, Kishitani S (2006) An alloplasmic male-sterile line of Brassica oleracea harboring the mitochondria from Diplotaxis muralis expresses a novel chimeric open reading frame, orf72. Plant Cell Physiol 47(4):549–553
Shiokai S, Shirasawa K, Sato Y, Nishio T (2010a) Improvement of the dot-blot-SNP technique for efficient and cost-effective genotyping. Mol Breed 25:179–185
Shiokai S, Kitashiba H, Nishio T (2010b) Prediction of the optimum hybridization conditions of dot-blot-SNP analysis using estimated melting temperature of oligonucleotide probes. Plant Cell Rep 29:829–834
Small ID, Peeters N (2000) The PPR motif: a TPR-related motif prevalent in plant organellar proteins. Trends Biochem Sci 25:46–47
Trendelkamp H, Uzunova MI, Ecke W (1999) Mapping a restorer gene for CMS tour 25-143 cytoplasm in rapeseed (Brassica napus L.). In: Proceedings of the 10th international rapeseed congress, Canberra, Australia
Van Ooijen JW (2006) JoinMap4.0 software for the calculation of genetic linkage maps in experimental populations. Kyazma BV, Wageningen
Wang X, Wang H, Wang J et al (2011) The genome of the mesopolyploid crop species Brassica rapa. Nat Genet 43:1035–1039. doi:10.1038/ng.919
Acknowledgments
This work was supported in part by the Program for Promotion of Basic and Applied Research for Innovations in Bio-oriented Industry (BRAIN), Japan. Ashutosh is a recipient of a research fellowship from the Japan Society for the Promotion of Science for Foreign Scientists.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Ashutosh, Sharma, B., Shinada, T. et al. Molecular mapping of a male fertility restorer locus of Brassica oleracea using expressed sequence tag-based single nucleotide polymorphism markers and analysis of a syntenic region in Arabidopsis thaliana for identification of genes encoding pentatricopeptide repeat proteins. Mol Breeding 30, 1781–1792 (2012). https://doi.org/10.1007/s11032-012-9761-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11032-012-9761-4