
Abstract
Infection and inflammation have been associated with the development of delirium for many centuries and there is a rapidly growing evidence base supporting the role of inflammation in exacerbating the neurological manifestations of both acute and chronic liver failure. Inflammation in the context of hepatic encephalopathy (HE) can arise directly within the brain itself resulting in astrocytic, microglial and neuronal dysfunction, impacting on the development of ‘brain failure’. Inflammation may also develop systemically and indirectly influence brain function. Systemic inflammation develops following liver injury, resulting in hyperammonemia and a ‘cytotoxic soup’ of pro-inflammatory mediators which are released into the circulation and modulate the impact of ammonia on the brain. The aim of this review is to summarise the current evidence base supporting the synergistic role of systemic inflammation and hyperammonemia in the pathogenesis of hepatic encephalopathy. Systemic inflammation and ammonia induce neutrophil degranulation and release reactive oxygen species into the peripheral circulation that may ultimately cross the blood brain barrier. Circulating endotoxin arising from the gut (bacterial translocation), superimposed sepsis, and hyperammonemia upregulate the expression of microbial pattern recognition receptors such as Toll-like receptors. The early recognition and management of systemic inflammation may not only facilitate improved outcomes in HE but supports the development of novel therapeutic strategies that reduce circulating endotoxemia and immune cell dysfunction.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
The role of ammonia in hepatic encephalopathy
Hepatic encephalopathy (HE) is a debilitating and life threatening consequence of liver disease complicating up to 25 % of presentations of acute liver failure (ALF) (Bernal et al. 2007). HE can arise across a spectrum of clinical severity, encompassing subtle loss of cognitive function, lethargy, depressed consciousness and coma. In liver cirrhosis between 60 and 80 % of patients are estimated to suffer with minimal hepatic encephalopathy (Ortiz et al. 2005).
Deranged nitrogen metabolism has been demonstrated to have a central role in the pathogenesis of HE. Portosystemic encephalopathy is a term which describes the dysregulation of the gut-liver axis in cirrhosis complicated by portal hypertension, allowing ammonia to bypass the liver into the systemic circulation. Ammonia is taken up by astrocytes in the brain and converted to glutamine where it exerts an osmotic effect and induces astrocytic swelling and brain edema (Butterworth 2002). Raised plasma ammonia levels are increasingly pronounced in patients with acute-on-chronic liver failure, ALF and in those with intracranial pressures >25 mmHg, respectively (Bernal et al. 2007; Clemmesen et al. 1999). However, in patients with cirrhosis, there is often a poor correlation between arterial plasma ammonia levels and the manifestation of HE (Shawcross et al. 2011). Moreover, the correlation between ammonia concentration and astrocyte swelling is not clear cut and may be modulated by the presence of both hyponatremia (Cordoba et al. 1998; Guevara et al. 2009) and the ability of the brain to buffer ammonia-induced increases in glutamine within the astrocytes by losing osmolytes such as myo-inositol (Shawcross et al. 2004a).
Inflammation, sepsis and it’s sequelae
The systemic inflammatory response syndrome (SIRS) occurs in response to a variety of severe clinical insults, both infectious and sterile, and is defined as the presence of two or more of the following features: 1) temperature >38 °C or <36 °C 2) heart rate >90 beats per minute 3) respiratory rate >20 breaths per minute or PaCO2 <32 mmHg and 4) white blood cell count >12,000/cu mm or <4,000/cu mm, or >10 % immature (band forms) (Bone et al. 1992). At the molecular level, SIRS describes a phenotype of a ‘cytokine storm’ with markedly raised pro-inflammatory cytokines TNF-α, IL-1β, IL-6, IL-12 and chemokines; this results in activation of neutrophils in the peripheral circulation (which themselves demonstrate increased translocation of NFκβ to the nucleus and the subsequent transcriptional upregulation of further pro-inflammatory cytokines in an spiralling inflammatory cascade) which can cause rise to sepsis-associated host tissue injury as a ‘bystander effect’ of inappropriate neutrophil activation. Clinical sequelae of this ‘cytokine storm’ range from peripheral vasodilatation to circulatory collapse and multiple organ failure, characterised by acute renal failure, cardiopulmonary compromise, coagulopathy, immunoparesis and encephalopathy.
Ammonia and inflammation are synergistic in the development and progression of encephalopathy
The association between systemic infection and impaired brain function was recognised >2,500 years ago; Hippocrates described patients with fever and abscesses who developed ‘inflammation of the mind’ which was termed ‘phrenitis’. This seminal observation was further explored by Galen in 200 AD, who first described the state of delirium which was later consolidated in 1892 by William Osler who published work on how infection impairs brain function in a condition that is now widely referred to as ‘septic encephalopathy’ (Shawcross et al. 2005).
It is becoming increasingly recognised that infection and inflammation play important roles in the pathogenesis of HE, acting in synergy with aberrant nitrogen metabolism (Pedersen et al. 2007; Wright and Jalan 2007). Nancy Rolando and colleagues observed a rapid progression in the severity of HE in those patients with ALF that had more marked inflammation (Rolando et al. 2000) and in a subsequent study from the US ALF group, progression of HE from mild to deeper stages was temporally associated with the development of infection, especially in those with acetaminophen-induced ALF (Vaquero et al. 2003). Likewise, in animal models (Tanaka et al. 2006; Marini and Broussard 2006; Cauli et al. 2007; Jover et al. 2006; Wright et al. 2007) and patients with cirrhosis (Shawcross et al. 2004b; Shawcross et al. 2007; Shawcross et al. 2011; Montoliu et al. 2011), there is mounting evidence for the role of SIRS in exacerbating the manifestation of HE throughout the spectrum of presentation from minimal (covert) to severe (grade 3–4) HE whereby the severity of neurocognitive impairment correlates with markers of inflammation.
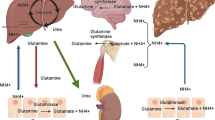
The relationship between systemic inflammation and ammonia is a synergistic one. In a bile duct ligated (BDL) rat model, ammonia-fed animals compared to those fed a normal chow diet had increased cerebral glutamine and brain water despite having identical levels of systemic inflammation (Jover et al. 2006). Furthermore, in another BDL rat study, intra-peritoneal administration of lipopolysaccharide (a major gram negative cell wall peptide) significantly increased frontal cerebral cortex water in sham-operated, ammonia-fed and BDL animals but this only led to pre-coma in the BDL animals. Cytotoxic brain edema and increases in frontal cortex nitrotyrosine were observed in the BDL and lipopolysaccharide-treated animals despite the integrity of the blood–brain barrier being preserved (Wright et al. 2007). These data support the conceptual framework that on a background of cirrhosis and hyperammonemia, superimposed systemic inflammation plays an important role in the development of HE [Fig. 1]. The observations circulating around the role of inflammation in HE also extend to prognostication whereby the SIRS score correlates with survival in severe HE; survival was independent however of ammonia concentration, liver biochemistry and the severity of liver disease as measured by the MELD score (Shawcross et al. 2011).

Immunological and biochemical alterations in the development of astrocytic edema and hepatic encephalopathy. (NH3 – ammonia; LPS – lipopolysaccharide; H2O - water)
Innate immune system dysregulation in liver failure
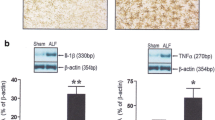
Patients with liver failure are functionally immunosuppressed and at increased risk of developing infection, a common precipitant of the development of HE and organ dysfunction. A host of neutrophil abnormalities have been described in ALF and cirrhosis (Shawcross et al. 2010). At King’s we have performed transmission electron microscopy on neutrophils isolated from patients with ALF and advanced cirrhosis with hyperammonemia which show morphologically increased nuclear to cytoplasmic ratios with vacuolation and migration of granules to surround the vacuoles, indicative of activation [unpublished data]. At the cellular level, neutrophils incubated ex vivo with 75 μmol ammonia swell and have impaired phagocytic activity, whilst ammonia-fed rats and patients with cirrhosis given an amino acid drink which temporarily increases blood ammonia levels, develop impaired neutrophil phagocytic activity with neutrophils spontaneously producing reactive oxygen species inducing oxidative stress (Shawcross et al. 2008). In patients with ALF and evidence of systemic inflammation, there is circulating neutrophil dysfunction akin to patients presenting with septic shock in the absence of liver disease; peak arterial ammonia concentration correlates with impaired neutrophil phagocytic activity which has been shown to be a marker of poor outcome in ALF (Taylor et al. 2012).
Neutrophil Toll-like receptor (TLR) 9 expression, part of the Toll-Like receptor family involved in recognition of pathogen-associated molecular patterns, correlates with the severity of HE (Vijay et al. 2012). Neutrophils appear to demonstrate a paradoxical role of aberrant activation contributing to systemic inflammation and bystander damage to host organs, whilst at the same time have impaired microbicidal capacity, which in turn predisposes to further infection and clinical decompensation of underlying disease.
Treatment modalities for hepatic encephalopathy
A paradoxical immunological conundrum in the prevention and management of HE therefore arises whereby it is desirable to augment innate immune system surveillance and killing of pathogens whilst simultaneously dampening the pro-inflammatory state of liver failure with any consequent bystander host-tissue organ damage and reducing serum ammonia levels.
Therapeutic strategies over the past 30 years have been directed at manipulation of ammonia metabolism and have centred around reducing ammonia generation and absorption in the gut. Ammonia lowering strategies have however, largely resulted in suboptimal clinical outcomes and it is generally acknowledged that therapies for HE should focus on managing any precipitating factors.
There is a wealth of emerging research directed at lowering the inflammatory burden in HE [Fig. 2]. The use of probiotics to manipulate the gut enterobiome have shown efficacy both at a cellular level, in normalising neutrophil function and clinically with significant improvement in minimal HE seen in 50 % of patients treated (Liu et al. 2004; Stadlbauer et al. 2008). Targeting of inflammation has been shown to be efficacious in the management of minimal HE in cirrhosis, with rifaximin identified as a promising therapeutic agent with an acceptable side effect profile (Bass et al. 2010). The therapeutic actions of rifaximin may be two-fold in reducing the burden of ammonia-producing enteric bacteria within the gut, as well as reducing systemic inflammation resulting from translocation of enteric bacteria across the congested, portal-hypertensive gut. Infusion of albumin which has antioxidant properties may have additional benefits over and above that of simple volume expansion, with reductions in the severity of HE (Jalan and Kapoor 2004), improved outcomes in those with spontaneous bacterial peritonitis and reduction in the incidence of hepato-renal syndrome (Sort et al. 1999). Albumin dialysis which filters off protein-bound (including endotoxin) and water-soluble (including ammonia) substances have been shown to be efficacious in the treatment of HE, however the mechanisms are not completely understood (Hassanein et al. 2007).

Hyperammonemia and immune system dysregulation contribute to the development of hepatic encephalopathy. Potential therapeutic interventions are highlighted in the blue boxes
T-regulatory cells have been shown to play a central role in the suppression of immune responses and can inhibit neutrophil function and promote their apoptosis after activation with lipopolysaccharide (Lewkowicz et al. 2006). Further understanding of the role of the innate and adaptive immune systems in the pathogenesis of HE and susceptibility to infection is required in order to optimise this delicate balance in the management of liver failure.
Conclusions
Infection and inflammation play a vital role in the precipitation and development of HE, acting in synergy with ammonia in the context of liver failure. This synergism is likely to be multifaceted including changes in cerebrovascular hemodynamics, pro-inflammatory cytokine release into the systemic circulation and immune system dysregulation. Early recognition and supportive management of SIRS can prevent the onset of HE and improve outcomes. Further understanding of the role of inflammation in the propagation of HE and multi-organ dysfunction will give rise to potential strategies directed at therapeutic manipulation of this phenomenon.
References
Bass NM, Mullen KD, Sanyal A, Poordad F, Neff G, Leevy CB, Sigal S, Sheikh MY, Beavers K, Frederick T, Teperman L, Hillebrand D, Huang S, Merchant K, Shaw A, Bortey E, Forbes WP (2010) Rifaximin treatment in hepatic encephalopathy. N Engl J Med 362:1071–1081
Bernal W, Hall C, Karvellas CJ, Auzinger G, Sizer E, Wendon J (2007) Arterial ammonia and clinical risk factors for encephalopathy and intracranial hypertension in acute liver failure. Hepatology 46:1844–1852
Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM, Sibbald WJ (1992) Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest 101:1644–1655
Butterworth RF (2002) Pathophysiology of hepatic encephalopathy: a new look at ammonia. Metab Brain Dis 17:221–227
Cauli O, Rodrigo R, Piedrafita B, Boix J, Felipo V (2007) Inflammation and hepatic encephalopathy: ibuprofen restores learning ability in rats with portacaval shunts. Hepatology 46:514–519
Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P (1999) Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology 29:648–653
Cordoba J, Gottstein J, Blei AT (1998) Chronic hyponatremia exacerbates ammonia-induced brain edema in rats after portacaval anastomosis. J Hepatol 29:589–594
Guevara M, Baccaro ME, Torre A, Gomez-Anson B, Rios J, Torres F, Rami L, Monte-Rubio GC, Martin-Llahi M, Arroyo V, Gines P (2009) Hyponatremia is a risk factor of hepatic encephalopathy in patients with cirrhosis: a prospective study with time-dependent analysis. Am J Gastroenterol 104:1382–1389
Hassanein TI, Tofteng F, Brown RS Jr, Mcguire B, Lynch P, Mehta R, Larsen FS, Gornbein J, Stange J, Blei AT (2007) Randomized controlled study of extracorporeal albumin dialysis for hepatic encephalopathy in advanced cirrhosis. Hepatology 46:1853–1862
Jalan R, Kapoor D (2004) Reversal of diuretic-induced hepatic encephalopathy with infusion of albumin but not colloid. Clin Sci (Lond) 106:467–474
Jover R, Rodrigo R, Felipo V, Insausti R, Saez-Valero J, Garcia-Ayllon MS, Suarez I, Candela A, Compan A, Esteban A, Cauli O, Auso E, Rodriguez E, Gutierrez A, Girona E, Erceg S, Berbel P, Perez-Mateo M (2006) Brain edema and inflammatory activation in bile duct ligated rats with diet-induced hyperammonemia: a model of hepatic encephalopathy in cirrhosis. Hepatology 43:1257–1266
Lewkowicz P, Lewkowicz N, Sasiak A, Tchorzewski H (2006) Lipopolysaccharide-activated CD4+CD25+ T regulatory cells inhibit neutrophil function and promote their apoptosis and death. J Immunol 177:7155–7163
Liu Q, Duan ZP, Ha DK, Bengmark S, Kurtovic J, Riordan SM (2004) Synbiotic modulation of gut flora: effect on minimal hepatic encephalopathy in patients with cirrhosis. Hepatology 39:1441–1449
Marini JC, Broussard SR (2006) Hyperammonemia increases sensitivity to LPS. Mol Genet Metab 88:131–137
Montoliu C, Cauli O, Urios A, Elmlili N, Serra MA, Giner-Duran R, Gonzalez-Lopez O, del Olmo JA, Wassel A, Rodrigo JM, Felipo V (2011) 3-nitro-tyrosine as a peripheral biomarker of minimal hepatic encephalopathy in patients with liver cirrhosis. Am J Gastroenterol 106:1629–1637
Ortiz M, Jacas C, Cordoba J (2005) Minimal hepatic encephalopathy: diagnosis, clinical significance and recommendations. J Hepatol 42(Suppl):S45–S53
Pedersen HR, Ring-Larsen H, Olsen NV, Larsen FS (2007) Hyperammonemia acts synergistically with lipopolysaccharide in inducing changes in cerebral hemodynamics in rats anaesthetised with pentobarbital. J Hepatol 47:245–252
Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R (2000) The systemic inflammatory response syndrome in acute liver failure. Hepatology 32:734–739
Shawcross DL, Balata S, Olde Damink SW, Hayes PC, Wardlaw J, Marshall I, Deutz NE, Williams R, Jalan R (2004a) Low myo-inositol and high glutamine levels in brain are associated with neuropsychological deterioration after induced hyperammonemia. Am J Physiol Gastrointest Liver Physiol 287:G503–G509
Shawcross DL, Davies NA, Williams R, Jalan R (2004b) Systemic inflammatory response exacerbates the neuropsychological effects of induced hyperammonemia in cirrhosis. J Hepatol 40:247–254
Shawcross DL, Olde Damink SW, Butterworth RF, Jalan R (2005) Ammonia and hepatic encephalopathy: the more things change, the more they remain the same. Metab Brain Dis 20:169–179
Shawcross DL, Wright G, Olde Damink SW, Jalan R (2007) Role of ammonia and inflammation in minimal hepatic encephalopathy. Metab Brain Dis 22:125–138
Shawcross DL, Wright GA, Stadlbauer V, Hodges SJ, Davies NA, Wheeler-Jones C, Pitsillides AA, Jalan R (2008) Ammonia impairs neutrophil phagocytic function in liver disease. Hepatology 48:1202–1212
Shawcross DL, Shabbir SS, Taylor NJ, Hughes RD (2010) Ammonia and the neutrophil in the pathogenesis of hepatic encephalopathy in cirrhosis. Hepatology 51:1062–1069
Shawcross DL, Sharifi Y, Canavan JB, Yeoman AD, Abeles RD, Taylor NJ, Auzinger G, Bernal W, Wendon JA (2011) Infection and systemic inflammation, not ammonia, are associated with Grade 3/4 hepatic encephalopathy, but not mortality in cirrhosis. J Hepatol 54:640–649
Sort P, Navasa M, Arroyo V, Aldeguer X, Planas R, Ruiz-Del-arbol L, Castells L, Vargas V, Soriano G, Guevara M, Gines P, Rodes J (1999) Effect of intravenous albumin on renal impairment and mortality in patients with cirrhosis and spontaneous bacterial peritonitis. N Engl J Med 341:403–409
Stadlbauer V, Mookerjee RP, Hodges S, Wright GA, Davies NA, Jalan R (2008) Effect of probiotic treatment on deranged neutrophil function and cytokine responses in patients with compensated alcoholic cirrhosis. J Hepatol 48:945–951
Tanaka S, Ide M, Shibutani T, Ohtaki H, Numazawa S, Shioda S, Yoshida T (2006) Lipopolysaccharide-induced microglial activation induces learning and memory deficits without neuronal cell death in rats. J Neurosci Res 83:557–566
Taylor NJ, Nishtala A, Vijay GK, Abeles RD, Auzinger G, Bernal W, Ma Y, Wendon JA and Shawcross DL (2012) Circulating neutrophil dysfunction in acute liver failure. Hepatol. doi:10.1002/hep.26102
Vaquero J, Polson J, Chung C, Helenowski I, Schiodt FV, Reisch J, Lee WM, Blei AT (2003) Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology 125:755–764
Vijay GKM, Abeles RD, Ramage S, Riva A, Ryan JM, Taylor NJ, Wendon JA, Chokshi S, Ma Y, Shawcross DL (2012) PMO-125 Neutrophil intracellular toll-like receptor (TLR) 9 expression serves as a biomarker that determines presence and severity of encephalopathy in acute liver failure and cirrhosis. Gut 61:A123–A124
Wright G, Jalan R (2007) Ammonia and inflammation in the pathogenesis of hepatic encephalopathy: pandora’s box? Hepatology 46:291–294
Wright G, Davies NA, Shawcross DL, Hodges SJ, Zwingmann C, Brooks HF, Mani AR, Harry D, Stadlbauer V, Zou Z, Williams R, Davies C, Moore KP, Jalan R (2007) Endotoxemia produces coma and brain swelling in bile duct ligated rats. Hepatology 45:1517–1526
Acknowledgments
Dr Shawcross is funded by a 5 year UK Department of Health HEFCE Clinical Senior Lectureship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tranah, T.H., Vijay, G.K.M., Ryan, J.M. et al. Systemic inflammation and ammonia in hepatic encephalopathy. Metab Brain Dis 28, 1–5 (2013). https://doi.org/10.1007/s11011-012-9370-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-012-9370-2