
Abstract
The concept of synergistic mechanisms as the pathophysiologic basis of hepatic encephalopathy started with the pioneering work of Les Zieve in Minneapolis some 60 years ago where synergistic actions of the liver-derived toxins ammonia, methanethiol, and octanoic acid were described. More recently, synergistic actions of ammonia and manganese, a toxic metal that is normally eliminated via the hepatobiliary route and shown to accumulate in brain in liver failure, on the glutamatergic neurotransmitter system were described. The current upsurge of interest in brain inflammation (neuroinflammation) in relation to the CNS complications of liver failure has added a third dimension to the synergy debate. The combined actions of ammonia, manganese and pro-inflammatory cytokines in brain in liver failure result in oxidative/nitrosative stress resulting from activation of glutamate (NMDA) receptors and consequent nitration of key brain proteins. One such protein, glutamine synthetase, the sole enzyme responsible for brain ammonia removal is nitrated and inactivated in brain in liver failure. Consequently, brain ammonia levels increase disproportionately resulting in alterations of brain excitability, impaired brain energy metabolism, encephalopathy and brain swelling. Experimental therapeutic approaches for which proof-of-principle has been established include the NMDA receptor antagonist memantine, N-acetyl cysteine (recently shown to have antioxidant properties at both hepatic and cerebral levels) and probiotics.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Andy walked into my office at the Research Centre at St.-Luc’s Hospital in Montreal one snowy afternoon to start a one-year sabbatical with us. It was during those heady days where investigators in the field of hepatic encephalopathy (HE) were divided into two opposing camps, the ammonia camp and the GABA camp. The ammonia crowd were convinced that it was the answer. After all, ammonia-lowering strategies were the only effective treatment at the time. Maybe they still are. Andy and I discussed the pros and cons on many occasions. The battle lines were drawn. We had just finished a 3 year series of studies in both animal models of HE and in human HE material and had failed to find any significant changes in the GABA system, its associated proteins or transporters. GABA was not the cause of HE; not alone anyway. Even then we referred to the existence of multiple factors and possible synergism.
The arguments for a major role of ammonia-related mechanisms went (and continue to go) something like this:
Arterial ammonia concentrations are increased two- to three-fold in cirrhotic patients with overt HE and may be increased up to 10-fold in acute liver failure where they present an independent risk factor for the development of intracranial hypertension and brain herniation (Clemmesen et al. 1999; Bernal et al. 2007). Increased brain ammonia has been confirmed in patients with mild HE using the technique of Positron Emission Tomography (PET) (Lockwood et al. 1991) and ammonia-lowering agents such as non-absorbable disaccharides and antibiotics remain the approach of choice for clinical management of HE in cirrhosis. Multiple putative mechanisms of action have been ascribed to the ammonia molecule in liver failure and they include direct effects of the ammonium ion on neural membranes, effects on brain energy metabolism (Lai and Cooper 1986) leading to oxidative/nitrosative stress as well as effects on the expression of genes coding for key brain proteins (Chastre et al. 2014).
Although impressive advances on ammonia-related mechanisms in HE have continued to emerge in recent years, it is clear that ammonia is not acting alone in producing the range of neurological and psychiatric symptoms that are characteristic of HE in cirrhosis. In particular, blood (particularly venous) ammonia concentrations are not a reliable predictor of the severity of HE in cirrhosis and ammonia-lowering drugs are only partially effective in the treatment of HE. These observations strongly suggest the existence of alternative or additional toxic factors produced in liver failure that gain access to the brain where they act alone or in concert with ammonia. The notion of “synergism” was incubating.
In an article published in 1989 entitled “Role of toxins and synergism in hepatic encephalopathy”, Leslie Zieve, a pioneer in the field of HE, starts with the intriguing statement that “The foremost hypothesis of pathogenesis of hepatic encephalopathy recognizes that in hepatic failure, toxins with coma-producing potential accumulate and depress neuronal function by affecting fundamental neurophysiologic processes such as post-synaptic inhibition and excitation and fundamental metabolic processes”. He goes on to suggest that, “while accumulating, the toxins interact synergistically with each other and with various augmenting endogenous metabolic abnormalities to intensify their cellular effects” (Zieve 1989).
Les Zieve went on to identify primary toxins with the potential to act synergistically and they included ammonia, mercaptans, phenols and fatty acids. The mercaptan story is particularly intriguing. Sixty years ago, methanethiol (MT) was isolated from the urine of a patient with liver failure (Challenger and Walshe 1955) and it was known at that time that small amounts of mercaptans readily induced coma in laboratory animals. It was subsequently demonstrated that serial changes in blood concentrations of MT were significantly correlated with clinical severity of HE in patients with cirrhosis (McClain et al. 1980). Synergism between ammonia and MT was suggested by findings that a sub-coma dose of MT reduced the dose of ammonia required to produce coma by 68 % and when combined with a sub-coma dose of ammonia, MT raised the incidence of coma from 0 % to 100 % (Zieve et al. 1974) (Fig. 1). The notion of toxin synergism in HE was born.

Synergism between ammonia and methanethiol, two neurotoxins implicated in the pathogenesis of HE. Figure indicates dose-response curves for coma-induction with ammonia in the presence or absence of a sub-coma dose (0.12 % by inhalation) of methanethiol (MT). From Zieve et al. 1974 with permission
In an editorial published in The Journal of Hepatology in 2004, Andy Blei, to whom this lecture is dedicated made the following statement and prediction: “Over the years, several theories have been proposed to explain the mechanisms responsible for HE. The search has been reinvigorated by a new perspective on synergism” (Blei 2004).
Indeed, the four decades since the pioneering studies of Les Zieve have witnessed the discovery of additional toxic substances in both the circulation and in the brain of patients with cirrhosis and HE. These toxins include manganese and pro-inflammatory cytokines and, in both cases, evidence of synergistic mechanisms of action with each other and with ammonia has been described.
Manganese
Excess manganese is normally removed via the hepatobiliary route and it is now well established that in cirrhosis, both circulating (Spahr et al. 1996) and brain (Pomier Layrargues et al. 1995) concentrations of manganese are significantly increased. In the brain, increased manganese is largely confined to basal ganglia structures such as globus pallidus and sustantia nigra giving rise to bilateral T1-weighted signal hyperintensities on magnetic resonance imaging (MRI) (Butterworth et al. 1995; Burkhard et al. 2003). Nigral manganese deposition has been suggested as the likely cause of a novel clinical entity known as Parkinsonism in cirrhosis (Butterworth 2013). It is well established that manganese has profound toxic effects primarily on the dopaminergic neurotransmitter systems of basal ganglia nuclei leading to a Parkinsonian syndrome similar to the one described in cirrhosis (Butterworth et al. 1995).
However, studies demonstrate that, in addition to the well-established toxic action on the dopamine system, manganese also has deleterious effects on the glutamatergic neurotransmitter system. Glutamate is the principal excitatory neurotransmitter of mammalian brain where glutamatergic fibre networks are widely distributed throughout. Manganese, in concentrations similar to those found in the brain of patients with cirrhosis and HE, causes decreased capacity for brain astrocytes to accumulate glutamate, a process that is essential for the inactivation of glutamate’s synaptic action (Hazell and Norenberg 1997). Ammonia has similar actions on glutamate uptake by the astrocyte and manganese and ammonia show synergistic actions resulting in a 63 % reduction in uptake of the transmitter (Fig. 2).

Synergistic effects of ammonia and manganese on the high affinity transport of glutamate by cultured cortical astrocytes. Figure indicates the effects of ammonia (5 mM) alone, manganese (100 uM) alone or a combination of ammonia and manganese on the high affinity uptake of the non-metabolizeable analogue of L-Glutamate (D-Aspartate) by primary cultures of rat cortical astrocytes compared to control medium. Differences significantly different from control are indicated by *p < 0.05, **p < 0.01 by ANOVA. From Hazell and Norenberg 1997 with permission
There are other examples of alterations of key brain processes that are affected by exposure to manganese results in alterations of cerebral function and synergism with ammonia. For example, both ammonia and manganese are activators of the gene coding for translocator protein (TLP), a mitochondrial protein expressed by neuroglia (astrocytes and microglia) and this has been reported in brain tissues from experimental animal models of HE in cirrhosis (Desjardins and Butterworth 2002) as well as in human HE brain using either classical biochemical techniques (Lavoie et al. 1990) or by PET (Cagnin et al. 2006). Activation of TLP results in stimulation of the uptake of cholesterol into the mitochondrion followed by increased synthesis of progesterone leading to brain accumulation of a novel series of compounds known as “neurosteroids”, one of which, allopregnanolone, a potent neuroinhibitory compound acting as a GABA receptor agonist, has been shown to be increased in brain in cirrhotic patients with HE (Ahboucha et al. 2005).
Pro-inflammatory cytokines
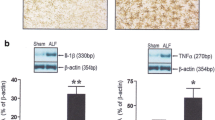
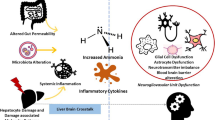
Systemic inflammation resulting from infection and/or hepatocyte cell death is common in liver failure and the acquisition of a systemic inflammatory response is a major predictor of HE (Rolando et al. 2000). Recent studies provide convincing evidence for a role of neuroinflammation (inflammation of the brain per se) in both acute liver failure and in cirrhosis (Butterworth 2013). This evidence includes activation of microglia, the immune-regulatory cells of the brain, as well as activation of the expression of genes coding for the pro-inflammatory cytokines tumour necrosis factor alpha (TNF-alpha) and the interleukins IL-1beta and IL-6 (Jiang et al. 2009a, 2009b). Signalling mechanisms linking the failing liver to cerebral dysfunction are multiple involving direct effects of pro-inflammatory molecules, recruitment of monocytes coupled to activation of microglia and direct effects of lactate generated by the toxic effects of ammonia on cellular glucose oxidation (Butterworth 2013).
Exposure of cultured astrocytes to ammonia and recombinant IL-1beta independently results in significant decreases in expression of genes coding for inducible nitric oxide synthase (iNOS) and hemoxygenase-1 (HO-1) two proteins implicated in oxidative/nitrosative stress (Chastre et al. 2010). Moreover, when ammonia and IL-1beta were combined, induction of these stress-related genes was found to be increased consistent with synergism. In a related study, treatment of experimental animals with ischemic liver failure with the antibiotic minocycline, an agent with dual properties of ammonia-lowering, inhibition of microglial activation and pro-inflammatory cytokine synthesis resulting in slowing of the progression of HE and brain edema (Jiang et al. 2009a).
Ammonia, manganese and pro-inflammatory cytokines have the potential to act synergistically to cause oxidative/nitrosative stress and to simultaneously limit the brain’s capacity to remove ammonia
According to the mechanisms summarized above, ammonia and manganese have the potential to act synergistically to inhibit high affinity glutamate transport into the perineuronal astrocyte and in so doing, lead to impaired removal of glutamate and its accumulation in the synaptic cleft. Indeed, such a mechanism has been described in the brains of animals with ALF (Michalak et al. 1996).This in turn initiates a cascade of mechanistic steps starting with the activation of glutamate(NMDA) receptors on neighbouring cells leading to activation of the nitric oxide synthase (NOS) signal transduction pathway as shown in Fig. 3.

Possible mechanisms implicated in the synergism between ammonia, manganese and proinflammatory cytokines related to the pathogenesis of HE in acute and chronic liver failure. Both ammonia and manganese inhibit high affinity glutamate transport by the astrocyte resulting in increased extracellular concentrations of glutamate leading to the activation of post-synaptic glutamate (NMDA) receptors, activation of the nitric oxide (NO) signal transduction pathway and nitration of glutamine synthetase (GS) protein. Since GS is the sole ammonia-detoxifying route in brain, this action results in further increases of brain ammonia and a vicious cycle. Neuroinflammation caused by microglial activation results independently in nitrosative stress and synergistic nitration of GS protein resulting in exacerbation of the loss of brain ammonia-removal capacity. The net result is further increases of brain ammonia with stimulation of the multiple mechanisms involving ammonia-induced accumulation of lactate, impending brain energy failure and neurotransmitter imbalance, brain edema and encephalopathy". LF; liver failure
Increases of brain cytokine synthesis following activation of microglia as described above has the capacity to magnify the problem and the combined actions of ammonia, manganese and pro-inflammatory cytokines resulting in further stimulation of the pathway shown in Fig. 3 leading ultimately to high levels of nitrosative stress. Studies in the rat model of ALF resulting from liver ischemia confirm the presence of inflammatory mediators including nitric oxide (NO) resulting from induction of several isoforms of nitric oxide synthase (NOS). Moreover they suggest that attenuation of HE severity and brain edema in this model by hypothermia involves both reduction of nitrosative stress (Jiang et al. 2009b) and an anti-inflammatory action (Jiang et al. 2009a).
One mechanism whereby nitrosative stress causes cellular dysfunction relates to its cause nitration of key proteins, a phenomenon known as protein tyrosine nitration (PTN). Studies using both in vitro and in vivo approaches demonstrate that the glutamine synthetase (GS) protein in brain is significantly nitrated as a consequence of exposure to ammonia (Schliess et al. 2002). GS is the sole enzyme responsible for ammonia detoxification in mammalian brain since the brain does not express all of the constituent enzymes of the urea cycle. Consequently, loss of GS capacity resulting from PTN in ALF would undoubtedly result in a loss of ammonia-removal capacity by the brain leading to further increases of brain ammonia and the start of a vicious cycle as shown in Fig. 3.
It should be borne in mind that the three elements of the cycle, namely ammonia, manganese and pro-inflammatory cytokines would exert their actions in a cell-selective and region-selective manner in the brain in ALF. Whereas ammonia and neuroinflammation appear to occur in a more generalized manner in ALF, the accumulation of manganese occurs principally in basal ganglia structures, particularly globus pallidus and striatum. The consequences of this selectivity are unknown at the present time.
A fuller understanding of mechanisms responsible for the pathogenesis of HE and, in particular a knowledge of the role of synergism, has the potential to result in novel treatment approaches some of which have already been subject to experimental evaluation and proof-of-principle established. Such agents include the non-competitive NMDA receptor antagonist memantine, currently under evaluation for treatment of neurodegenerative disorders and previously shown to be beneficial for the prevention of the CNS complications of ALF in a rodent model (Vogels et al. 1997).As outlined above, minocycline prevents neuroinflammation and simultaneously lowers circulating ammonia leading to slowing of HE progression and prevention of brain edema in the same model. A study of the mechanisms responsible for the beneficial effect of N-acetyl cysteine in non-acetaminophen-induced ALF in a mouse model of ALF provided evidence for beneficial actions at both hepatic and cerebral loci involving both antioxidant and anti-inflammatory mechanisms (Bemeur et al. 2010).Translation of these potentially-interesting leads into the clinic is anxiously awaited.
Alas, Andy left too soon. Way ahead of his time. But the concept of synergism survives and becomes more complex and multifactorial with each new discovery. It was the pioneering work of Les Zieve and the enthusiasm of Andy that will no doubt bring success one day. We miss you, Andy, all of us.
References
Ahboucha S, Pomier Layrargues G, Mamer O, Butterworth RF (2005) Increased brain concentrations of a neuroinhibitory steroid in human hepatic encephalopathy. Ann Neurol 58:169–170
Bemeur C, Vaquero J, Desjardins P, Butterworth RF (2010) N-acetyl cysteine attenuates cerebral complications of non-acetaminophen-induced acute liver failure in mice: antioxidant and anti-inflammatory mechanisms. Metab Brain Dis 25:241–249
Bernal W, Hall C, Karvellas CJ, et al. (2007) Arterial ammonia and clinical risk factors for encephalopathy and intracranial hypertension in acute liver failure. Hepatology 4:1844--1852
Blei AT (2004) Infection, inflammation and hepatic encephalopathy: synergism redefined. J Hepatol 40:327–330
Burkhard PR, Delavelle J, Du Pasquier R, Spahr L (2003) Chronic parkinsonism associated with cirrhosis: a distinct subset of acquired hepatocerebral degeneration. Arch Neurol 60:521–528
Butterworth RF (2013) Parkinsonism in cirrhosis: pathogenesis and current therapeutic options. Metab Brain Dis 28:261–267
Butterworth RF, Spahr L, Fontaine S, Layrargues GP (1995) Manganese toxicity, dopaminergic dysfunction and hepatic encephalopathy. Metab Brain Dis 10:259–267
Cagnin A, Taylor-Robinson SD, Forton DM, Banati RB (2006) In vivo imaging of “peripheral benzodiazepine sites” in patients with hepatic encephalopathy. Gut 55:547–553
Challenger F, Walshe JM (1955) Methyl mercaptan in relation to fetor hepaticus. Biochem J 59:372–375
Chastre A, Jiang W, Desjardins P, Butterworth RF (2010) Ammonia and pro-inflammatory cytokines modify expression of genes coding for astrocytic proteins implicated in brain edema in acute liver failure. Metab Brain Dis 25:17–21
Chastre A, Belanger M, Nguyen BN, Butterworth RF (2014) Lipopolysaccharide precipitates hepatic encephalopathy and increases blood-brain barrier permeability in mice with acute liver failure. Liver Int 34(3):353–61
Clemmesen JO, Larsen FS, Kondrup J, et al. (1999) Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentrations. Hepatol 29:648–653
Desjardins P, Butterworth RF (2002) The “peripheral-type” benzodiazepine (omega-3) receptor in hyperammonemic disorders. Neurochem Int 41:109--114
Hazell AS, Norenberg MD (1997) Manganese decreases glutamate uptake in cultured astrocytes. Neurochem Res 22:1443–1447
Jiang W, Desjardins P, Butterworth RF (2009a) Direct evidence for central proinflammatory mechanisms in rats with experimental acute liver failure: protective effect of hypothermia. J Cereb Blood Flow Metab 29:944–952
Jiang W, Desjardins P, Butterworth RF (2009b) Hypothermia attenuates oxidative/nitrosative stress, encephalopathy and brain edema in acute (ischemic) liver failure. Neurochem Int 55:124–128
Lai JCK, Cooper AJL (1986) Brain alpha-ketoglutarate dehydrogenase complex: kinetic properties, regional distribution and effects of inhibitors. J Neurochem 47:1376–1386
Lavoie J, Pomier Layrargues G, Butterworth RF (1990) Increased densities of peripheral-type benzodiazepine receptors in brain autopsy samples from cirrhotic patients with hepatic encephalopathy. Hepatol 11:874–878
Lockwood AH, Yap EW, Wong WH (1991) Cerebral ammonia metabolism in patients with severe liver disease and minimal hepatic encephalopathy. J Cereb Blood Flow Metab 11:337–341
McClain CJ, Zieve L, Doizaki WM, et al. (1980) Blood methanethiol in alcoholic liver disease with and without hepatic encephalopathy. Gut 21:318–323
Michalak A, Rose C, Butterworth J, Butterworth RF (1996) Neuroactive amino acids and glutamate (NMDA) receptors in frontal cortex of rats with experimental acute liver failure. Hepatol 24:908–913
Pomier Layrargues G, Spahr L, Butterworth RF (1995) Increased manganese concentrations in pallidum of cirrhotic patients. Lancet 345:735
Rolando N, Wade J, Davalos M, et al. (2000) The systemic inflammatory response syndrome in acute liver failure. Hepatology 32:734--739
Schliess F, Gorg B, Fischer R, et al. (2002) Ammonia induces MK-801-sensitive nitration and phosphorylation of protein tyrosine residues in rat astrocytes. FASEB J:739–741
Spahr L, Butterworth RF, Fontaine S et al. (1996) Increased blood manganese in cirrhotic patients: relationship to pallidal magnetic resonance signal hyperintensity and neurological symptoms, Hepatol 24:1116–1120
Vogels BA, Maas MA, Daalhuisen J, et al. (1997) Memantine, a non-competitive NMDA receptor antagonist improves hyperammonemia-induced encephalopathy and acute liver failure encephalopathy in rats. Hepatol 25:820–827
Zieve L (1989) Role of toxins and synergism in hepatic encephalopathy. In: Butterworth RF, Layrargues GP (eds) Hepatic encephalopathy: pathophysiology and treatment. Humana Press, Clifton, NJ, p 141--156
Zieve L, Doizaki WM, Zieve FJ (1974) Synergism between mercaptans and ammonia or fatty acids in the production of coma: a possible role for mercaptans in the pathogenesis of hepatic coma. J Lab Clin Med 83:16–28
Author information
Authors and Affiliations
Corresponding author
Additional information
This review article was based on the Andy Blei Lecture delivered at the 2014 biennial meeting of the International Society for Hepatic Encephalopathy and Nitrogen Metabolism (ISHEN) in London, UK.
Rights and permissions
About this article

Cite this article
Butterworth, R.F. Pathogenesis of hepatic encephalopathy in cirrhosis: the concept of synergism revisited. Metab Brain Dis 31, 1211–1215 (2016). https://doi.org/10.1007/s11011-015-9746-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-015-9746-1