
Abstract
Barth syndrome is a rare X-linked genetic disease classically characterized by cardiomyopathy, skeletal myopathy, growth retardation, neutropenia, and 3-methylglutaconic aciduria. It is caused by mutations in the tafazzin gene localized to chromosome Xq28.12. Mutations in tafazzin may result in alterations in the level and molecular composition of the mitochondrial phospholipid cardiolipin and result in large elevations in the lysophospholipid monolysocardiolipin. The increased monolysocardiolipin:cardiolipin ratio in blood is diagnostic for the disease, and it leads to disruption in mitochondrial bioenergetics. In this review, we discuss cardiolipin structure, synthesis, and function and provide an overview of the clinical and cellular pathophysiology of Barth Syndrome. We highlight known pharmacological management for treatment of the major pathological features associated with the disease. In addition, we discuss non-pharmacological management. Finally, we highlight the most recent promising therapeutic options for this rare mitochondrial disease including lipid replacement therapy, peroxisome proliferator-activated receptor agonists, tafazzin gene replacement therapy, induced pluripotent stem cells, mitochondria-targeted antioxidants and peptides, and the polyphenolic compound resveratrol.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Barth syndrome (MIM302060) (abbrev. BTHS) is a rare X-linked genetic recessive disorder first described by Dr. Peter Barth in 1983 [1]. BTHS is mainly manifest in infancy and early childhood, and it is characterized primarily by cardiomyopathy, skeletal myopathy, growth retardation, neutropenia, and often 3-methylglutaconic aciduria (3-MGA) [2, 3]. The major cause of death in patients with BTHS is heart failure. On the other hand, neutropenia (cyclic, chronic, or intermittent) is the second leading cause of death in BTHS patients due to a high risk of infection [4]. The cardiovascular disease component of BTHS requires careful assessment and management. However, infections associated with the disease require urgent intervention.
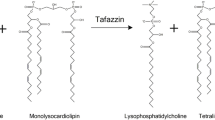
BTHS is caused by mutations in the tafazzin (TAZ) gene localized to chromosome Xq28.12 [1,2,3,4,5]. Over 100 different mutations in the TAZ gene have been described. The TAZ gene encodes the protein tafazzin (Taz), a transacylase enzyme involved in the remodeling and maturation of the mitochondrial phospholipid cardiolipin (CL). Taz transfers fatty acyl groups from phospholipids such as phosphatidylcholine and phosphatidylethanolamine to monolysocardiolipin (MLCL) to produce CL [6]. Thus, TAZ mutations result in CL remodeling impairments which lead to reductions in CL, accumulation of MLCL, mitochondrial dysfunction, and the development of BTHS [7]. BTHS is the only known genetic disorder that is related directly to impairment in CL remodeling.
Cardiolipin structure and biosynthesis
CL (1,3-bis(sn-3’-phosphatidyl)-sn-glycerol) is a specific phospholipid of mitochondrial membranes. It is an anionic phospholipid and is composed of two phosphatidic acid molecules linked by a glycerol moiety [8]. CL was isolated from bovine heart by Mary Pangborn [9]. It exhibits a conical structure with a small polar head group and a large hydrophobic tail. CL is an essential phospholipid of the inner mitochondrial membrane (IMM) where it comprises approximately 15–20% of the IMM phospholipid mass [10]. Unlike other phospholipids, CL is predominantly localized to the IMM and exhibits a unique dimeric molecular structure: three glycerol groups, two phosphate moieties, and four acyl chains [11] (Fig. 1a). This unique structure of CL introduces tension into mitochondrial membranes, and it is believed that this characteristic structure of CL plays an important role in its functional role in mitochondrial membranes. Moreover, the negative charges of CL facilitate its interaction with the outer mitochondrial membrane (OMM) and IMM proteins, and this explains why large quantities of CL are found in cristae membranes [12, 13]. In functioning mitochondria cristae membranes exhibit a charge. During oxidative phosphorylation, electrons are transferred through the electron transport chain (ETC), and protons are pumped out of the mitochondrial matrix. The pumping of protons out of the matrix creates an electrochemical gradient across the membrane which results in a negative charge on the inner side of the cristae membrane [14].

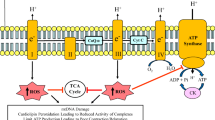
Mitochondria in normal and BTHS cells. In normal mitochondria, CL aids in the assembly of the respiratory chain complexes into supercomplex structure to facilitate electron transfer during the oxidative phosphorylation process (a). In BTHS mitochondria, with CL deficiency, the respiratory chain complexes are destabilized causing release of cytochrome C (Cty c) and increase reactive oxygen species (ROS) production resulting in impairment of oxidative phosphorylation and decrease in ATP production (b)
The molecular species of CL differ between tissues. For example, mammalian cardiac mitochondrial CL consists of approximately 80–90% linoleic acid (C18:2), the most abundant CL species being tetralinoleoyl-CL (L4CL). The tissue-specific acyl chain composition of CL is important for proper mitochondrial function [15]. For example, enrichment in L4CL is critical for oxidative phosphorylation and ETC function in the heart. Decreasing L4CL in the CL cardiac profile results in mitochondrial dysfunction [15, 16]. Previous studies showed that disruption of the specific acyl chain composition of CL leads to a decrease in CL mass and plays a major role in the development of cardiac pathophysiology [17, 18]. Since L4CL is essential for optimal mitochondrial function in the heart, replacing linoleic acid with other fatty acids leads to a decrease in the activity of the proteins in IMM due to changes in membrane characteristics. For example, high levels of docosahexaenoic acid (DHA) (22:6) in CL acyl chain composition result in disturbed protein-lipid interactions due to increase in the fluidity of the mitochondrial membranes [19].
De novo biosynthesis of nascent mammalian CL begins with formation of cytidine-5’-diphosphate-1,2-diacyl-sn-glycerol (CDP-DG) from phosphatidic acid and cytidine triphosphate catalyzed by the mitochondrial assembly and maintenance protein Tam41 [20, 21], otherwise known as CDP-DG synthetase. In the next step, phosphatidylglycerol phosphate (PGP) is synthesized through transfer of a phosphatidyl group from CDP-DG to the sn-1 position of glycerol-3-phosphate which is catalyzed by PGP synthase. PGP is then rapidly dephosphorylated by protein tyrosine phosphatase mitochondrion-1 to form phosphatidylglycerol. The last step in de novo biosynthesis of nascent CL is the condensation of phosphatidylglycerol with another molecule of CDP-DG catalyzed by CL synthase [22, 23].
Subsequent to de novo biosynthesis nascent CL undergoes remodeling by deacylation and subsequent resynthesis. Nascent CL may be deacylated to MLCL by phospholipase A2s [188]. Mature CL is then resynthesized (remodeled) by either Taz (described above), MLCL acyltransferase-1/trifunctional protein alpha subunit (MLCLAT-1/HADHA) or acyl-CoenzymeA:lysocardiolipin acyltransferase-1 (ALCAT1). Taz and MLCLAT-1/HADHA resynthesize CL in mitochondria. ALCAT1 resynthesizes small fractions of MLCL to mature CL in the mitochondria-associated membrane [24]. Both MLCLAT-1/HADHA and ALCAT1 are acyltransferases which transfer acyl chains from fatty acyl-Coenzyme A to MLCL to form mature CL [6, 25, 26]. In BTHS lymphoblasts, a compensatory upregulation of MLCLAT-1 was reported [27]. Transfection of BTHS lymphoblasts with a MLCLAT-1 construct resulted in elevated CL and enhanced mitochondrial function by reducing mitochondrial reactive oxygen species (ROS), improving basal mitochondrial respiration and proton leak [28]. Unlike MLCLAT-1, ALCAT1 is linked to pathogenic CL remodeling leading to increased oxidative stress and mitochondrial dysfunction [29].
Cardiolipin function
CL is essential for mitochondrial structure and function. It plays an important role in mitochondrial cristae organization and biogenesis, mitochondrial membrane architecture, mitochondrial bioenergetics and oxidative phosphorylation, lipid–protein interactions, and mitochondrial supercomplexes assembly [30]. Mitochondrial cristae contain a significant amount of CL and provide a large surface area for mitochondrial bioenergetics reactions such as oxidative phosphorylation for ATP production and mitochondrial respiration activity (Fig. 1a). In addition, CL is important for mitochondrial fission and fusion and cellular apoptosis [24]. CL is essential for proper function of mitochondrial respiratory supercomplexes which are composed of complex I (NADH-ubiquinone oxidoreductase), complex III (ubiquinone-cytochrome c oxidoreductase), complex IV (cytochrome c oxidase), and complex V (ATP synthase) [31,32,33]. Impairments in CL biosynthesis, remodeling, and/or metabolism may lead to metabolic diseases, including cardiovascular diseases and type 2 diabetes [34, 35], and of course the life-threatening genetic disorder BTHS. Deficiency in CL resulting from TAZ mutations affects mitochondrial biogenesis and leads to a significant decrease in ATP synthesis. Moreover, mitochondrial morphology and IMM organization are disrupted by CL deficiency [36].
CL exhibits two negatively charged phosphates. Therefore, it induces negative membrane curvature (Fig. 1a). It thus has the ability to trap protons at the IMM and interact with proteins in the OMM and IMM [37]. CL is required for the ADP/ATP carrier activity and 6 molecules of CL are tightly bound to it [38]. The absence of CL synthase, with a resulting loss in CL, was shown to affect mitochondrial function by disrupting mitochondrial membrane potential and result in impairments in ADP/ATP carrier activity on the IMM [39]. The IMM proton gradient is disrupted by decreased CL content, and this affects the IMM microenvironment and reduces protein carrier and respiratory enzymes activity in the IMM leading to membrane proton gradient disruption [39, 40]. The unique conical shape of CL and its negative charge allow CL to interact with different proteins such as prohibitins, stomatin-like 2 protein, mitochondrial contact site and cristae organizing system (MICOS), and microtubule-associated protein 1 light-chain 3 (MAP1LC3) [41, 42]. Prohibitins are proteins found in the IMM involved in phospholipid homeostasis and cellular signaling. CL molecular composition was shown to be altered in HEK293 cells by knockdown of prohibitin-2 [41, 43]. MICOS is a large heterogeneous protein that is present in cristae junctions and plays an important role in cristae formation. It interacts with CL and cellular membranes. MAP1LC3 acts as a marker for autophagosome recruitment [41]. As indicated above, CL plays an essential role in apoptosis. In the intrinsic apoptotic pathway (mitochondrial-mediated apoptosis), CL interacts with caspase-8 and recruits caspase-8 to the OMM [44]. CL is also required for the recruitment of intrinsic apoptotic factors such as tBid from cytosol to the IMM [45]. In addition, CL plays an important role in mitophagy, a selective mechanism to eliminate damaged mitochondria by autophagy through interaction with other proteins such as MAP1LC3 [42].
As indicated above, CL is critical for mitochondrial supercomplexes stability, their optimal function and structure, and CL deficiency can lead to dissociation of supercomplexes and a decrease in energy production [46]. CL thus acts as the glue that holds mitochondrial respiratory supercomplexes together [47] (Fig. 1a). BTHS results in dissociation of supercomplexes (Fig. 1b). In addition, reduction in supercomplexes formation and activity is observed in many tissues of Taz knockdown (TazKD) mice, an animal model of BTHS [48]. Mitochondrial reactive ROS production increases when supercomplexes are disrupted and this leads to CL peroxidation. The CL molecular structure is very sensitive to lipid peroxidation and CL peroxidation initiates apoptosis through the release of cytochrome c [49]. In addition, since CL is localized to the IMM where generation of ROS occurs, this itself makes CL susceptible to peroxidation [24]. An imbalance between mitochondrial ROS and antioxidant enzymes such as superoxide dismutase may lead to a significant increase in mitochondrial and cellular damage [50].
Diagnosis of Barth syndrome
BTHS was previously diagnosed clinically by the triad of cardiomyopathy, neutropenia, and elevated 3-MGA levels in plasma and urine. Approximately 70% of BTHS patients exhibit cardiomyopathy [51]. However, some patients with cardiomyopathy, growth delay, and muscle weakness were initially not diagnosed with BTHS because they had normal levels of 3-MGA in urine. In many BTHS patients, 3-MGA is increased from 5- to 20-fold [52, 53]. 3-MGA is a CoA intermediate in the metabolism of L-leucine. However, increasing urinary excretion of 3-MGA is independent of the metabolism of leucine in BTHS individuals. Because BTHS patients have both elevated 3-MGA and hypocholesterolemia, it is possible that defects of sterol and isoprenoid metabolism may be associated with elevated 3-MGA levels. These defects might cause overflow of mevalonate carbon via the mevalonate shunt pathway to 3-methyl glucaconate [54, 186]. In addition, decreased cholesterol biosynthesis in BTHS patients (see below) can induce hydroxymethylglutaryl -Coenzyme A (HMG-CoA) reductase resulting in increased HMG-CoA levels through the cholesterol biosynthesis pathway. Thus, the HMG-CoA could be dehydrated to 3-MGA by 3-MGA hydrase leading to increased 3-MGA concentrations in the urine [55]. However, no studies have been reported that validate the above in BTHS. In fact, secondary 3-methylglutaconic aciduria occurs in numerous inborn errors of metabolism that do not manifest in hypocholesterolemia. For example, the lack of such a shunt was demonstrated in Smith-Lemli-Opitz syndrome, a genetic deficiency in the last enzyme of the cholesterol synthetic pathway, 7-dehydrocholesterol reductase [187]. An alternative route posits that 3-MGA arises in three steps from mitochondrial acetyl-CoA under conditions of mitochondrial dysfunction [185]. However, it is still unknown how mitochondrial dysfunction leads to elevated 3-MGA levels. It has been suggested that impairment in the oxidative phosphorylation system disturbs the NADP/NADPH ratio. Therefore, NADP- and NADPH-dependent enzymes such as 3-MGA hydrase will be affected and cause elevation in 3-MGA levels [54]. Thus, additionally measuring 3-MGA is not sufficient for a BTHS diagnosis. A practical test for diagnosis is the measurement of the MLCL:CL ratio in blood specimen spots. Measuring CL alone is not adequate for diagnosis as some BTHS patients have near normal levels of CL but an abnormal MLCL:CL ratio. Thus, an elevated MLCL:CL ratio measurement is regarded as a 100% specific and sensitive test to diagnose BTHS [56]. A final definitive test used for diagnosis of BTHS, once the elevated MLCL:CL ratio is established, is the sequencing of the TAZ gene and confirmation of the mutation. Interestingly, there appears to be no correlation between genotype and phenotype of the disease even among families [57].
Clinical phenotypes in BTHS
Cardiac effects
Due to loss in CL, cardiomyopathy is the most significant problem in BTHS patients [184]. There is a wide variability of cardiac phenotypes of BTHS, including dilated cardiomyopathy, left ventricular non-compaction, endocardial fibroelastosis, ventricular arrhythmias, and sudden cardiac death [58]. Heart failure and arrhythmias in BTHS patients occur due to damage in heart muscle and tissues around the heart (cardiomyopathy). Cardiomyopathy develops in BTHS patients as a result of CL deficiency that cause mitochondrial dysfunction [59]. Mitochondria generate most of the energy needed by the cardiac tissue and are the primary source of the energy in cardiomyocytes. Mitochondria produce approximately 90% of 6 kg of ATP produced in 1 day through oxidative phosphorylation. Mitochondria are highly present in cardiac myocytes and occupy about 35% of the cardiac myocyte [60]. Oxidative phosphorylation begins with the citric acid cycle products, NADH and FADH2. Electrons are delivered to complex I and complex III in the ETC from NADH and FADH2, respectively. Transferring the electrons from NADH and FADH2 is the process of oxidation. Then electron transfer from complex III to complex IV through the cytochrome c carrier occurs. The final electron acceptor is oxygen. Oxygen is essential for oxidative phosphorylation to receive electrons from the complexes. Electron flux drives proton pumping in complexes I, III, and IV leading to the accumulation of protons in the mitochondrial intermembrane space resulting in a difference in the electrochemical gradient. The electrical charge, thus, differs between the two sides of the IMM. The positive charge on the outside of the IMM is due to proton accumulation, and the negative charge inside of the IMM due to loss of protons. Protons are then moved from the outside of the IMM through the F1FO-ATP synthase and produce ATP (phosphorylation) [61]. During mitochondrial respiration, physiological amounts of ROS are produced primarily by complexes I and III, and about 0.2–2% of oxygen is incompletely reduced and converted to superoxide anion (O2−). Normally, superoxide is detoxified by the mitochondrial antioxidant enzymes including glutathione peroxidase (GPx), manganese superoxide dismutase (Mn-SOD), and catalase. Uncontrolled ROS production results in mitochondrial structural and functional damage. Oxidative stress is associated with mitochondrial ultrastructural changes to mtDNA, proteins, and lipids leading to mitochondrial dysfunction and heart diseases [62]. The oxidative phosphorylation process is impaired in BTHS patients. Decreasing CL mass in BTHS mitochondria results in instability of ETC complexes, acceleration of ROS formation, and alterations in mitochondrial biogenesis and dynamics [60] (Fig. 1b). A recent study examined the maximum capacity of superoxide/H2O2 production and the ex vivo rate of superoxide/H2O2 production in each of the 11 sites that generate mitochondrial oxidants in heart and skeletal muscle mitochondria of wild-type and TazKD mice [189]. It was determined that despite reduced oxidative capacity, superoxide/H2O2 production was indistinguishable between TazKd mice and wild-type littermates. This study raised questions about the involvement of mitochondrial oxidants in the pathology of BTHS. In another study, no differences were observed in oxidative phosphorylation coupling efficiency or membrane potential in TazKD mice compared to wild type but dysregulation of CoA-dependent intermediary metabolism was observed [190]. In addition, dysregulation of Taz in HeLa cells was shown to result in an alteration of mitophagy suggesting that the mitophagic pathway may be involved in BTHS pathology [191]. In spite of these observations, mitochondrial dysfunction remains the core cause of cardiomyopathy and heart failure in BTHS patients as cardiac metabolism is strongly dependent on oxidative phosphorylation as a source of energy. TAZ mutations in these patients result in CL deficiency. Since CL is essential for mitochondrial cristae morphology, oxidative phosphorylation process, and normal mitochondrial structure and function (as indicated above), CL deficiency has been implicated in the pathophysiology of the heart failure and cardiac arrhythmias in BTHS patients. Cardiac arrhythmias also occur in BTHS patients and are the result of improper electrical impulses throughout the heart causing irregular, too fast, or too slow heartbeat. The signs or symptoms of heart arrhythmias include tachycardia, bradycardia, fluttering in the chest, chest pain, sweating, fainting, and breathing difficulties. There are different types of arrhythmias, including atrial fibrillation, atrial flutter, supraventricular tachycardia, Wolff-Parkinson-white syndrome, ventricular tachycardia, and ventricular fibrillation.
Neutropenia
Neutropenia is a major hematological abnormality in patients with BTHS and may cause severe infection. As indicated above, although cardiomyopathy is the major symptom of BTHS, some patients do not have cardiomyopathy at diagnosis but exhibit infections from infancy [4]. The molecular mechanisms underlying neutropenia in BTHS are unclear. Although BTHS lymphocytes showed increase in mitochondria size and mass, reduced cristae formation and variable cristae distribution [63], neutrophils in BTHS patients exhibit normal motility, killing activity, phagocytosis, mitochondrial shape, and mass [64]. However, neutrophil clearance may be increased by tissue macrophages because of the increase of phosphatidylserine exposure on their surface. Phosphatidylserine is normally an intracellular membrane phospholipid, and its exposure on the plasma membrane is a key apoptosis marker. A previous study showed that increasing ROS in BTHS neutrophils induced phosphatidylserine exposure and enhanced neutrophil clearance [63]. Human HL60 myeloid progenitor cells and U937 myeloid cells have been used as in vitro models to study the impact of Taz deficiency. Knockdown of the TAZ gene in human HL60 cells or U937 cells using a shRNA approach increased the number of annexin V-positive cells, caspase-3 activity and cytochrome c release from mitochondria [65]. In U937 cells, these markers of apoptosis were reduced by inhibition of caspase activity using zVAD-fink, a specific caspase inhibitor.
Skeletal muscle metabolic dysfunction
Skeletal muscle atrophy and cardiac dysfunction in patients with BTHS result in impairments in fatty acid oxidation and elevated amino acid utilization [66, 67]. The alteration in myocardial and whole-body substrate metabolism may worsen the pathology of chronic heart failure in these patients. Patients with BTHS have higher proteolytic rate, whole body per unit of fat-free mass and lower skeletal muscle mass than normal boys [68]. Alteration in CL remodeling and fatty acid composition may cause alteration in substrate metabolism, and this could contribute to the cardiomyopathy and skeletal myopathy in patients with BTHS [18]. Growth delay and decrease of the oxidative phosphorylation in skeletal muscle during exercise may alter the metabolism of fatty acid and proteins in skeletal muscle. Therefore, other substrates such as glucose and amino acids are used to produce the energy if fatty acid oxidation is not efficient enough to produce energy. Interestingly, Cade et al. [68] showed no differences in fatty acid kinetics between adolescents and young adults with BTHS and healthy subjects. In addition, patients with BTHS had the same fatty acid oxidation rate during base line and hyperinsulinemia compared to healthy controls when being measured in bed rest. They suggested that fatty acid oxidation deficits occur during activity or exercises when increased mitochondrial respiration is required. Mitochondrial fatty acid oxidation might be enough during a rest period; however, daily living and activities that need high-energy production cause fatty acid oxidation deficiency in BTHS mitochondria [68]. Increasing levels of serum fatty acid in BTHS patients may a result in impaired fatty acid oxidation in skeletal muscle and altered substrate metabolism such as amino acids. Patients with BTHS exhibited a remarkable increase in basal hepatic glucose production, similar absolute glucose utilization, and a higher glucose disposal rate by insulin stimulation compared to controls [68]. In patients with BTHS, amino acids are used to increase energy production [69]. In these patients, proteolysis rate is increased, involvement of amino acids in the citric acid cycle is decreased, and metabolism of amino acids and proteins is altered. A significant decrease in plasma arginine, ornithine, and citrulline was observed in patients with BTHS compared to controls suggesting that an alteration in amino acid substrate utilization may occur to increase energy production in these patients [68]. The breaking down of cardiac and skeletal muscle protein is increased in patients with BTHS in order to increase energy production. This may, in part, explain the low muscle mass and cardiac dysfunction in these patients [69]. Patients with BTHS also exhibit high glucose disposal rates compared to age-matched healthy controls [68]. We previously showed that glucose transporter-3 protein levels were increased 40% in BTHS lymphoblasts compared to age-matched controls [70]. In addition, phosphorylated adenosine monophosphate kinase (pAMPK) was increased 100%, and the ratio of pAMPK:AMPK elevated 58% in BTHS lymphoblasts compared to controls. AMPK activation and its phosphorylation were also shown to be elevated in neonatal ventricular fibroblasts prepared from TazKD mice [71]. Thus, the increased glucose transporter-3 protein observed in BTHS lymphoblasts may be related to the increase of AMPK phosphorylation [70]. Kiebish et al. [72] showed that glycolysis was increased in TazKD mice cardiomyocytes through increased basal extracellular acidification rate when compared to control cardiomyocytes. Indeed, ATP production from oxidative phosphorylation is significantly impaired in patients with BTHS due to loss of CL in their mitochondria. Therefore, increased glycolysis in BTHS cells may be used as an alternative source to increase ATP production. We observed that radiolabeled glucose incorporation into triacylglycerol (TG) was increased 90% in BTHS lymphoblasts compared to age-matched control cells [70]. This was accompanied by a 29% increase in diacylglycerol acyltransferase-2 activity which may contribute to the increased synthesis of TG. This increase in glucose uptake and its incorporation into TG may possibly explain why some BTHS patients exhibit a higher percentage fat mass if this is the case in other tissues.
The cognitive phenotype
As a multi-system disorder, an early study identified cognitive defects in BTHS patients [3]. In that study, data were collected from 5 kindergarten or first-grade boys with BTHS and all had significantly lower visual spatial skills, but comparable reading-related skills, when compared with boys of similar age or grade level. Using a psychoeducational assessment battery administered to 15 boys with BTHS, it was subsequently reported that although boys with BTHS had age-appropriate performance on all measures of reading-related skills, their performance on mathematics and visual spatial tasks was significantly lower than boys in the comparison group [73]. In addition, specific aspects of visual short-term memory also differed from available norms. A study of 19 patients revealed that the math difficulties in school-aged boys with BTHS were not evident in preschool years but appeared to emerge by kindergarten [74]. It was suggested that executive functions may underlie or mediate math performance of the boys with BTHS who were deficient in mathematics, suggesting that BTHS is associated with math difficulties but not with math learning disability. In a more recent study, compared to healthy controls, children with BTHS were rated as having more internalizing and externalizing symptoms, social problems, loneliness, and lower independent functioning [75]. In the most comprehensive study to date, the majority of individuals and parents reported normative levels of psychological functioning with younger age being associated with poorer health-related quality of life in some domains [76]. In that study, increased levels of internalizing symptoms were associated with poorer psychosocial functioning in individuals with BTHS. As a result of these studies, management of the cognitive profile of BTHS now warrants educational support during the early school-age years [74]. Brain CL profile shows diverse acyl species compared to other tissues like heart, liver, and skeletal muscle. In these tissues, CL molecular species predominantly have linoleic acid as a major type of acyl chain. However, the brain is rich in fatty acids and contains more polyunsaturated fatty acids such as arachidonic and docosahexaenoic acid, and this results in diverse acyl species of CL [77, 78]. Therefore, the availability of different acyl substrates in the brain could result in diversity of brain CL acyl species compared to other tissues. To address potential mechanisms for the cognitive phenotype, we recently characterized brain CL metabolism and cognitive function in TazKD mice [79]. The brain of TazKD mice exhibited reduced total CL levels and a 19-fold accumulation of MLCL compared to wild-type littermate controls and a markedly distinct profile of CL and MLCL molecular species. Mitochondrial complex I was significantly elevated in the monomeric and supercomplex forms with TAZ deficiency, and this corresponded with elevated mitochondrial state I respiration, attenuated spare capacity, and elevated ROS production. While motor function remained normal in TazKD mice, they showed significant memory deficiency based on novel object recognition tests. These results correlated with reduced synaptophysin protein levels and derangement of the neuronal CA1 layer in the hippocampus. In addition, TazKD mice brains exhibited elevated activation of microglia compared to littermate controls. This study indicated that Taz-mediated remodeling of CL contributes significantly to the expansive distribution of CL molecular species in the brain, plays a key role in brain mitochondrial respiratory activity, maintains normal cognitive function, and identified the hippocampus as a potential therapeutic target for the cognitive phenotype of BTHS.
Hypocholesterolemia
In an observational, cross-sectional study of 34 patients, hypocholesterolemia was present in 24% and decreased low-density lipoprotein cholesterol in 56% [80]. The reason for the hypocholesterolemia in BTHS patients has received little attention. We have hypothesized that the mild hypocholesterolemia observed in BTHS patients could be related to altered cholesterol metabolism. We examined this in age-matched control lymphoblasts and those from two BTHS patients that were incubated in the presence or absence of serum to induce cholesterol synthesis [81]. The pool size of cholesterol, HMG-CoA reductase (HMGR) activity and expression, and cholesterol de novo biosynthesis were examined. Although total cholesterol levels were similar, BTHS lymphoblasts had a reduced ability to respond to increased demand for cholesterol biosynthesis in the absence of serum because of an already elevated level of synthesis under standard culture conditions [81]. Regulation may occur at the level of sterol response element-binding protein-2 (SREBP-2) processing as Insig1 mRNA levels were increased in BTHS lymphoblasts compared to control (Fig. 2a, b). Downstream synthesis of cholesterol from mevalonate was unaltered in one of the patients compared to his mother (Fig. 2c). We have also observed reduced plasma cholesterol levels in TazKD mice which are lean compared to wild-type controls [82]. In that study, increased de novo CL synthesis, specifically in the liver, allowed for optimal mitochondrial oxidative phosphorylation and fatty acid oxidation. The excess in fatty acids was directed toward liver fatty acid oxidation and away from storage in the liver, adipose, and skeletal muscle. These findings may help to explain why some Barth Syndrome boys remain lean in spite of an increased caloric intake. Interestingly, expression of the Taz gene in Chinese hamster ovary cells appeared to increase cholesterol and cholesterol ester synthesis from acetate, HMGR enzyme activity, and the mRNA expression of HMGR and SREBP-2 (Fig. 3a–c). The above studies implicate the involvement of Taz in the regulation of cholesterol metabolism and support a previous observation that CL is required for optimal cholesterol synthesis [83].

Insig1 expression and cholesterol synthesis from mevalonate in BTHS lymphoblasts. Cells from an age-matched control and two BTHS patients (∆TAZ1, ∆TAZ2) were incubated in the absence (open bars) or presence (closed bars) of serum for 24 h. a. Relative gene expression of Insig1 mRNA was determined as in [81]. b. The relative gene expression of Insig1 expressed as a percent difference between serum-free and serum incubation conditions as in [81]. Data represent the mean + SD of three experiments. *p < 0.05 compared to control; bp < 0.05 compared to ∆TAZ1. c. Lymphoblasts from a BTHS boy and his mother were incubated for 24 h with 0.1 µM RS-[2-14C] mevalonate and radioactivity incorporated into cholesterol determined as in [81]. Data represent the mean + SD of three experiments. The level of significance was determined using Student's t test. (data courtesy of Dr. Kristin D. Hauff)

Expression of Taz gene in CHO cells increases cholesterol synthesis. Control (C) and CHO cells overexpressing the Taz gene (+ TAZ) were incubated with 0.1 µM [14C] acetate for 24 h and radioactivity incorporated into cholesterol (CH) and cholesterol ester (CHE) (a), HMGR reductase activity (b), and HMGR and SREBP-2 mRNA expression (c) determined as in [81]. a and c, Data are the mean, n = 2 experiments. b, Data are the mean + SD of n = 3, *p < 0.05. The level of significance was determined using Student's t test. (data courtesy of Dr. Kristin D. Hauff)
Pharmacological management of BTHS
Pharmacological management of heart failure
Currently, there is no cure or specific treatment for BTHS. Generally, the management of BTHS depends on the signs and symptoms of the condition (Fig. 4). The management of heart failure in most BTHS cases is mainly dependent on standard heart failure medications including beta blockers such as carvedilol, angiotensin-converting enzyme inhibitors, Angiotensin II receptor blockers, or diuretics [4, 58, 84, 85]. In some cases of heart failure patients require advanced therapies which include digoxin [84, 86] and vasodilators such as hydralazine, or inotropes such as dobutamine [58]. Anticoagulants drugs such as heparin and warfarin are recommended in heart failure BTHS patients with stroke, atrial fibrillation, or diabetes mellitus. In advanced heart failure cases, pharmacological intervention alone may be not effective to treat the heart failure. Therefore, surgical support techniques may be needed such as mitral valve replacement and heart transplantation [87, 88].

Pharmacological management of BTHS
Pharmacological management of cardiac arrhythmia
There are several classes of antiarrhythmic drugs used to treat BTHS (Fig. 4). Class I includes sodium channel blockers including lidocaine, flecainide, and procainamide. Class II includes beta blockers such as metoprolol. Class III includes potassium channel blockers such as amiodarone and sotalol. Class IV includes calcium channel blockers such as verapamil. Class V is drugs for variable mechanisms and includes digoxin, adenosine, and magnesium sulfate. These medications work by altering cardiac electrical conduction and improving the heart rhythms [89, 90]. In some BTHS patients, the antiarrhythmic drugs are not sufficient to be used alone to manage the arrhythmia. Therefore, implantable cardioverter defibrillator or pacemakers are needed in patients with bradycardia or heat block [88, 90]. Some BTHS patients with heart failure need surgical intervention in addition to pharmacological therapy. Mitral valve replacement, cardiac transplant, ventricular assistance device, or implantable cardioverter defibrillator has been reported to be used with patients who were suffering from ventricular arrhythmias to prevent sudden death [58]. However, these devices should be used with caution for patients with BTHS because they might be a source of infection especially in patients with neutropenia. Berlin Heart EXCOR has been used as a ventricular assisting device with BTHS patients who need heart transplant [91, 92]. However, that device may cause a serious infection in BTHS patients with neutropenia [93]. Dedieu et al. [94] reported a successful case of heart transplantation which used mechanical support Berlin Heart EXCOR for 251 days. Berlin Heart EXCOR was used as bridge to heart transplant for 8 months in a three-year-old boy with BTHS. Although he had intermittent severe neutropenia, the ventricular assist device was used successfully without serious infection. In this particular case, the boy was diagnosed with BTHS at the age of 3 months. By the age of two years, he had severe cardiomyopathy and depended on milrinone infusion. At age three, a Berlin Heart EXCOR was used as a bridge to heart transplant. The neutrophil count was tested daily, and granulocyte colony-stimulating factor (G-CSF) was used to manage the neutropenia. For Berlin Heart device, flucloxacillin and amikacin were used intravenously as prophylactic antibiotics. He had infection at the cannulation site after 11 days of Berlin Heart insertion, and piptazobactam and teicoplanin were used to treat the infection. This case study provided a successful example of how to use a ventricular assist device for a substantial period of time (251 days) without neutropenia complications. However, neutrophil count tests and prophylactic antibiotics are still important for management of the neutropenia in these cases.
Pharmacological management of neutropenia
The usual medication to treat the neutropenia in BTHS is G-CSF (Fig. 4). G-CSF works by stimulating the bone marrow to form and maturate the neutrophils. It is used in BTHS patients with cyclic, chronic, or intermittent neutropenia. Possible side effects of G-CSF include headache, bleeding, nausea, and pain in the legs, arms, or back [89, 95]. In addition to G-CSF, prophylactic antibiotics are also used to prevent recurrent infections in patients with neutropenia. Recurrent fever and infections in BTHS patients with neutropenia can be treated with G-CSF and a low dose of penicillin V or trimthoprim-sulfamethoxazole as prophylactic antibiotics for 3 days or a week [95]. Since BTHS patients have a high risk to develop infectious diseases due to neutropenia, using vaccines is considered necessary for these patients in order to protect them from infections by improving their immune system [89]. Cyclic, chronic, or intermittent unexplained neutropenia in boys is an important sign of BTHS [95]. Woiewodski et al. [56] reported a case of a boy who was born with BTHS that was not diagnosed until reaching the age of 10 years. This patient had normal cardiac function, normal 3-MGA, and normal plasma amino acids. However, he had severe neutropenia at 4 months of age with recurrent infection and growth delay. He was also diagnosed with microcytic anemia at 2 years of age. He had a follow-up care with a hematological and infectious disease unit until he was 9 years old. At 10 years of age, he exhibited abnormal pain, neutropenia, significant decrease in systolic function, and enlargement in left atrium and ventricle. He was treated with heart failure medications: carvidilol, digoxin, and angiotensin-converting enzyme inhibitor. BTHS was finally confirmed in this case by identification of a TAZ gene mutation. Therefore, the presence of neutropenia in male patients, especially in infants with muscle weakness or growth delay, may be an important sign of BTHS.
Additional therapy for BTHS
Nutritional and feeding support in BTHS
BTHS patients may require dietary intervention with nutritional supplements (Fig. 5). These include oral supplements such as L-carnitine, arginine, cornstarch, intravenous amino acid, magnesium, potassium, and multivitamins. Some patients with BTHS have deficiencies in potassium, magnesium, minerals, and vitamins. Those deficiencies can be improved by supplementation with multivitamins and minerals [89]. Cornstarch may help prevent hypoglycaemia and decrease muscle protein loss when given before bedtime. In some cases, corticosteroids (oral or topical) help alleviate the pain of mouth ulcers [58]. Arginine supplementation is used to enhance growth rate [96]. Nutritional supplements are very important for individuals with BTHS especially for patients who are suffering from diarrhea. Diarrhea in BTHS patients could result from infectious diarrhea or non-infectious diarrhea. Infectious diarrhea occurs as a result of bacterial and viral infections. However, non-infectious diarrhea may occur as a result of low blood cholesterol levels. Hypocholesterolemia in BTHS patients may cause watery diarrhea due to decrease in fat absorption. Dietary fats are absorbed by bile acids, and bile acids are formed from cholesterol in the liver. BTHS patients may have lower rates of cholesterol synthesis. Therefore, the bile acid formation may be decreased and fat absorption reduced [97]. Diarrhea in BTHS patients can be managed by intravenous fluids [98]. Diarrhea should be carefully managed in these patients, and electrolyte deficiencies should be replaced.

Non-pharmacological management of BTHS
Some BTHS patients have low plasma levels of carnitine [89]. Therefore, L-carnitine supplementation may be recommended for these patients. L-Carnitine plays a vital role in energy production and fat metabolism and an important role in fatty acid movement across the mitochondrial membrane. L-Carnitine is given to patients who have low total plasma carnitine concentrations only. The initial pharmacological doses of L-carnitine is 30 mg/kg/day [99]. Some BTHS patients are able to tolerate up to 100 mg/kg/day without serious side effects [52]. Early studies showed that oral L-carnitine supplementation improved cardiac function and muscle strength [89]. However, Ostman-Smith et al. [99] noted that there were no clinical advantages for the use of pharmacological concentrations of L-carnitine. On the contrary, they suggested that L-carnitine might deteriorate cardiac and mitochondrial function when it is used at high concentrations (> 100 mg/kg/day).
Various feeding strategies are recommended for BTHS patients who have difficulty in feeding, are finicky eaters, or have sensory sensitivities. These include gastronomy tubes, nasogastric tubes, preeminent nipples, Haberman feeders, supplemental nutrition system, eating smaller portions of food, and introducing new foods to their meals [58]. Muscle weakness and growth delay in patients with BTHS may be attributed to feeding problems. Feeding problems may also contribute to cardiac dysfunction and fatigability. A previous study showed approximately 50% of BTHS patients refused food and selected specific types of food, and approximately 70% had problems with swallowing [100]. Feeding with tubes such as gastrostomy tubes or nasogastric tubes are recommended for 1/3 patients with BTHS [51]. This behavior appears early in boys with BTHS before the age of 6 months and continues after infancy. In addition, some BTHS boys have a taste and smell sensitivity that may contribute to food selectivity and food refusal [57]. The difficultly in transitioning from tube feeding to oral feeding may develop taste and smell sensitivity and lead to disliking the food [101].
Physical therapy in BTHS
In some BTHS cases with muscle weakness, patients require physiotherapy to improve their movement or require the use of wheelchairs (Fig. 5). Previous studies showed that muscle tone can be improved with physiotherapy [63, 102, 103]. Oxygen uptake during a peak of exercise (VO2 peak) is used to measure oxidative function in both cardiac and skeletal muscles. Therefore, it is a good marker to measure clinical outcome in BTHS patients during exercise intervention. Patients with BTHS showed extreme limitation in VO2 peak. VO2 peak reduction in patients with BTHS may occur as a result of cardiac and skeletal muscle dysfunction and oxidative phosphorylation impairment. A small cohort study reported a reduction in cardiac and skeletal muscle bioenergetics in children, adolescents, and young adults with BTHS after exercise compared to age-matched healthy controls [104]. In contrast, healthy children showed an increase in oxidative metabolism compared to healthy adults during exercise. Exercise intolerance in BTHS patients was related to impairments in cardiac and skeletal muscle bioenergetics [104]. In general, endurance exercise is safe, and it is recommended as important therapy to improve physical performance and quality of life for patients with mitochondrial myopathies [105]. The TazKD mouse model exhibits similar phenotypes to patients with BTHS including cardiac dysfunction, mitochondrial dysfunction, abnormal cristae formation, decrease in L4CL, exercise intolerance, and decreased skeletal muscle contraction [106]. Soustek et al. [107] examined exercise intolerance and the effect of endurance exercise training on cardiac and skeletal muscle function on young TazKD mice. Three-month-old TazKD mice were trained on swim endurance exercise. Mitochondrial DNA was evaluated by measuring the expression of mitochondrial transcription factor A. A significant increase in mitochondrial DNA in trained TazKD mice was observed compared to age-matched untrained mice. In addition, ROS and intracellular pH were significantly decreased in exercise trained mice compared to untrained animals. Mitochondrial antioxidant ability is increased during exercise, and therefore, exercise could result in a reduction in ROS [108]. Mitochondrial respiratory complexes I, III, and IV activities were significantly decreased in TazKD mice compared to wild-type controls. Interestingly, mitochondrial complex III activity was significantly increased after endurance exercise training in cardiac tissues but not in skeletal muscle tissues of TazKD mice compared to untrained animals. However, complex IV activity was significantly reduced in trained TazKD mice. In young TazKD mice (2 months of age), cardiac mitochondria showed normal cristae formation. However, skeletal muscle mitochondria exhibited abnormal cristae formation. Therefore, tissue specificity may explain the significant increase in mitochondrial complex III activity in heart but not in skeletal muscle. Endurance exercise in TazKD mice did not appear to accelerate cardiac hypertrophy in these mice. Therefore, exercise may be considered useful for mitochondrial myopathies patients including BTHS patients [107].
In healthy individuals, endurance exercise training (aerobic exercise) improves cardiac and skeletal muscle function, mitochondrial function, and quality of life. In addition, endurance exercise training in non-BTHS patients with heart failure enhances systolic function, peak oxygen consumption, decreases hospitalization, and improves quality of life [109]. In contrast, endurance exercise training in patients with BTHS did not show improvement in cardiac function, skeletal function, or quality of life. However, endurance exercise training was safe even in BTHS patients with life-threatening arrhythmia and enhanced non-significantly oxygen consumption. Interestingly, endurance exercise training in non-BTHS syndrome patients with other mitochondrial myopathies improved peak oxygen consumption, mitochondrial function, ATP production, skeletal muscle oxygen utilization, but did not appear to improve cardiac function [110]. Therefore, pathogenesis of BTHS may contribute to the decrease of oxygen consumption improvement in skeletal muscle during exercising compared to other mitochondrial myopathies. Unlike BTHS, other mitochondrial myopathies have heterogeneous mutations in mitochondrial DNA. Therefore, endurance exercise may increase non-defective mitochondria and improve skeletal muscle oxygen consumption. However, due to homogenous mutation in mitochondria in BTHS, mutant mitochondria might be increased during endurance exercise and result in decreased oxidative phosphorylation and oxygen utilization in skeletal muscle [109]. In contrast, resistance exercise training may help to increase muscle strength and improve muscle weakness and muscle mass in patients with BTHS.
Exercise intolerance may be related to cardiac and skeletal muscle impairment and the decrease of ATP production [111]. Endurance exercise training is safe and increases exercise tolerance in normal individuals with heart failure and increases exercise tolerance approximately 5% in BTHS patients [109, 112, 113]. However, exercise tolerance was increased by approximately 15–25% in non-BTHS patients with cardiomyopathies. Unlike endurance exercise, resistance exercise increases muscle contraction and depends on glycolytic metabolism which is non-oxidative metabolism. Endurance exercise targets mainly oxidative muscle fibers. Therefore, endurance exercise may increase production of dysfunctional mitochondria in BTHS patients rather than improve exercise tolerance. However, resistance exercise training may improve muscle weakness in patients with BTHS because it depends on glycolytic more than oxidative metabolism [111, 114]. Resistance exercise training for 12 weeks in adolescents and young adults with BTHS significantly improved muscle strength, increased leucine formation rate (a biochemical marker for protein catabolism), increased muscle mass, decreased fat mass, increased whole-body lean mass, enhanced bone mineral density, and improved lower extremity torque and power production compared to age-matched, untrained BTHS patients [111].
Academic and psychological support
Specific academic education programs are recommended for about 33% of BTHS boys [52] (Fig. 5). In addition, another study suggested that students with BTHS need specific requirements such as class room desks, additional books, extra breaks, more interactive classes, and advisors [58]. Thus, some BTHS students require psychological support at school by specific psychologists [58, 115].
Promising therapeutic options for Barth syndrome
Lipid replacement therapy
The rationale behind lipid replacement therapy is to replace damaged lipids in cell membranes, in some cases by administrating a functional oral supplement [116]. The administered lipid would then be absorbed and transported to tissues and subsequently to intracellular membranes by lipid transport systems. For example, phospholipids may be protected from hydrolysis and enzymatic reactions by complexing phospholipid micelles or liposomes with fructooligosaccharides. Lipid replacement has been explored as a potential therapy for BTHS (Fig. 6). Valianpour et al. [117] showed that addition of linoleic acid to the growth culture media of cultured skin BTHS fibroblasts increased L4CL levels in these cells. Knockdown of the TAZ gene in HL60 cells using TAZ short-hairpin RNA (shRNA) increased apoptosis, and when these cells were incubated with L4CL in a water-soluble nanodisk delivery system, the apoptotic response was attenuated [65]. Exogenous administration of CL to restore mitochondrial CL inside cells has also been evaluated using lipid nanodisks containing CL. CL nanodisk addition restored intracellular CL levels in TAZ knockdown HL60 cells [118]. TazKD mice exhibit abnormal mitochondrial structure, high MLCL levels, elevated tissue MLCL:CL ratios, and skeletal and cardiac muscle defects due to CL deficiency [106, 119, 120]. At 8 months of age, TazKD mice develop cardiomyopathy and a significant increase in the left ventricular dysfunction. Ikon et al. [121] investigated whether exogenous administration of CL nanodisks in vivo to TazKD mice would restore the CL profile in these animals. CL nanodisks were administrated by intraperitoneal injection for 10 weeks. They injected 90 mg/kg CL nanodisks once a week (bolus injection) or injected 18 mg/kg CL nanodisks daily (daily injection). In contrast to what was reported in HL60 cells, CL nanodisk treatment did not alter the CL profile of TazKD mice. The impact of CL nanodisks administration on mitochondrial structure and ventricular dysfunction leading to cardiomyopathy was subsequently not examined.

Promising therapeutic options for BTHS
In intact cells, mitochondrial function may be assessed by examining the rate of ATP production, the proton leak rate, the coupling efficiency, the maximum respiratory rate, the respiratory control ratio, and the spare respiratory capacity [122]. We previously showed that BTHS lymphoblasts exhibit a decrease in Taz protein, CL mass, reduced Complex I containing supercomplexes, increased ROS production, and a decrease in MLCLAT-1/HADHA protein [28]. We examined whether expression of a MLCLAT-1 construct in BTHS lymphoblasts could restore CL levels and mitochondrial function. Previous studies have shown that CL is required to support mitochondrial supercomplexes stability and that ROS production is increased when mitochondrial supercomplexes are disrupted as a result of CL deficiency [46, 123, 124]. We observed that transfection of BTHS lymphoblasts with a MLCLAT-1 construct decreased superoxide production and improved mitochondrial basal respiration but did not restore mitochondrial supercomplexes nor completely restore CL mass to control levels [28]. Basal respiration in BTHS lymphoblasts is significantly increased in comparison to healthy lymphoblasts [28]. When BTHS lymphoblasts were transfected with the MLCLAT-1 construct, basal respiration was significantly decreased to near the levels observed in control lymphoblasts. BTHS lymphoblasts additionally exhibit a high proton leak due to damage in mitochondrial membrane structure. Transfection of BTHS with the MLCLAT-1 construct reduced the proton leak to that near of healthy lymphoblasts. Finally, expression of the MLCLAT-1 construct appeared to partially improve the growth characteristics of BTHS lymphoblasts (Fig. 7a, b). The results suggested that transfection of BTHS lymphoblasts with an MLCLAT-1 expression construct compensates, but not completely, for loss of mitochondrial respiratory function. Since increasing L4CL in vitro in BTHS models has been shown to improve mitochondrial function, lipid replacement therapies may potentially be a new therapeutic approach for treatment of BTHS patients.

Improvement of growth in BTHS lymphoblasts expressing a MLCLAT-1 construct. Lymphoblasts from a 9-year-old BTHS patient (Exon 2, c.171del. a, frameshift) were incubated for 24 h in the absence (a) or presence (b) of a MLCLAT-1 construct as described in [28]. a and b are photomicrographs of cells at 40X magnification. Arrows indicate large groupings of cells (image courtesy of Dr. Edgard M. Mejia)
Peroxisome proliferator-activated receptor agonists
Peroxisome proliferator-activated receptors (PPARs) are nuclear receptors which exist as three isoforms including PPARα, PPARγ, and PPARβ/δ [125]. These three isoforms have different physiological functions, specific tissue distribution, bind different ligands, and are expressed in diverse organs. PPARα is mainly expressed in liver, skeletal muscle, cardiomyocytes, and brown adipose tissue. PPARγ is expressed mainly in adipose tissue, liver, bone marrow, lymphocytes, and monocytes. PPARβ/δ is expressed mainly in liver, kidney, and skeletal muscle [125]. PPARα plays a central role in fatty acid oxidation, β-oxidation, and lipid homeostasis in different tissues, including heart muscle, liver, skeletal muscle, and brown adipose tissues [126]. Bezafibrate is a peroxisome proliferator-activated receptor alpha (PPARα) pan-agonist used in hyperlipidemic patients. The pharmacological effects of bezafibrate include activating PPAR-gamma coactivator 1-alpha (PGC-1α) signaling and promoting oxidative metabolism gene transcription.
Bezafibrate treatment has been shown to decrease myocardial infarction incidence and reduce the risk of cardiac mortality [127]. In addition, bezafibrate has been used for patients with carnitine palmitoyl transferase-II deficiency [128]. In animal studies, the activation of PPARα improves heart function in post-ischemic attack and reduces left ventricular dysfunction in a cardiomyopathy model that is induced by tachycardia. Huang et al. [129] treated TazKD mice with bezafibrate in chow at weaning. Isoproterenol, a β-adrenergic agonist which accelerates cardiac dysfunction, was then administered to 2.5-month-old TazKD mice and wild-type mice. In 7-month-old TazKD mice, bezafibrate treatment ameliorated isoproterenol-induced left ventricular dysfunction. In addition, bezafibrate treatment improved cardiac mitochondrial biogenesis in these mice by enhancing activity of mitochondrial electron transport chain complexes and increasing mtDNA. In that study, bezafibrate improved mitochondrial biogenesis in different tissues, including heart, spleen, liver, and skin. Interestingly, although bezafibrate increased mitochondrial citrate synthase activity, it did not alter Taz protein expression. The authors concluded that the PGC-1α signaling pathway is a suitable therapeutic option to improve cardiomyopathy in BTHS patients (Fig. 6). Another study showed long-term treatment with bezafibrate for 7 months in TazKD mice improved cardiomyopathy, enhanced systolic function and left ventricular ejection fraction, and upregulated genes of PPAR downstream that were involved in fatty acid metabolism, mitochondrial ETC proteins, glucose, and protein metabolism [130]. As indicated above, exercise capacity is significantly impaired in TazKD mice. Long-term treatment with bezafibrate for 4 months improved exercise capacity in TazKD mice when combined with voluntary exercise on the treadmill. However, bezafibrate treatment itself failed to improve exercise capacity in these TazKD mice. The ongoing CARDIOMAN clinical trial will examine the role of bezafibrate on cardiac improvement in BTHS patients.
Tafazzin gene replacement therapy
In BTHS, mutation in the TAZ gene is responsible for impairing CL remodeling and the accumulation of MLCL which results in mitochondrial dysfunction and an inefficient ATP production [15, 131]. The effects of Taz gene replacement therapy on cardiac and skeletal muscle and mitochondrial function were recently examined in TazKD mice. Neonatal and adult TazKD mice were treated with three different adeno-associated viral (AAV) vectors under the desmin, cytomegalovirus, or native Taz promoters [132]. AAV was used for gene therapy to deliver Taz since AAV persists for long periods of time, provides stable gene transfer, and has a minimal effect on immune stimulation since it is a non-pathogenic virus [133]. Taz gene expression and distribution, muscle strength and mice activity, cardiac function, mitochondrial function including oxygen consumption, structure of mitochondria, and activity of electron transport chain complexes were examined at 5 months of age post AAV-Taz administration to TazKD mice [132]. Interestingly, the three promoters exhibited different impact on Taz gene expression levels. The cytomegalovirus promoter provided ubiquitous expression of Taz. In contrast, the desmin promoter provided a high level of Taz gene expression in cardiac and skeletal muscle with lower expression levels in other tissues including the liver. The native Taz promoter resulted in Taz gene expression in all tissues. Exercise tolerance and whole-body activity were significantly improved in TazKD mice that were treated with AAV-Taz vector. Cardiac function was enhanced in all Taz gene promoter-treated mice. In addition, fatigability was significantly decreased in all Taz gene promoter-treated mice. Mitochondrial structure, numbers, and cristae formation are altered in tissues of TazKD mice in comparison to wild-type mice. Electron micrograph images showed improvement in mitochondrial size and numbers, increased mitochondrial width, improved cristae formation, enhanced mitochondrial localization between sarcomers in the heart in all AAV-Taz-treated groups compared to untreated mice. In addition to improvement of mitochondrial structure, mitochondrial function was improved in all Taz gene therapy groups. TazKD mice heart mitochondria exhibit a decrease in oxygen consumption rates and state 3 level respiration compared to wild-type control mice. Mitochondrial respiration and oxygen consumption rates were significantly improved in all AAV-Taz-treated groups. TazKD mice also exhibit impairments in Complex I, Complex II, and Complex III activity compared to wild-type controls. In all Taz-AAV gene therapy groups, Complex I, Complex II, and Complex III activities were improved. However, complex III activity was not significantly enhanced. All three vectors increased Taz gene expression and restored cardiac and skeletal muscle function. However, the AAV- desmin Taz vector was the most effective in improving cardiac mitochondrial structure and function, whole-body function, and exercise tolerance in both groups of neonates and in adult TazKD mice when compared to the cytomegalovirus and native Taz promoters. The above preclinical data on Taz gene replacement therapy provide strong evidence for an effective potential therapeutic option for patients with BTHS (Fig. 6).
Stem cells
The use of cardiac stem cells as a potential therapeutic option for BTHS has gained attention as the major cause of death in patients with BTHS is heart failure (Fig. 6). Pluripotent cells such as human embryonic stem cells are characterized with self-renewal and can differentiate into any cell type. However, the most significant problem with human embryonic stem cell therapy is immunologic rejection [134]. Different stem cell types have been targeted to improve cardiac performance, including myoblasts and cardiac stem cells. Myoblasts harvested from peripheral muscle have a regenerative advantage. They have been introduced into scarred tissue in preclinical heart failure models to improve cardiac function [135]. Due to the regenerative capacity of myoblasts, they may produce new tissues with contractile activity in the scarred area of the heart and may stimulate cardiomyocyte maturation. The main adverse effect of myoblast therapy is ventricular arrhythmia in patients treated with myoblasts. However, the ventricular arrhythmia can be prevented by amiodarone administration. Cardiac stem cells which are present in cardiac tissue may also be used to improve cardiac function. There are two types of cardiac stem cells which can be used to improve left ventricular function including c-kit + cells and cardiosphere-derived stem cells. A previous cohort study of heart failure patients used c-kit + cells to treat patients with left ventricular dysfunction [136]. C-kit + cells were delivered into patients using coronary artery bypass graft surgery. This cohort study reported that 23 of 81 patients had a 30.3–35.9% improvement in ejection fraction after 1 month of cardiac stem cell therapy and a 38.5% improvement after 4 months. Another study used cardiosphere-delivered stem cells in patients with myocardial infarction [137]. Cardiosphere-delivered stem cells were delivered into patients using intracoronary infusion. This study showed that patients who were treated with stem cells had lower left ventricular mass and scarred mass compared to untreated patients.
Patient mature somatic cells can be reprogrammed to generate induced pluripotent stem cells (iPSCs) which can be differentiated into variety types of cells, including cardiac cells, liver cells, and neurons [135]. The major advantage of iPSCs is that the cells are patient specific and avoid immunologic rejection. iPSCs models are used to study the pathophysiological mechanisms of a disease, the possible therapeutic target, and perform drug screening [135]. An in vitro model of cardiomyocytes may also aid in the understanding of the pathophysiology of specific genetic diseases [138]. Several cardiac diseases have been studied using iPSCs, including dilated and hypertrophic cardiomyopathy, catecholaminergic polymorphic ventricular tachycardia, hypoplastic left heart syndrome, and now BTHS. Patient-specific BTHS iPSCs (BTHS-iPSCs) were differentiated into cardiomyocytes to study two distinct mutations of the TAZ gene, a frame shift mutation and a missense mutation [138]. Using this ex vivo model, abnormal structure and function of cardiomyocytes linked to TAZ gene mutations were confirmed. In addition, BTHS-iPSCs cardiomyocytes exhibited abnormal mitochondrial structure and function, including smaller mitochondrial size, fragmented mitochondria, higher basal oxygen consumption, and impaired mitochondrial electron transport. Three different therapeutic options for BTHS were examined using these BTHS-iPSCs cardiomyocytes. These included bromoenol lactone which is a mitochondrial phospholipase A2 inhibitor, arginine plus cysteine amino acids which promote anaplerosis, and linoleic acid which is an unsaturated fatty acid used as a precursor of mature CL [139]. When BTHS-iPSCs cardiomyocytes were treated with linoleic acid and bromoenol lactone, the ratio of MLCL:CL ratio decreased. Thus, the combination of these two therapeutic compounds may potentially restore CL in these cells. In contrast, arginine plus cysteine did not alter the MLCL:CL ratio. In addition, linoleic acid and arginine plus cysteine significantly increased ATP production in BTHS-iPSCs cardiomyocytes. However, bromoenol lactone treatment alone did not significantly increase ATP production in these cells [139]. Treatment of BTHS-iPSCs cardiomyocytes with linoleic acid improved mitochondrial basal oxygen consumption rates with little change on maximal mitochondrial respiration capacity. In contrast, bromoenol and arginine plus cysteine had no effect on mitochondrial basal oxygen consumption or maximal mitochondrial respiration capacity. In addition, sarcomere organization and contractile deficits of BTHS-iPSCs cardiomyocytes were improved with linoleic acid treatment. These data indicated that linoleic acid addition was more effective in restoring function in BTHS-iPSCs cardiomyocytes than bromoenol lactone or arginine plus cysteine [139].
Mitochondria-targeted antioxidants
Mitochondria are a major source of ROS in cells, and ROS is produced from enzymatic reactions within the mitochondria, including pyruvate dehydrogenase, glycerol-3-phosphate dehydrogenase, cytochrome b5 reductase, and aconitase [140]. ROS may also be produced from cellular enzymatic reactions including NADPH oxidase, uncoupled nitric oxide synthase, and xanthine oxidase [141]. Mitochondrial antioxidant enzymes that protect the mitochondria from high levels of ROS include gluthione peroxidase, manganese superoxide dismutase (Mn-SOD), and thioredoxine peroxidase [141, 142]. Mitochondrial ROS is increased in Taz knockdown neonatal cardiac fibroblasts [71]. In addition, knockdown of Taz gene in cardiomyocytes in vitro decreased ATP production, induced cardiomyocyte hypertrophy, decreased contractile activity, and increased cell death. The effect of the mitochondria-targeted antioxidant Mito-TEMPO on mitochondrial function was examined in Taz knockdown cardiomyocytes [143]. The Taz gene was knocked down in 9-week-old mice cardiomyocytes using Taz shRNA adenovirus, and cardiomyocytes were then treated plus or minus 25 µM Mito-TEMPO overnight. Taz gene expression and CL levels were decreased and MLCL increased in Taz knockdown cardiomyocytes. In addition, mitochondrial ROS production was increased and intracellular ATP production decreased. Treatment of these cardiomyocytes with Mito-TEMPO significantly decreased mitochondrial ROS production and enhanced ATP production. AMPK and Janus kinase (JAK) activation are involved in cardiomyocyte hypertrophy [144, 145]. In Taz knockdown cardiomyocytes, AMPK and JAK phosphorylation were significantly increased [143, 144]. AMPK and JAK activation induced by Taz knockdown were significantly attenuated by Mito-TEMPO treatment.
BTHS patients have cardiac and skeletal muscle dysfunction due to TAZ gene mutation [51] and TazKD mice show decrease in skeletal and cardiac muscle contraction and enhanced cardiac hypertrophy. Treatment of TazKD mice cardiomyocytes with Mito-TEMPO improved cardiomyocyte contractile activity, as assessed by measuring sarcomere shortening, and attenuated cardiomyocyte hypertrophy, as assessed by a decrease in cell surface area [143]. Decreased CL content in the IMM causes dissociation of cytochrome c and its release into the cytoplasm. Cytochrome c release is increased in the cytoplasm of cells of BTHS patients due to CL deficiency in their mitochondria. Mitochondrial cytochrome c content was shown to be decreased in TazKD mice cardiomyocytes [146]. Increased cytoplasmic cytochrome c reduces cell survival and promotes cell death through apoptosis. Mito-TEMPO treatment of TazKD mice cardiomyocytes increased mitochondrial cytochrome c content and enhanced cell survival [143]. Another study showed that the increased production of mitochondrial ROS, as assessed using MitoSOX, in BTHS-iPSC cardiomyocytes was decreased by Mito-TEMPO treatment [139]. In addition, Mito-TEMPO treatment improved sarcomere organization and contractile deficits in these cells (Fig. 6). The potential effectiveness of other antioxidants including N-acetylcysteine, Mito Q-10, and melatonin in TazKO mice are currently being explored.
Mitochondria-targeted peptides
The main source of energy generation inside cells is the ETC, especially in high-energy demanding tissues such as the heart, liver, and skeletal muscle. Therefore, any abnormality in the mitochondrial ETC causes mitochondrial dysfunction and decreases energy production. The mitochondria is considered a potential target of medications to treat mitochondrial diseases such as heart failure, ischemic heart disease, and metabolic disorders [147]. A class of compounds that increases mitochondrial electron transport and enhances mitochondrial bioenergetics by targeting the mitochondrial ETC that is the Szeto-Schiller (SS) peptide family [148] (Fig. 6). SS-02 peptide shows high affinity to the mitochondria, especially to the IMM. In general, the mitochondrial potential gradient allows cationic compounds to penetrate into the mitochondrial matrix. However, SS-02 penetration does not depend on the mitochondrial potential gradient, and it does not cause mitochondrial depolarization. SS-31 and SS-20 are peptides, which are analogous to SS-02 and target the IMM [149]. SS-31 is also referred to as Bendavia, MTP-13, and Elamipretide. SS-31 is a synthetic lipophilic tetrapeptide (D-Arg-2′6′- dimethyl Tyr- Lys-Phe-NH2) with high cell permeability properties. It acts on all cell types such as cardiomyocytes, macrophages, neuron, renal, and endothelial cells. SS-31 interacts with CL by hydrophobic or electrostatic bond. SS-31 was the first compound developed as therapeutic agent to protect CL and aid in mitochondrial bioenergetics restoration [150]. This polar peptide does not depend on cell receptors or transporter mechanisms. It penetrates the cell by a diffusion mechanism [151]. SS-31 interacts only with an ionic phospholipids (e.g., CL and phosphatidylserine), but not with zwitterionic phospholipids (phosphatidylethanolamine and phosphatidylcholine). Therefore, SS-31 primarily targets CL in the IMM because the amount of phosphatidylserine in the IMM is negligible [152]. SS-31 and SS-20 compounds thus bind selectively to CL in the IMM. Because CL is mainly localized to the IMM, targeting CL with these peptides can target mitochondrial respiratory complexes and may help to increase energy production and restore mitochondrial function. These peptides are the first compounds used to target phospholipids on membranes rather than target proteins [148].
In BTHS, CL alteration and remodeling impairment lead to mitochondrial respiratory complexes dysfunction, severely inhibits oxidative phosphorylation, and increases in ROS production [153,154,155]. Hence, therapies that reduce ROS generation are highly warranted for BTHS. A previous study reported a significant decrease in infarct size in the hearts of rabbits subjected to ischemia–reperfusion injury after Bendavia administration [156]. Bendavia was effective if added immediately after reperfusion but did not affect the infarct size if administered after 10 min of reperfusion indicating that its therapeutic timing is very important to reduce the infarction. Bendavia treatment did not change heart rate, arterial blood pressure, or systolic blood pressure but decreased ROS production. ROS production promotes mitochondrial membrane potential collapse during early reperfusion. Thus, mitochondria are not able to maintain production of ATP resulting in heart dysfunction and arrhythmia under these conditions. Blocking the mitochondrial permeability transition pore may help to maintain mitochondrial membrane potential [157, 158]. Bendavia added immediately after ischemia–reperfusion maintained the mitochondrial permeability transition pore [156]. Mitochondrial calcium is increased when ATP levels are reduced and this occurs in ischemia. The increase in calcium promotes mitochondrial ROS production and CL peroxidation [150]. A previous study showed SS-31 increased ATP production and maintained mitochondrial cristae formation in vitro by preventing CL peroxidation [159]. Cytochrome c in mitochondria has been shown to have a major role in cellular metabolism and apoptosis by interacting with CL. There are two types of interaction between CL and cytochrome c. First is the electrostatic interaction where cytochrome c exists in close proximity to mitochondrial respiratory complexes to produce maximum electron transfer. The second interaction is hydrophobic. In this interaction, cytochrome c might be unfolded and electron carrier changes to a peroxidase [159]. In vitro, SS-31 interacts with CL and markedly inhibits hydrophobic interaction between cytochrome c and CL and prevents unfolding of cytochrome c. Thus, the peroxidase activity of mitochondrial cytochrome c is inhibited by SS-31. SS-31 was also shown to inhibit mitochondrial peroxidase activity in isolated mitochondria [159]. Previous studies have reported the beneficial effect of SS-31 in different diseases such as neurodegenerative disease, metabolic disorder, chronic renal disease, and mitochondrial toxicity that is induced by drugs [150].
SS-31 and heart failure: Patients with heart failure show abnormal mitochondrial structure and cristae formation with a decrease in mitochondrial respiratory complexes activity. In addition, in these patients and in heart failure animal studies, CL biosynthesis and remodeling is altered. Therefore, SS-31 might improve mitochondrial bioenergetics and restore mitochondrial respiratory complexes activity by protecting CL [150]. A previous study showed that SS-31 improved cardiac hypertrophy and diastolic function in a hypertensive cardiomyopathy mouse model [160]. Another study reported a beneficial effect of SS-31 in improving cardiac hypertrophy and systolic dysfunction in a classic transverse aortic constriction (TAC) rat model. In this model, the left ventricular mass is increased twofold, the left ventricle is significantly dilated, and myocardial fibrosis is significantly increased after 4 weeks of TAC. Furthermore, TAC induces mitochondrial dysfunction and oxidative phosphorylation impairment. Treatment with SS-31 completely prevented the myocardial fibrosis [161].
SS-31 and skeletal muscle atrophy: Skeletal muscle weakness is also a major problem in patients with BTHS. Proteolysis is increased, and protein synthesis is decreased in patients with muscle atrophy. SS-31 treatment in animal models of muscle atrophy was shown to attenuate the skeletal muscle weakness and muscle atrophy [150]. Skeletal muscle proteases are activated by increased production of ROS, and ROS production is increased in muscle atrophy due to the increase in calcium uptake from the mitochondria and the decrease of mitochondrial respiratory function and the elevation in fatty acid peroxidation [150]. A previous study reported that the SS-31 treatment in a mechanical ventilation mouse model prevented diaphragmatic contractile dysfunction and muscle atrophy [162]. Moreover, calpain, 20S roteosome, and caspase-3 activation were prevented with SS-31 treatment. Another study showed the protective effect of SS-31 on skeletal muscle atrophy in a mouse immobilization model [163]. Immobilization of mice or rats induces skeletal muscle atrophy, a significant increase in mitochondrial H2O2, a significant decrease in state 3 respiration, an increase in calpain and caspase-3 activation, and an upregulation of proteasomes. Interestingly, SS-31 does not affect mitochondrial respiration activity or ROS production in normal mice [150]. However, administration of SS-31 prevented the state 3 respiration reduction, ROS production, proteolytic activation, and prevented muscle atrophy induced by immobilization. Szetro and Schiller [163] summarized the pharmacokinetic properties of SS-31 in vitro and in vivo in different animal species. Interestingly, ex vivo studies have reported that elamipretide binds with MLCL and CL with the same ratio and its actual mechanism of action may be related to a peptide binding dependent on surface charge density and not the identity of particular component lipids [164]. The results of the ongoing TAZ POWER clinical trial with elamipretide will determine whether the drug will gain approval for use as a mitochondrial therapy for BTHS [165].
Resveratrol
Resveratrol (RESV) is a small polyphenolic compound found in several plant species [166,167,168]. We previously observed that exogenously added RESV increased CL in isolated H9c2 cardiac myocytes and that dietary RESV feeding increased L4CL in the hearts of spontaneously hypertensive rats [169] (Fig. 6). RESV effects many signaling pathways that may improve cardiac function including AMPK, nicotinamide adenine dinucleotide (NAD) +-dependent deacetylase, and sirtuin-1 (SIRT1) and sirtuin-3 (SIRT3) [170, 171]. RESV decreases the circulating levels of free fatty acids leading to decreased free fatty acid-induced lipotoxicity [172]. Moreover, RESV affects lipid metabolism by decreasing circulating low-density lipoproteins levels through increased low-density lipoprotein receptor expression [173]. Activation of SIRT1 and SIRT3 by RESV improves mitochondrial function by increasing fatty acid oxidation, decreasing mitochondrial ROS production, and improving mitochondrial respiration [172]. In addition, RESV activates AMPK leading to phosphorylated downstream proteins that are involved in metabolic process. AMPK is a key regulator of mitochondrial function and cellular homeostasis [174]. Activation of AMPK improves fatty acid and glucose uptake, glycolysis, and mitochondrial biogenesis through increased expression of PGC1α [175]. In addition to the anti-lipotoxic effects of RESV, RESV has anti-oxidant properties and can decrease mitochondrial oxidative stress and improve cardiac function. Increased mitochondrial ROS results in lipid peroxidation, including peroxidation of CL, which may lead to release of cytochrome c and initiation of apoptosis. RESV decreases mitochondrial ROS production by increasing expression of antioxidant enzymes [169]. In heart, RESV activates nuclear SIRT1 leading to an induction of superoxide dismutase 2 and a decrease oxidative damage [176]. It is well known that mitochondrial dysfunction is associated with accumulation of lipotoxic lipids [169]. In addition, accumulation of these lipotoxic lipids in the heart is known to alter and impair cardiac myocyte structure and function [177, 178].
We recently observed that high-fat feeding of TazKD mice accelerated the cardiac dysfunction in these mice, and this was characterized by impaired systolic and diastolic function, mitochondrial respiratory dysfunction, elevated fibrosis, oxidative damage, and steatosis [179]. These mice developed a hypertrophic lipotoxic cardiomyopathy in the absence of obesity. Both low-fat (6% w/w fat) as well as high-fat (23% fat w/w) fed TazKD mice accumulated cardiac TG and exhibited increased fatty acid synthase protein expression. The accumulation of TG and development of heart dysfunction were accompanied by a significant elevation in toxic lipid species (cholesterol, N:16 ceramide, and diglyceride) in high-fat fed TazKD mice. Treatment of these mice with RESV attenuated the cardiomyopathy via reducing oxidative damage and steatosis through improved complex I respiration and reduced fatty acid synthase protein levels [179]. The ability of RESV to act directly on the heart to reverse the cardiac lipotoxicity could be separated from the anti-obesity effects of RESV on the heart. These studies provided the first direct evidence that CL deficiency itself promotes the development of a hypertrophic lipotoxic cardiomyopathy. In addition, these studies provided preclinical evidence for RESV as a treatment option for attenuating lipotoxic cardiomyopathies regardless of the metabolic phenotype and improving cardiovascular outcomes in BTHS.
Conclusion
In summary, although BTHS is considered a rare genetic disease, many studies have been performed to understand the pathogenesis of the disease and develop effective therapies. However, developing effective therapies for patients with BTHS is particularly challenging due to the small number of BTHS patients and the high variability of patient phenotypes [180]. Current management of BTHS relies on treating the symptoms and complications, and early diagnosis of BTHS is important for survival of the patients. An elevated MLCL:CL ratio in blood spot specimens is the most reliable biochemical marker for rapid BTHS diagnosis. In addition, family history, cardiologic investigations, electromyography, and hematologic features are also important for diagnosing BTHS. The mitochondrial phospholipid CL plays a key role in the pathogenesis of a variety of human diseases, including BTHS. As indicated above, CL is critical for mitochondrial morphology and bioenergetics, cristae formation, mitochondrial fission and fusion, optimal mitochondrial electron transport activity, mitochondrial protein interaction, proton trapping for oxidative phosphorylation, apoptosis, mitophagy, and mitochondrial supercomplexes stability. Consequently, it is not unexpected that the dysfunctionality of CL remodeling due to the Taz gene mutation is responsible for the structurally and functionally abnormal mitochondria observed in BTHS patients. Therefore, specifically targeting mitochondria and in particular CL may provide novel therapeutic options for treating BTHS. Unique and novel approaches such as phosphokinome analysis of patient cells may aid in the identification of new targets for treatment [181]. In addition, the use of other non-mammalian model organisms including S. cerevisiae [182, 183, 192, 193] as well as the impact of Taz deficiency on other membrane components, such as plasmalogens [194], will further our understanding of this unique and complicated disease.
References
Barth PG, Scholte HR, Berden JA et al (1983) An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J Neurol Sci 62:327–355. https://doi.org/10.1016/0022-510X(83)90209-5
Kelley RI, Cheatham JP, Clark BJ et al (1991) X-linked dilated cardiomyopathy with neutropenia, growth retardation, and 3-methylglutaconic aciduria. J Pediatr 119:738–747. https://doi.org/10.1016/S0022-3476(05)80289-6
Mazzocco MMM, Henry AE, Kelly RI (2007) Barth syndrome is associated with a cognitive phenotype. J Dev Behav Pediatr 28:22–30. https://doi.org/10.1097/01.DBP.0000257519.79803.90
Rigaud C, Lebre A-S, Touraine R et al (2013) Natural history of Barth syndrome: A national cohort study of 22 patients. Orphanet J Rare Dis 8:70–70. https://doi.org/10.1186/1750-1172-8-70
Barth PG, Valianpour F, Bowen VM et al (2004) X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): An update. Am J Med Genet 126A:349–354
Xu Y, Malhotra A, Ren M, Schlame M (2006) The enzymatic function of Tafazzin. J Biol Chem 281:39217–39224. https://doi.org/10.1074/jbc.M606100200
Kulik W, van Lenthe H, Stet FS et al (2008) Bloodspot assay using HPLC–tandem mass spectrometry for detection of Barth syndrome. Clin Chem 54:371. https://doi.org/10.1373/clinchem.2007.095711
Lewis RNAH, McElhaney RN (2009) The physicochemical properties of cardiolipin bilayers and cardiolipin – containing lipid membranes. Biochim Biophys Acta Biomembr 1788:2069–2079. https://doi.org/10.1016/j.bbamem.2009.03.014
Pangborn MC (1947) The composition of cardiolipin. J Biol Chem 168:351–361
Osman C, Voelker DR, Langer T (2011) Making heads or tails of phospholipids in mitochondria. J Cell Biol 192:7–16
Cheng P, Hatch GM (1995) Inhibition of cardiolipin biosynthesis in the hypoxic rat heart. Lipids 30:513–519. https://doi.org/10.1007/BF02537025
Mejia EM, Hatch GM (2016) Mitochondrial phospholipids: Role in mitochondrial function. J Bioenerg Biomembr 48:99–112. https://doi.org/10.1007/s10863-015-9601-4
Renner LD, Weibel DB (2011) Cardiolipin microdomains localize to negatively curved regions of Escherichia coli membranes. Proc Natl Acad Sci USA 108:6264–6269. https://doi.org/10.1073/pnas.1015757108
Zorova LD, Popkov VA, Plotnikov EY et al (2018) Mitochondrial membrane potential. Anal Biochem 552:50–59. https://doi.org/10.1016/j.ab.2017.07.009
Schlame M, Ren M, Xu Y et al (2005) Molecular symmetry in mitochondrial cardiolipins. Chem Phys Lipid 138:38–49. https://doi.org/10.1016/j.chemphyslip.2005.08.002
Hoch FL (1992) Cardiolipins and biomembrane function. Biochim Biophys Acta Biomembr 1113:71–133. https://doi.org/10.1016/0304-4157(92)90035-9
Sparagna GC, Chicco AJ, Murphy RC, Bristow MR (2007) Loss of cardiac tetralinoleoyl cardiolipin in human and experimental heart failure. J Lipid Res 48:1559–1570
Vreken P, Valianpour F, Nijtmans LG et al (2000) Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem Biophys Res Commun 279:378–382. https://doi.org/10.1006/bbrc.2000.3952
Stanley WC, Khairallah RJ, Dabkowski ER (2012) Update on lipids and mitochondrial function: Impact of dietary n-3 polyunsaturated fatty acids. Curr Opin Clin Nutr Metab Care 15:122–126. https://doi.org/10.1097/MCO.0b013e32834fdaf7
Bratschi MW, Burrowes DP, Kulaga A et al (2009) Glycerol-3-phosphate acyltransferases gat1p and gat2p are microsomal phosphoproteins with differential contributions to polarized cell growth. Eukaryot Cell 8:1184–1196. https://doi.org/10.1128/EC.00085-09
Tamura Y, Harada Y, Nishikawa S et al (2013) Tam41 is a CDP-diacylglycerol synthase required for cardiolipin biosynthesis in mitochondria. Cell Metab 17:709–718. https://doi.org/10.1016/j.cmet.2013.03.018
Hatch GM (2004) Cell biology of cardiac mitochondrial phospholipids. Biochem Cell Biol 82:99–112. https://doi.org/10.1139/o03-074
Davidson JB, Stanacev NZ (1971) Biosynthesis of cardiolipin in mitochondria isolated from guinea pig liver. Biochem Biophys Res Commun 42:1191–1199. https://doi.org/10.1016/0006-291X(71)90032-5
Ye C, Shen Z, Greenberg ML (2016) Cardiolipin remodeling: A regulatory hub for modulating cardiolipin metabolism and function. J Bioenerg Biomembr 48:113–123. https://doi.org/10.1007/s10863-014-9591-7
Cao J, Liu Y, Lockwood J et al (2004) A novel cardiolipin-remodeling pathway revealed by a gene encoding an endoplasmic reticulum-associated Acyl-CoA: Lysocardiolipin Acyltransferase (ALCAT1) in mouse. J Biol Chem 279:31727–31734. https://doi.org/10.1074/jbc.M402930200
Taylor WA, Hatch GM (2003) Purification and characterization of monolysocardiolipin acyltransferase from pig liver mitochondria. J Biol Chem 278:12716–12721. https://doi.org/10.1074/jbc.M210329200
Taylor WA, Hatch GM (2009) Identification of the human mitochondrial linoleoyl-coenzyme A monolysocardiolipin acyltransferase (MLCLAT-1). J Biol Chem 284:30360–30371. https://doi.org/10.1074/jbc.M109.048322
Mejia EM, Zegallai H, Bouchard ED et al (2018) Expression of human monolysocardiolipin acyltransferase-1 improves mitochondrial function in Barth syndrome lymphoblasts. J Biol Chem 293:7564–7577. https://doi.org/10.1074/jbc.RA117.001024
Li J, Romestaing C, Han X et al (2010) Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metab 12:154–165. https://doi.org/10.1016/j.cmet.2010.07.003
Aprikyan AA, Khuchua Z (2013) Advances in the understanding of Barth syndrome. Br J Haematol 161(3):330–338. https://doi.org/10.1111/bjh.12271
Eble KS, Coleman WB, Hantgan RR, Cunningham CC (1990) Tightly associated cardiolipin in the bovine heart mitochondrial ATP synthase as analyzed by 31P nuclear magnetic resonance spectroscopy. J Biol Chem 265:19434–19440
Fry M, Green DE (1981) Cardiolipin requirement for electron transfer in complex I and III of the mitochondrial respiratory chain. J Biol Chem 256:1874–1880
Robinson NC (1993) Functional binding of cardiolipin to cytochrome c oxidase. J BioenergBiomembr 25:153–163
Sugamura K, Keaney John F (2011) Reactive oxygen species in cardiovascular disease. Free Radical Biol Med 51:978–992. https://doi.org/10.1016/j.freeradbiomed.2011.05.004
Ha H, Hwang I, Park JH, Lee HB (2008) Role of reactive oxygen species in the pathogenesis of diabetic nephropathy. Diabetes Res Clin Pract 82:S42–S45. https://doi.org/10.1016/j.diabres.2008.09.017
Xu Y, Sutachan JJ, Plesken H et al (2005) Characterization of lymphoblast mitochondria from patients with Barth syndrome. Lab Invest 85:823–830. https://doi.org/10.1038/labinvest.3700274
Khalifat N, Rahimi M, Bitbol A et al (2014) Interplay of packing and flip-flop in local bilayer deformation. How phosphatidylglycerol could rescue mitochondrial function in a cardiolipin-deficient yeast mutant. Biophys J 107(4):879–890. https://doi.org/10.1016/j.bpj.2014.07.015
Beyer K, Klingenberg M (1985) ADP/ATP carrier protein from beef heart mitochondria has high amounts of tightly bound cardiolipin, as revealed by phosphorus-31 nuclear magnetic resonance. Biochemistry 24(15):3821–3826. https://doi.org/10.1021/bi00336a001
Jiang F, Ryan MT, Schlame M et al (2000) Absence of cardiolipin in the crd1 null mutant results in decreased mitochondrial membrane potential and reduced mitochondrial function. J Biol Chem 275:22387–22394. https://doi.org/10.1074/jbc.M909868199
Basu Ball W, Baker CD, Neff JK et al (2018) Ethanolamine ameliorates mitochondrial dysfunction in cardiolipin-deficient yeast cells. J Biol Chem 293:10870–10883. https://doi.org/10.1074/jbc.RA118.004014
Schlame M, Greenberg ML (2017) Biosynthesis, remodeling and turnover of mitochondrial cardiolipin. Biochim Biophys Acta Mol Cell Biol Lipids 1862:3–7. https://doi.org/10.1016/j.bbalip.2016.08.010
Chu CT, Bayır H, Kagan VE (2014) LC3 binds externalized cardiolipin on injured mitochondria to signal mitophagy in neurons. Autophagy 10:376–378. https://doi.org/10.4161/auto.27191
Tatsuta T, Model K, Langer T (2005) Formation of membrane-bound ring complexes by prohibitins in mitochondria. Mol Biol Cell 16:248–259. https://doi.org/10.1091/mbc.e04-09-0807
Gonzalvez F, Schug ZT, Houtkooper RH et al (2008) Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J Cell Biol 183:681–696. https://doi.org/10.1083/jcb.200803129
Lutter M, Fang M, Luo X et al (2000) Cardiolipin provides specificity for targeting of tBid to mitochondria. Nat Cell Biol 2:754–756. https://doi.org/10.1038/35036395
Pfeiffer K, Gohil V, Stuart RA et al (2003) Cardiolipin stabilizes respiratory chain supercomplexes. J Biol Chem 278:52873–52880. https://doi.org/10.1074/jbc.M308366200
Zhang M, Mileykovskaya E, Dowhan W (2002) Gluing the respiratory chain together: Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J Biol Chem 277:43553–43556. https://doi.org/10.1074/jbc.C200551200
Huang Y, Powers C, Madala SK et al (2015) Cardiac metabolic pathways affected in the mouse model of barth syndrome. PLoS ONE 10:e0128561–e0128561. https://doi.org/10.1371/journal.pone.0128561
Shidoji Y, Hayashi K, Komura S et al (1999) Loss of molecular interaction between cytochrome c and cardiolipin due to lipid peroxidation. Biochem Biophys Res Commun 264:343–347. https://doi.org/10.1006/bbrc.1999.1410
Ray PD, Huang B-W, Tsuji Y (2012) Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 24:981–990. https://doi.org/10.1016/j.cellsig.2012.01.008
Roberts AE, Nixon C, Steward CG et al (2012) The Barth syndrome registry: Distinguishing disease characteristics and growth data from a longitudinal study. Am J Medi Genet Part A 158A(11):2726–2732. https://doi.org/10.1002/ajmg.a.35609
Clarke SLN, Bowron A, Gonzalez IL et al (2013) Barth syndrome. Orphanet J Rare Dis 8:23–23. https://doi.org/10.1186/1750-1172-8-23
Schmidt MR, Birkebaek N, Gonzalez I, Sunde L (2004) Barth syndrome without 3-methylglutaconic aciduria. Acta Paediatr 93:419–421. https://doi.org/10.1111/j.1651-2227.2004.tb02974.x
Wortmann SB, Kluijtmans LA, Engelke UFH et al (2012) The 3-methylglutaconic acidurias: What’s new? J Inherit Metab Dis 35:13–22. https://doi.org/10.1007/s10545-010-9210-7
Kelley RI, Kratz L (1995) 3-methylglutaconic acidemia in Smith-Lemli-Opitz syndrome. Pediatr Res 37:671–674. https://doi.org/10.1203/00006450-199505000-00020
Woiewodski L, Ezon D, Cooper J, Feingold B (2017) Barth syndrome with late-onset cardiomyopathy: A missed opportunity for diagnosis. J Pediatr 183:196–198. https://doi.org/10.1016/j.jpeds.2016.12.070
Thompson WR, DeCroes B, McClellan R et al (2016) New targets for monitoring and therapy in Barth syndrome. Genet Med 18:1001–1010. https://doi.org/10.1038/gim.2015.204
Reynolds S (2015) Successful management of Barth syndrome: A systematic review highlighting the importance of a flexible and multidisciplinary approach. J Multidiscip Healthc 8:345–358. https://doi.org/10.2147/JMDH.S54802
Dudek J, Hartmann M, Rehling P (2019) The role of mitochondrial cardiolipin in heart function and its implication in cardiac disease. Biochim Biophys Acta Mol Basis Dis 1865:810–821. https://doi.org/10.1016/j.bbadis.2018.08.025
Dudek J, Maack C (2017) Barth syndrome cardiomyopathy. Cardiovasc Res 113:399–410. https://doi.org/10.1093/cvr/cvx014
El-Hattab AW, Scaglia F (2016) Mitochondrial cardiomyopathies. Front Cardiovasc Med 3:25–25. https://doi.org/10.3389/fcvm.2016.00025
Gustafsson ÅB, Gottlieb RA (2008) Heart mitochondria: Gates of life and death. Cardiovasc Res 77:334–343. https://doi.org/10.1093/cvr/cvm005
Finsterer J, Frank M (2013) Haematological features in Barth syndrome. Curr Opin Hematol 20(1):36–40. https://doi.org/10.1097/moh.0b013e32835a01d9
Kuijpers TW (2004) Neutrophils in Barth syndrome (BTHS) avidly bind annexin-V in the absence of apoptosis. Blood 103(10):3915–3923. https://doi.org/10.1182/blood-2003-11-3940
Makaryan V, Kulik W, Vaz FM et al (2012) The cellular and molecular mechanisms for neutropenia in Barth syndrome. Eur J Haematol 88:195–209. https://doi.org/10.1111/j.1600-0609.2011.01725.x
Bergmann SR, Herrero P, Sciacca R et al (2001) Characterization of altered myocardial fatty acid metabolism in patients with inherited cardiomyopathy. J Inherit Metab Dis 24:657–674
Dávila-Román VG, Vedala G, Herrero P et al (2002) Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J Am Coll Cardiol 40:271–277. https://doi.org/10.1016/S0735-1097(02)01967-8
Cade WT, Spencer CT, Reeds DN et al (2013) Substrate metabolism during basal and hyperinsulinemic conditions in adolescents and young-adults with Barth syndrome. J Inherit Metab Dis 36:91–101. https://doi.org/10.1007/s10545-012-9486-x
Norrelund H, Wiggers H, Halbirk M et al (2006) Abnormalities of whole body protein turnover, muscle metabolism and levels of metabolic hormones in patients with chronic heart failure. J Intern Med 260:11–21. https://doi.org/10.1111/j.1365-
Mejia EM, Zinko JC, Hauff KD et al (2017) Glucose uptake and triacylglycerol synthesis are increased in Barth syndrome lymphoblasts. Lipids 52:161–165. https://doi.org/10.1007/s11745-017-4232-7
He Q et al (2005) Tafazzin knockdown interrupts cell cycle progression in cultured neonatal ventricular fibroblasts. Am J Physiol Heart CircPhysiol 305(9):H1332–H1343
Kiebish MA, Yang K, Liu X et al (2013) Dysfunctional cardiac mitochondrial bioenergetic, lipidomic, and signaling in a murine model of Barth syndrome. J Lipid Res 54:1312–1325. https://doi.org/10.1194/jlr.M034728
Raches D, Mazzocco MM (2012) Emergence and nature of mathematical difficulties in young children with Barth syndrome. J Dev Behav Pediatr 33:328–335
Ferreira, C. et al. (2014). In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2016.
Storch EA, Keeley M, Merlo LJ et al (2009) Psychosocial functioning in youth with Barth syndrome. Child Health Care 38:137–156. https://doi.org/10.1080/02739610902813344
Jacob ML et al (2017) Psychosocial functioning in Barth syndrome: Assessment of individual and parental adjustment. Child Health Care 46:66–92
Cheng H, Mancuso DJ, Jiang X et al (2008) Shotgun lipidomics reveals the temporally dependent, highly diversified cardiolipin profile in the mammalian brain: Temporally coordinated postnatal diversification of cardiolipin molecular species with neuronal remodeling. Biochemistry 47:5869–5880. https://doi.org/10.1021/bi7023282
Houtkooper RH, Turkenburg M, Poll-The BT et al (2009) The enigmatic role of tafazzin in cardiolipin metabolism. Biochim Biophys Acta Biomembr 1788:2003–2014. https://doi.org/10.1016/j.bbamem.2009.07.009
Cole LK, Kim JH, Amoscato AA et al (2018) Aberrant cardiolipin metabolism is associated with cognitive deficiency and hippocampal alteration in tafazzin knockdown mice. Biochim Biophys Acta Mol Basis Dis 1864:3353–3367. https://doi.org/10.1016/j.bbadis.2018.07.022
Spencer CT, Bryant RM, Day J et al (2006) Cardiac and clinical phenotype in Barth syndrome. Pediatrics 118:e337. https://doi.org/10.1542/peds.2005-2667
Hauff KD, Hatch GM (2010) Reduction in cholesterol synthesis in response to serum starvation in lymphoblasts of a patient with Barth syndrome. This paper is one of a selection of papers published in this special issue entitled “Second International Symposium on Recent Advances in Basic, Clinical, and Social Medicine” and has undergone the Journal’s usual peer review process. Biochem Cell Biol 88:595–602. https://doi.org/10.1139/O09-186
Cole LK, Mejia EM, Vandel M et al (2016) Impaired cardiolipin biosynthesis prevents hepatic steatosis and diet-induced obesity. Diabetes 65:3289–3300. https://doi.org/10.2337/db16-0114
Hauff KD, Choi S-Y, Frohman MA, Hatch GM (2009) Cardiolipin synthesis is required to support human cholesterol biosynthesis from palmitate upon serum removal in Hela cells. Can J Physiol Pharmacol 87:813–820. https://doi.org/10.1139/Y09-055
Bachou T, Giannakopoulos A, Trapali C et al (2009) A novel mutation in the G4.5 (TAZ) gene in a Greek patient with Barth syndrome. Blood Cells Mol Dis 42:262–264. https://doi.org/10.1016/j.bcmd.2008.11.004
Aljishi E, Ali F (2010) Barth syndrome: An X-linked cardiomyopathy with a novel mutation. Indian J Pediatr 77:1432–1433. https://doi.org/10.1007/s12098-010-0222-y
Finsterer J (2019) Barth syndrome: Mechanisms and management. Appl Clin Genet 12:95–106. https://doi.org/10.2147/TACG.S171481
Mangat J, Lunnon-Wood T, Rees P et al (2007) Successful cardiac transplantation in Barth syndrome – single-centre experience of four patients. Pediatr Transplant 11:327–331. https://doi.org/10.1111/j.1399-3046.2006.00629.x
Lei M, Wu L, Terrar DA, Huang CL (2018) Modernized classification of cardiac antiarrhythmic drugs. Circulation 138(17):1879–1896. https://doi.org/10.1161/circulationaha.118.035455
Barth Syndrome Foundation. Medications used in Barth syndrome. 2018. Available from: https://www.barthsyndrome.org/file_download/inline/9339f952-f5cf-4f51-958a-4c89941b2b04. Accessed 10 Feb 2019.
Ferreira C, Thompson R, Vernon H. Barth Syndrome. 2014 Oct 9. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK247162/
Cassidy J, Haynes S, Kirk R et al (2009) Changing patterns of bridging to heart transplantation in children. J Heart Lung Transplant 28:249–254. https://doi.org/10.1016/j.healun.2008.11.912
Karimova A, Van Doorn C, Brown K et al (2011) Mechanical bridging to orthotopic heart transplantation in children weighing less than 10 kg: Feasibility and limitations. Eur J Cardiothorac Surg 39:304–309. https://doi.org/10.1016/j.ejcts.2010.05.015
Holman WL, Park SJ, Long JW et al (2004) Infection in permanent circulatory support: Experience from the REMATCH trial. J Heart Lung Transplant 23:1359–1365. https://doi.org/10.1016/j.healun.2003.09.025
Dedieu N, Giardini A, Steward CG et al (2012) Successful mechanical circulatory support for 251 days in a child with intermittent severe neutropenia due to Barth syndrome. Pediatr Transplant 17(2):E46–E49. https://doi.org/10.1111/petr.12027
Steward CG, Groves SJ, Taylor CT et al (2019) Neutropenia in Barth syndrome: Characteristics, risks, and management. Curr Opin Hematol 26:6–15. https://doi.org/10.1097/MOH.0000000000000472
Jefferies JL (2013) Barth syndrome. Am J Med Genet C Semin Med Genet 163C:198–205. https://doi.org/10.1002/ajmg.c.31372
Kelley R (2008) Management of diarrheal illness in patients with Barth syndrome. https://www.barthsyndrome.org.uk/userfiles/Factsheets/ManagementofDiarrheainBTHSFactSheet.pdf. Accessed 10 May 2020
Barth syndrome foundation. Management of diarrheal illness in patients with Barth syndrome. 2008. Available from: https://www.barthsyndrome.org/file_download/inline/d60a606e-fc62-40b0-bdf0-736edfa5d11a. Accessed 2 March 2019
Ostman-Smith I, Brown G, Johnson A, Land JM (1994) Dilated cardiomyopathy due to type II X-linked 3-methylglutaconic aciduria: Successful treatment with pantothenic acid. Br Heart J 72:349–353. https://doi.org/10.1136/hrt.72.4.349
Reynolds S, Kreider CM, Meeley LE, Bendixen RM (2015) Taste perception and sensory sensitivity: Relationship to feeding problems in boys with Barth Syndrome. J Rare Disord 3:1–9
Mason SJ, Harris G, Blissett J (2005) Tube feeding in infancy: Implications for the development of normal eating and drinking skills. Dysphagia 20(1):46–61. https://doi.org/10.1007/s00455-004-0025-2
Sabater-Molina M, Guillén-Navarro E, García-Molina E et al (2013) Barth syndrome in adulthood: A clinical case. Revista Española de Cardiología (English Edition) 66(1):68–70. https://doi.org/10.1016/j.rec.2012.05.012
Singh HR, Yang Z, Siddiqui S et al (2009) A novel Alu-mediated Xq28 microdeletion ablates TAZ and partially deletes DNL1L in a patient with Barth syndrome. Am J Med Genet Part A 149A(5):1082–1085. https://doi.org/10.1002/ajmg.a.32822
Bashir A, Bohnert KL, Reeds DN et al (2017) Impaired cardiac and skeletal muscle bioenergetics in children, adolescents, and young adults with Barth syndrome. Physiol Rep 5:13130. https://doi.org/10.14814/phy2.13130
Jeppesen TD, Schwartz M, Olsen DB et al (2006) Aerobic training is safe and improves exercise capacity in patients with mitochondrial myopathy. Brain 129(12):3402–3412. https://doi.org/10.1093/brain/awl149
Acehan D, Vaz F, Houtkooper RH et al (2011) Cardiac and skeletal muscle defects in a mouse model of human Barth syndrome. J Biol Chem 286:899–908. https://doi.org/10.1074/jbc.M110.171439
Soustek MS, Baligand C, Falk DJ et al (2015) Endurance training ameliorates complex 3 deficiency in a mouse model of Barth syndrome. J Inherit Metab Dis 38:915–922. https://doi.org/10.1007/s10545-015-9834-8
Radak Z, Zhao Z, Koltai E et al (2013) Oxygen consumption and usage during physical exercise: The balance between oxidative stress and ROS-dependent adaptive signaling. Antioxid Redox Signal 18:1208–1246. https://doi.org/10.1089/ars.2011.4498
Cade WT, Reeds DN, Peterson LR et al (2017) Endurance exercise training in young adults with Barth syndrome: A pilot study. JIMD Rep 32:15–24. https://doi.org/10.1007/8904_2016_553
Taivassalo T, Shoubridge EA, Chen J et al (2001) Aerobic conditioning in patients with mitochondrial myopathies: Physiological, biochemical, and genetic effects. Ann Neurol 50(2):133–141. https://doi.org/10.1002/ana.1050
Bittel AJ, Bohnert KL, Reeds DN et al (2018) Reduced muscle strength in Barth syndrome may be improved by resistance exercise training: A pilot study. JIMD Rep 41:63–72. https://doi.org/10.1007/8904_2018_102
Romualdo B, Demetrios G, Giovanni C, Augusto P (1999) Randomized, controlled trial of long-term moderate exercise training in chronic heart failure. Circulation 99:1173–1182. https://doi.org/10.1161/01.CIR.99.9.1173
De Maeyer C, Beckers P, Vrints CJ, Conraads VM (2013) Exercise training in chronic heart failure. Ther Adv Chronic Dis 4:105–117. https://doi.org/10.1177/2040622313480382
Consitt LA, Dudley C, Saxena G (2019) Impact of endurance and resistance training on skeletal muscle glucose metabolism in older adults. Nutrients 11:2636. https://doi.org/10.3390/nu11112636
Hanke SP, Gardner AB, Lombardi JP et al (2012) Left ventricular noncompaction cardiomyopathy in Barth syndrome: An example of an undulating cardiac phenotype necessitating mechanical circulatory support as a bridge to transplantation. Pediatr Cardiol 33:1430–1434. https://doi.org/10.1007/s00246-012-0258-z
Nicolson GL, Ash ME (2014) Lipid replacement therapy: A natural medicine approach to replacing damaged lipids in cellular membranes and organelles and restoring function. Biochim Biophys Acta Biomembr 1838:1657–1679. https://doi.org/10.1016/j.bbamem.2013.11.010
Valianpour FF, Wanders R, Overmars H (2002) Linoleic acid supplementation of Barth syndrome fibroblasts restores cardiolipin levels: Implications for treatment. J Lipid Res 44:560–566
Ikon N, Su B, Hsu F-F et al (2015) Exogenous cardiolipin localizes to mitochondria and prevents TAZ knockdown-induced apoptosis in myeloid progenitor cells. Biochem Biophys Res Commun 464:580–585. https://doi.org/10.1016/j.bbrc.2015.07.012
Soustek MS, Falk DJ, Mah CS et al (2011) Characterization of a transgenic short hairpin RNA-induced murine model of Tafazzin deficiency. Hum Gene Ther 22:865–871. https://doi.org/10.1089/hum.2010.199
Phoon CKL, Acehan D, Schlame M et al (2012) Tafazzin knockdown in mice leads to a developmental cardiomyopathy with early diastolic dysfunction preceding myocardial noncompaction. J Am Heart Assoc 1:jah3-e00455. https://doi.org/10.1161/JAHA.111.000455
Ikon N, Hsu F-F, Shearer J et al (2018) Evaluation of cardiolipin nanodisks as lipid replacement therapy for Barth syndrome. J Biomed Res 32:107–112. https://doi.org/10.7555/JBR.32.20170094
Brand MD, Nicholls DG (2011) Assessing mitochondrial dysfunction in cells. Biochem J 437:575–575
McKenzie M, Lazarou M, Thorburn DR, Ryan MT (2006) Mitochondrial respiratory chain supercomplexes are destabilized in Barth syndrome patients. J Mol Biol 361:462–469. https://doi.org/10.1016/j.jmb.2006.06.057
Paradies G, Petrosillo G, Pistolese M, Ruggiero FM (2002) Reactive oxygen species affect mitochondrial electron transport complex I activity through oxidative cardiolipin damage. Gene 286:135–141. https://doi.org/10.1016/S0378-1119(01)00814-9
Grygiel-Górniak B (2014) Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications–a review. Nutr J 13:17–17. https://doi.org/10.1186/1475-2891-13-17
Tyagi S, Gupta P, Saini AS et al (2011) The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J Adv Pharm Technol Res 2:236–240. https://doi.org/10.4103/2231-4040.90879
Tenenbaum A, Motro M, Fisman EZ et al (2005) Bezafibrate for the secondary prevention of myocardial infarction in patients with metabolic syndrome. Arch Intern Med 165:1154–1160. https://doi.org/10.1001/archinte.165.10.1154
Bonnefont JP, Bastin J, Laforêt P et al (2010) Long-term follow-up of bezafibrate treatment in patients with the myopathic form of carnitine palmitoyltransferase 2 deficiency. Clin Pharmacol Ther 88(1):101–108. https://doi.org/10.1038/clpt.2010.55
Huang Y, Powers C, Moore V et al (2017) The PPAR pan-agonist bezafibrate ameliorates cardiomyopathy in a mouse model of Barth syndrome. Orphanet J Rare Dis 12:49–49. https://doi.org/10.1186/s13023-017-0605-5
Schafer C, Moore V, Dasgupta N et al (2018) The effects of PPAR stimulation on cardiac metabolic pathways in Barth syndrome mice. Front Pharmacol 9:318–318. https://doi.org/10.3389/fphar.2018.00318
Xu Y, Kelley RI, Blanck TJJ, Schlame M (2003) Remodeling of cardiolipin by phospholipid transacylation. J Biol Chem 278:51380–51385. https://doi.org/10.1074/jbc.M307382200
Suzuki-Hatano S, Saha M, Rizzo SA, Witko RL et al (2019) AAV-mediated TAZ gene replacement restores mitochondrial and cardioskeletal function in Barth syndrome. Hum Gene Ther 30(2):139–154. https://doi.org/10.1089/hum.2018.020
Schnepp BC, Clark KR, Klemanski DL et al (2003) Genetic fate of recombinant adeno-associated virus vector genomes in muscle. J Virol 77:3495–3504. https://doi.org/10.1128/jvi.77.6.3495-3504.2003
Swijnenburg R-J, Schrepfer S, Govaert JA et al (2008) Immunosuppressive therapy mitigates immunological rejection of human embryonic stem cell xenografts. Proc Natl Acad Sci USA 105:12991–12996. https://doi.org/10.1073/pnas.0805802105
Liang P, Du J (2014) Human induced pluripotent stem cell for modeling cardiovascular diseases. Regen Med Res 2:4–4. https://doi.org/10.1186/2050-490X-2-4
Povsic TJ, O’Connor CM, Henry T et al (2011) A double-blind, randomized, controlled, multicenter study to assess the safety and cardiovascular effects of skeletal myoblast implantation by catheter delivery in patients with chronic heart failure after myocardial infarction. Am Heart J 162:654-662.e1. https://doi.org/10.1016/j.ahj.2011.07.020
Makkar RR, Smith RR, Cheng K et al (2012) Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): A prospective, randomised phase 1 trial. Lancet 379:895–904. https://doi.org/10.1016/S0140-6736(12)60195-0
Saric A, Andreau K, Armand A-S et al (2016) Barth syndrome: From mitochondrial dysfunctions associated with aberrant production of reactive oxygen species to pluripotent stem cell studies. Front Genet 6:359–359. https://doi.org/10.3389/fgene.2015.00359
Wang G, McCain ML, Yang L et al (2014) Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat Med 20:616–623. https://doi.org/10.1038/nm.3545
Addabbo F, Montagnani M, Goligorsky MS (2009) Mitochondria and reactive oxygen species. Hypertension 53:885–892. https://doi.org/10.1161/HYPERTENSIONAHA.109.130054
Takimoto E, Kass DA (2007) Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension 49(2):241–248. https://doi.org/10.1161/01.hyp.0000254415.31362.a7
Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE (2009) Mitochondria and reactive oxygen species. Free Radical Biol Med 47:333–343. https://doi.org/10.1016/j.freeradbiomed.2009.05.004
He Q, Harris N, Ren J, Han X (2014) Mitochondria-targeted antioxidant prevents cardiac dysfunction induced by tafazzin gene knockdown in cardiac myocytes. Oxid Med Cell Longev 2014:654198–654198. https://doi.org/10.1155/2014/654198
He Q (2010) Tafazzin knockdown causes hypertrophy of neonatal ventricular myocytes. Am J Physiol Heart Circ Physiol 299(1):210–216. https://doi.org/10.1152/ajpheart.00098.2010
Liu C, Cao F, Tang Q-Z et al (2010) Allicin protects against cardiac hypertrophy and fibrosis via attenuating reactive oxygen species-dependent signaling pathways. J Nutr Biochem 21:1238–1250. https://doi.org/10.1016/j.jnutbio.2009.11.001
Ott M, Robertson JD, Gogvadze V et al (2002) Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci USA 99:1259–1263. https://doi.org/10.1073/pnas.241655498
Hersh SP (2010) Mitochondria: An emerging target for therapeutics. Clin Pharmacol Ther 87(6):630–632. https://doi.org/10.1038/clpt.2010.22
Szeto HH, Birk AV (2014) Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clin Pharmacol Ther 96:672–683. https://doi.org/10.1038/clpt.2014.174
Zhao K, Zhao G-M, Wu D et al (2004) Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem 279:34682–34690. https://doi.org/10.1074/jbc.M402999200
Szeto HH (2014) First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol 171:2029–2050. https://doi.org/10.1111/bph.12461
Zhao K, Luo G, Zhao G-M et al (2003) Transcellular transport of a highly polar 3+ net charge opioid tetrapeptide. J Pharmacol Exp Ther 304:425. https://doi.org/10.1124/jpet.102.040147
Birk AV, Chao WM, Bracken C et al (2014) Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br J Pharmacol 171:2017–2028. https://doi.org/10.1111/bph.12468
Paradies G, Ruggiero FM, Petrosillo G, Quagliariello E (1997) Age-dependent decline in the cytochrome c oxidase activity in rat heart mitochondria: Role of cardiolipin. FEBS Lett 406:136–138. https://doi.org/10.1016/S0014-5793(97)00264-0
Lesnefsky EJ, Gudz TI, Moghaddas S et al (2001) Aging decreases electron transport complex III activity in heart interfibrillar mitochondria by alteration of the cytochrome c binding site. J Mol Cell Cardiol 33:37–47. https://doi.org/10.1006/jmcc.2000.1273
Petrosillo G, Matera M, Casanova G et al (2008) Mitochondrial dysfunction in rat brain with aging: Involvement of complex I, reactive oxygen species and cardiolipin. Neurochem Int 53:126–131. https://doi.org/10.1016/j.neuint.2008.07.001
Brown DA, Hale SL, Baines CP et al (2014) Reduction of early reperfusion injury with the mitochondria-targeting peptide bendavia. J Cardiovasc Pharmacol Ther 19:121–132. https://doi.org/10.1177/1074248413508003
Brown DA, O’Rourke B (2010) Cardiac mitochondria and arrhythmias. Cardiovasc Res 88:241–249. https://doi.org/10.1093/cvr/cvq231
Piot C, Croisille P, Staat P et al (2008) Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med 359:473–481. https://doi.org/10.1056/NEJMoa071142
Birk AV, Liu S, Soong Y et al (2013) The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J Am Soc Nephrol 24:1250–1261. https://doi.org/10.1681/ASN.2012121216
Dai D-F, Chen T, Szeto H et al (2011) Mitochondrial targeted antioxidant Peptide ameliorates hypertensive cardiomyopathy. J Am Coll Cardiol 58:73–82. https://doi.org/10.1016/j.jacc.2010.12.044
Dai D-F, Hsieh EJ, Chen T et al (2013) Global proteomics and pathway analysis of pressure-overload-induced heart failure and its attenuation by mitochondrial-targeted peptides. Circ Heart Fail 6:1067–1076. https://doi.org/10.1161/CIRCHEARTFAILURE.113.000406
Levine S, Budak MT, Dierov J, Singhal S (2011) Inactivity-induced diaphragm dysfunction and mitochondria-targeted antioxidants: New concepts in critical care medicine*. Crit Care Med 39:1844–1845
Szeto HH, Schiller PW (2011) Novel therapies targeting inner mitochondrial membrane–from discovery to clinical development. Pharm Res 28:2669–2679. https://doi.org/10.1007/s11095-011-0476-8
Mitchell W, Ng EA, Tamucci JD et al (2020) The mitochondria-targeted peptide SS-31 binds lipid bilayers and modulates surface electrostatics as a key component of its mechanism of action. J Biol Chem 295:7452–7469. https://doi.org/10.1074/jbc.RA119.012094
Barth Syndrome Foundation.Summary of stealth BioTherapeutics’s Webinar On clinical trial TAZPOWER.2018. Available from: https://www.barthsyndrome.org/research/clinicaltrials/tazpower.html. Accessed 16 March 2019
Salehi B, Mishra AP, Nigam M et al (2018) Resveratrol: A double-edged sword in health benefits. Biomedicines 6:91. https://doi.org/10.3390/biomedicines6030091
Albertoni G, Schor N (2015) Resveratrol plays important role in protective mechanisms in renal disease – mini-review. Brazilian J Nephrol 37:106–114
Gambini J, Inglés M, Olaso G et al (2015) Properties of resveratrol: In vitro and in vivo studies about metabolism, bioavailability, and biological effects in animal models and humans. Oxid Med Cell Longev 2015:837042. https://doi.org/10.1155/2015/837042
Dolinsky VW, Cole LK, Sparagna GC, Hatch GM (2016) Cardiac mitochondrial energy metabolism in heart failure: Role of cardiolipin and sirtuins. Biochim Biophys Acta Mole Cell Biol Lipids 1861:1544–1554. https://doi.org/10.1016/j.bbalip.2016.03.008
Bagul PK, Katare PB, Bugga P et al (2018) SIRT-3 modulation by resveratrol improves mitochondrial oxidative phosphorylation in diabetic heart through deacetylation of TFAM. Cells 7:235. https://doi.org/10.3390/cells7120235
Pan H, Finkel T (2017) Key proteins and pathways that regulate lifespan. J Biol Chem 292:6452–6460. https://doi.org/10.1074/jbc.R116.771915
Huang D-D, Shi G, Jiang Y et al (2020) A review on the potential of Resveratrol in prevention and therapy of diabetes and diabetic complications. Biomed Pharmacother 125:109767. https://doi.org/10.1016/j.biopha.2019.109767
Yashiro T, Nanmoku M et al (2012) Resveratrol increases the expression and activity of the low density lipoprotein receptor in hepatocytes by the proteolytic activation of the sterol regulatory element-binding proteins. Atherosclerosis 220:369–374
Herzig S, Shaw RJ (2018) AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol 19:121–135. https://doi.org/10.1038/nrm.2017.95
Zong H, Ren JM, Young LH et al (2002) AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci USA 99:15983–15987. https://doi.org/10.1073/pnas.252625599
Tanno M, Kuno A, Yano T et al (2010) Induction of manganese superoxide dismutase by nuclear translocation and activation of SIRT1 promotes cell survival in chronic heart failure. J Biol Chem 285:8375–8382. https://doi.org/10.1074/jbc.M109.090266
Schrauwen P, Schrauwen-Hinderling V, Hoeks J, Hesselink MKC (2010) Mitochondrial dysfunction and lipotoxicity. Biochim Biophys Acta Mol Cell Biol Lipids 1801:266–271. https://doi.org/10.1016/j.bbalip.2009.09.011
Wende AR, Abel ED (2010) Lipotoxicity in the heart. Biochim Biophys Acta 1801:311–319. https://doi.org/10.1016/j.bbalip.2009.09.023
Cole LK, Mejia EM, Sparagna GC et al (2020) Cardiolipin deficiency elevates susceptibility to a lipotoxic hypertrophic cardiomyopathy. J Mol Cell Cardiol 144:24–34. https://doi.org/10.1016/j.yjmcc.2020.05.001
Hauff KD, Hatch GM (2006) Cardiolipin metabolism and Barth syndrome. Prog Lipid Res 45:91–101. https://doi.org/10.1016/j.plipres.2005.12.001
Agarwal P, Cole LK, Chandrakumar A et al (2018) Phosphokinome analysis of Barth syndrome lymphoblasts identify novel targets in the pathophysiology of the disease. Int J Mol Sci 19:2026. https://doi.org/10.3390/ijms19072026
Raja V, Greenberg ML (2014) The functions of cardiolipin in cellular metabolism-potential modifiers of the Barth syndrome phenotype. Chem Phys Lipid 179:49–56. https://doi.org/10.1016/j.chemphyslip.2013.12.009
Gaspard GJ, McMaster CR (2015) Cardiolipin metabolism and its causal role in the etiology of the inherited cardiomyopathy Barth syndrome. Chem Phys Lipid 193:1–10. https://doi.org/10.1016/j.chemphyslip.2015.09.005
Ikon N, Ryan RO (2017) Barth syndrome: Connecting cardiolipin to cardiomyopathy. Lipids 52:99–108. https://doi.org/10.1007/s11745-016-4229-7
Ikon N, Ryan RO (2016) On the origin of 3-methylglutaconic acid in disorders of mitochondrial energy metabolism. J Inherit Metab Dis 39:749–756. https://doi.org/10.1007/s10545-016-9933-1
Gibson KM, Sherwood WG, Hoffman GF, Stumpf DA, Dianzani I, Schutgens RB, Barth PG, Weismann U, Bachmann C, Schrynemackers-Pitance P, Verloes A, Narisawa K, Mino M, Ohya A, Kelley RI (1991) Phenotypic heterogeneity in the syndromes of 3-methylglutaconic aciduria. J Pediatr 118:885–90. https://doi.org/10.1016/s0022-3476(05)82199-7
Roullet JB, Merkens LS, Pappu AS, Jacobs MD, Winter R, Connor WE, Steiner RD (2012) No evidence for mevalonate shunting in moderately affected children with Smith-Lemli-Opitz syndrome. J Inherit Metab Dis 35:859–869. https://doi.org/10.1007/s10545-012-9453-6
Hsu YH, Dumlao DS, Cao J, Dennis EA (2013) Assessing phospholipase A2 activity toward cardiolipin by mass spectrometry. PloS ONE 8:e59267. https://doi.org/10.1371/journal.pone.0059267
Goncalves RLS, Schlame M, Bartelt A, Brand MD, Hotamışlıgil GS (2020) Cardiolipin deficiency in Barth syndrome is not associated with increased superoxide/H 2 O 2 production in heart and skeletal muscle mitochondria. FEBS Lett. https://doi.org/10.1002/1873-3468.13973
Le CH, Benage LG, Specht KS, Li Puma LC, Mulligan CM, Heuberger AL, Prenni JE, Claypool SM, Chatfield KC, Sparagna GC, Chicco AJ (2020) Tafazzin deficiency impairs CoA-dependent oxidative metabolism in cardiac mitochondria. J Biol Chem 295:12485–12497. https://doi.org/10.1074/jbc.RA119.011229
Petit PX, Ardilla-Osorio H, Penalvia L, Rainey NE (2020) Tafazzin mutation affecting cardiolipin leads to increased mitochondrial superoxide anions and mitophagy inhibition in Barth syndrome. Cells 9:2333. https://doi.org/10.3390/cells9102333
Li Y, Lou W, Grevel A, Böttinger L, Liang Z, Ji J, Patil VA, Liu J, Ye C, Hüttemann M, Becker T, Greenberg ML (2020) Cardiolipin-deficient cells have decreased levels of the iron-sulfur biogenesis protein frataxin. J Biol Chem 295:11928–11937. https://doi.org/10.1074/jbc.RA120.013960
Ghosh S, Basu Ball W, Madaris TR, Srikantan S, Madesh M, Mootha VK, Gohil VM (2020) An essential role for cardiolipin in the stability and function of the mitochondrial calcium uniporter. Proc Natl Acad Sci 117:16383–16390. https://doi.org/10.1073/pnas.2000640117
Bozelli JC Jr, Lu D, Atilla-Gokcumen GE, Epand RM (2020) Promotion of plasmalogen biosynthesis reverse lipid changes in a Barth Syndrome cell model. Biochim Biophys Acta Mol Cell Biol Lipids 1865:158677. https://doi.org/10.1016/j.bbalip.2020.158677
Acknowledgements
The authors wish to thank Dr. Kristin D. Hauff and Dr. Edgard M. Mejia for the data in Figs. 2, 3, and 7. The work conducted for Figs. 2, 3, and 7 was performed under biosafety approval from the University of Manitoba. This work was supported by grants from HSFC, NSERC, CHRIM, and the BSF to GMH. HZ was supported by a traineeship from the Canadian Bureau for International Education and the Libyan North American Scholarship Program. GMH is the Canada Research Chair in Molecular Cardiolipin Metabolism.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zegallai, H.M., Hatch, G.M. Barth syndrome: cardiolipin, cellular pathophysiology, management, and novel therapeutic targets. Mol Cell Biochem 476, 1605–1629 (2021). https://doi.org/10.1007/s11010-020-04021-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-020-04021-0