
Abstract
Therapies for mitochondrial diseases has been largely limited to supportive and symptomatic therapies; however, in the last decade, advances in understanding the causes and pathomechanisms of these diverse disorders have enabled development of novel treatment strategies. Here, we highlight current use of dietary supplements and exercise therapy as well as emerging treatments in preclinical and clinical trial stages of development. Broad-spectrum therapies that may be applied multiple diseases include: activation of mitochondrial biogenesis, regulation of mitophagy and mitochondrial dynamics, bypass of mitochondrial biochemical defects, mitochondrial replacement therapy, and hypoxia. Tailored disease-specific therapies in development include: scavenging of toxic compounds, deoxynucleoside therapy, cell replacement therapies, viral-mediated gene-delivery, shifting heteroplasmy of mitochondrial DNA pathogenic variants, and stabilization of mitochondrial transfer RNAs.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

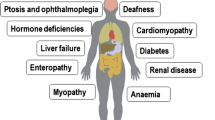

11.1 Introduction
Mitochondrial disorders (MDs) represent a heterogeneous group of inborn errors of the oxidative phosphorylation (OXPHOS) system in mitochondria, where most of the cell’s ATP is generated. This metabolic pathway is under the dual genetic control of the mitochondrial and nuclear genomes.
The genetic complexity only partially accounts for the clinical heterogeneity of these disorders. Onset varies widely from childhood to adulthood, even among members of the same family. Virtually every organ system can be affected by mitochondrial dysfunction (in particular brain, skeletal and cardiac muscle) and as a consequence, MDs are often multisystemic (DiMauro et al. 2013).
Taken together, MDs have an estimated prevalence greater than 1:5000 individuals (Schaefer et al. 2008). The 10 most common pathogenic mitochondrial DNA (mtDNA) mutations alone have an estimated incidence of 1 in 200 infants (Elliott et al. 2008). Given their prevalence and their progressive nature, often worsening over many decades, these disorders cause substantial morbidity among both pediatric and adult populations.
Currently, only supportive care is available for the vast majority of patients with MDs (Pfeffer et al. 2012; Kerr 2013). However, extraordinary progress has been made in recent years in understanding the pathogenesis of these disorders (Hirano et al. 2018). Based on this knowledge, therapeutic strategies have been proposed and experimental evidence is increasing in in vitro and in vivo studies.
Numerous challenges albeit hamper the translation of these therapies from bench to bedside. The main challenges are caused by the genetic, biochemical, and phenotypic variability of MDs. This heterogeneity, for instance, makes it difficult to collect a sufficient number of patients to conduct reliable natural history studies, clinical trials, and to identify universally validated outcome measures.
In this chapter, we will discuss separately current therapeutic approaches, including supportive therapy, the results of previous clinical trials, and emerging therapies that have shown promising results at preclinical and clinical level.Footnote 1
11.2 Current Treatments in Clinical Practice
11.2.1 Supportive Treatments
Supportive treatments are often the only available option in the management of mitochondrial patients when a specific therapy is lacking. For the vast majority of patients, therapy is limited to either preventing or treating the complications of MDs. Nevertheless, supportive measures are extremely important and can significantly improve quality of life and survival in this group of patients. Symptomatic approaches often require a multidisciplinary team for early recognition and treatment of complications/manifestations.
Epileptic seizures commonly occur in MDs, such as, for example, Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS), Myoclonic epilepsy with ragged red fibers (MERRF) and Leigh syndrome. Most available anticonvulsants can be used, except sodium valproate that can cause carnitine deficiency and may precipitate hepatic failure in Alpers syndrome (Rahman 2012). Myoclonic epilepsy in MERRF can be treated effectively with levetiracetam, clonazepam, or zonisamide (DiMauro and Hirano 1993). Dystonia, often present in Leigh syndrome, can benefit of anti-dystonia oral medications (e.g. anticholinergic, neuroleptics) or botulinum toxin injection.
Heart is often affected in MDs. Conduction defects are commonly present in patients with Kearns-Sayre syndrome (KSS), but also in Leber hereditary optic neuropathy (LHON), and m.3243A>G mutation. Timely placement of a pacemaker can be lifesaving in KSS patients. Cardiomyopathy can also occur and it is estimated to affect 20–40% of children with MDs (Scaglia et al. 2004). Hypertrophic cardiomyopathy is the most common manifestation, being present in 50% of mitochondrial patients with cardiac involvement (Finsterer and Kothari 2014). MDs presenting cardiomyopathy can be due to defects in respiratory chain complexes subunits and assembly factors, mitochondrial tRNAs, rRNAs, ribosomal proteins, translation factors, mtDNA maintenance, CoQ10 synthesis, or defect of lipid milieau, like Barth syndrome (El-Hattab and Scaglia 2016). Cardiac involvement should be closely monitored with by a cardiologist and pharmacologically or surgically treated when necessary.
Endocrine dysfunction can be present in MDs, and hormone replacement can be necessary (insulin, thyroxine, growth hormone). In the case of diabetes mellitus, for instance in patients with MELAS and Maternally Inherited Diabetes-Deafness (MIDD) syndrome, diet and low doses of insulin/oral hypoglycemic drugs are usually sufficient to maintain the euglycemic state. Metformin should be avoided because it can cause lactic acidosis (Murphy et al. 2008).
Gastrointestinal (GI) problems are also common in patients with mitochondrial disorders and include dysphagia, weight loss, constipation, pseudo-obstruction, nausea, failure to thrive. Among syndromic MDs, Mitochondrial neurogastrointestinal encephalopathy (MNGIE) is the one with prevalent GI involvement (Garone et al. 2011). Adequate nutrition can be achieved with hypercaloric nutritional supplements but may require PEG (percutaneous or parenteral nutrition endoscopic gastrostomy) feeding in severe cases (Finsterer and Frank 2017).
Other supportive therapies include electrolyte replacement, renal dialysis or transplantation in patients with renal involvement, uncommon but described in patients with mitochondrial DNA or nuclear DNA mutations (O’Toole 2014); blood transfusion in case of anemia in Pearson syndrome or other forms of anemia; respiratory support for restrictive lung disease; and psychological support for patients and their families.
Non-pharmacologic approaches, include the use of hearing aids or cochlear implants for patients with hearing loss (Sinnathuray et al. 2003), and eyelid surgery for ptosis, as a mean to improve not only the vision but also the psychological wellbeing and social interaction of the patient.
11.2.2 Pharmacological Approaches
Multiple vitamins and cofactors are often used in patients with mitochondrial disorders, although these therapies are not yet standardized or definitively proven to be effective. The dietary supplements are used with different purposes, such as: (1) increase the respiratory chain flux (CoenzymeQ10 [CoQ10], riboflavin), (2) serve as antioxidants (e.g. CoQ10, idebenone, alpha-lipoic acid, vitamins C and E), and/or act as cofactors (e.g. riboflavin, thiamine), or (3) function as mitochondrial substrates (L-carnitine). Accumulation of reactive oxygen species (ROS) as a toxic byproduct of a mitochondrial respiratory chain dysfunction may lead to cellular damage contributing to the pathogenesis of MDs. Moreover, a transgenic murine model overexpressing a catalase targeted to mitochondria extended life span (Schriner et al. 2005). Based on this rational, antioxidants have been frequently used in the treatment of mitochondrial patients. CoQ10 is the most commonly utilized and many clinical trials have been investigating its efficacy and that of its analogues, like idebenone and EPI-743, in different MDs (see Sect. 11.3). Other antioxidants like vitamins C and E might also be beneficial in patients with MDs. An example is an analogue of vitamin E, trolox ornithylamide hydrochloride, when applied to fibroblasts from patients with Leigh syndrome reduced ROS levels and an increased activities of mitochondrial complexes I and IV, and citrate synthase (Blanchet et al. 2015). The efficacy of antioxidants in patients with MDs nonetheless remains controversial. A Cochrane review of mitochondrial therapies has found little evidence supporting the use of any vitamin or cofactor (Pfeffer et al. 2012). However, benefits of various agents (riboflavin, alpha-lipoic acid, etc) have been anecdotally reported. Consensus recommendations from the Mitochondrial Medicine Society recently reported (Parikh et al. 2015) aimed to standardize treatment options for mitochondrial patients. According to these recommendations, patients with primary mitochondrial disorders, and not only CoQ10 deficiency, should be offered CoQ10 in its reduced form (ubiquinol), and plasma or leukocyte levels should be monitored to assess adherence to treatment.
In addition, alpha-lipoic acid (ALA) and riboflavin are frequently offered to mitochondrial patients. L-carnitine should be administered when deficient. Folinic acid should be given to mitochondrial patients when deficient and the central nervous system is involved. Moreover, supplements should be given starting with one supplement at the time, avoiding “cocktails” initially.
Effective treatment of acute stroke-like episodes or their prevention has not been established. Open-label studies suggest that treatment of acute mitochondrial stroke-like episodes with intravenous (IV) arginine hydrochloride, a precursor of nitric oxide, is beneficial for patient with the m.3243 A>G mutation in MTTL1 (Koenig et al. 2016). IV arginine administration should be considered in acute stroke-like episodes associated with other primary mitochondrial disorders. Open-label studies also suggest that daily oral arginine to prevent strokes should be considered in MELAS patients with the m.3243 A>G mutation (Parikh et al. 2015).
Lastly, some drugs should be avoided in patients with mitochondrial dysfunction, or used with caution: valproic acid, statins, metformin, high-dose acetaminophen, and specific antibiotics (i.e. aminoglycosides, linezolid, tetracycline, azithromycin, erythromycin).
A survey on the use of dietary supplements was recently conducted by the North American Mitochondrial Disease Consortium (NAMDC) (Karaa et al. 2016). The survey pointed out how the majority of patients takes four or more dietary supplements, despite the recent recommendation of the Mitochondrial Medicine Society. Even though no or minor side effects were reported, and patients noted overall some improvements, the economic burden to the families was considerable; 90% of patients purchased the supplements out-of-pocket. The authors conclude that this burden and the potential side effects are not justifiable, considering the lack of evidence for using these “cocktails”. Importantly, this survey reveal an increase interest towards patients’ perception of their care and quality of life; this approach is fundamental to determine reliable outcome measures to use in future well-conducted clinical trials.
11.2.3 Exercise
Exercise has been proven beneficial in some patients with MDs (Voet et al. 2013; Tarnopolsky 2014). In particular, aerobic endurance training can increase mitochondrial mass, by stimulating mitochondrial biogenesis, and increase muscle mitochondrial enzyme activities and muscle strength. Endurance training has been proven beneficial and safe in trials of patients with mitochondrial DNA mutations (Taivassalo et al. 2001, 2006). A combination of progressive endurance with or without resistance exercise should be recommended to mitochondrial patients (Parikh et al. 2015).
11.3 Clinical Trials
In 2012 a systematic review by the Cochrane collaboration evaluated 1335 studies comparing pharmacological and non-pharmacological treatments for MDs (Pfeffer et al. 2012). Only 12 studies were selected for inclusion in the review, the most common reason for exclusion being lack of randomization/blinding and presence of methodological biases. The primary outcome measures included any change in muscle strength or neurological features. Secondary outcome measures included quality life evaluation, biochemical biomarkers (e.g. lactic acidosis), and negative outcomes. The 12 studies investigated the effects of CoQ10, dichloroacetate (DCA), creatinine, dimethylglycine, whey-based cysteine and combination therapy of creatine, α-lipoic acid and CoQ10. Dramatic effects were not observed in any of these studies, and one trial assessing the effects of DCA in MELAS patients had to be terminated because of toxicity (NCT00068913). Several clinical trials are currently underway or have been recently completed, but the results were not published for most of them and are still unclear. The majority of the studies focused on patients with MELAS and LHON, which could be studied in relatively large cohorts. Many other trials analyzed less homogeneous cohort of patients, including, for instance, patients with similar phenotype (i.e. mitochondrial myopathy) but different genetic background. A summary of the studies is reported in Table 11.1. We will briefly discuss some examples.
MELAS Syndrome
MELAS is one of the most frequent maternally inherited mitochondrial disorders. The pathogenesis of this disorder is not completely understood and results from different factor. Energy failure due to faulty mitochondria is a common feature of MDs as well as the overproduction of ROS. Different approaches have been studied in order to reduce the oxidative stress in MDs and in particular in MELAS patients. Idebenone is a short-tail ubiquinone synthetic analog, which is more water-soluble compared to CoQ10 and act as antioxidant. Several studies have been conducted to assess the efficacy and safety of idebenone in Friedreich Ataxia (FA), and proven that idebenone is in fact well tolerated and may stabilize neurological features in this disorder (Mariotti et al. 2003; Di Prospero et al. 2007a, b; Meier et al. 2012; Lynch et al. 2010). A phase 2a, randomized, double blind, placebo-control, dose-finding study in patients with MELAS syndrome has recently been completed and showed that the primary endopoint did not reach statistical significance (NCT00887562). KH176 is a small molecule derived from Vitamin E and is a potent ROS scavenger. After the completion of a dose escalating clinical trial with KH176 in healthy individual that has demonstrated good tolerability and a promising pharmacokinetic profile (NCT02544217), a randomized trial studied 18 adults with the m.3243A>G mutation (NCT02909400). Twice daily oral 100mg KH176 was well-tolerated and appeared safe. Primary outcome (gait measures) did not reach statistical significance, but possible positive effects on alterness and mood were noted (Janssen et al. 2019). Moreover, bezafibrate is an activator of the transcription factor peroxisomal proliferator receptors (PPARs), that in turn when activated promote transcription of mitochondrial genes. Its efficacy is being evaluated in patients with m.3243A>G mutation (NCT02398201) and evidence of myopathy, but results are not available at the moment. In addition to energy failure and ROS accumulation, there has been growing evidence that nitric oxide (NO) deficiency play a central role in the pathogenesis of the stroke-like episodes (El-Hattab et al. 2016). Arginine is the substrate of nitric oxide synthase, which produces NO, therefore arginine is a promising treatment for MELAS patients. Multiple open-label trials have been conducted (NCT01603446, JMA-IIA00023, and JMA-IIA00025) and have shown the efficacy of chronic oral administration (Koga et al. 2006, 2007; Rodan et al. 2015) and acute intravenous administration (Koga et al. 2005, 2006) of arginine in patients with MELAS syndrome, although a placebo-controlled randomized clinical trial has not yet been conducted. Furthermore, preliminary evidence has been provided for the effect of citrulline in stroke-like episodes in MELAS patients (El-Hattab et al. 2016) and a Phase 1, open-label study has been conducted (NCT01339494), although data are not available yet. Other molecules investigated in clinical trials for patients with MELAS include pyruvate (JMA-IIA00093), taurine (UMIN000011908), and supplemental medium chain triglycerides (NCT01252979); however, those results have never been reported.
Leber Hereditary Optic Neuropathy (LHON)
LHON is a mitochondrial disorder characterized by painless, subacute visual loss affecting the central visual field in one eye, followed by similar symptoms in the other eye typically with 2 or 3 months of delay. Three mtDNA mutations are commonly associated with LHON: m.3460G>A in MT-ND1, m.11778G>A in MT-ND4, or m.14484T>C in MT-ND6 (Yu-Wai-Man and Chinnery 1993). Many therapeutic approaches have been tested in patients with LHON, the majority of which are focused on the use of antioxidants. In particular, Idebenone has been evaluated in a clinical trial of LHON and, even if primary endpoints did not reach statistical significance, a possible beneficial effect has been shown in a subgroup of patients with discordant visual acuity at baseline (Klopstock et al. 2011) (NCT00747487). Its use has been recently approved for the treatment of this disease in Europe. MTP-131 (elamipretide) is a small molecule targeting inner mitochondrial membrane that has been demonstrated to correct excessive ROS and increase ATP synthesis in pre-clinical studies (Szeto 2014); this molecule has entered clinical trials for LHON (NCT02693119), as well as for mitochondrial myopathy and Barth Syndrome (NCT02367014, NCT02976038, NCT02805790). Although the preliminary results of phase I and II studies of this compound for mitochondrial myopathy were promising, the phase III study failed to reach primary endpoints. Further assessments of elamipretide for Barth syndrome are ongoing (NCT03098797). Curcumin, a derivate of the spice turmeric (Curcuma longa), has also displayed antioxidant properties and a trial has been completed in patients with LHON, but no results are available (NCT00528151). Lastly, AAV-mediated allotopic ND4 gene therapy has been attempted by three groups in patients with LHON (NCT02161380; NCT02064569; NCT02652767; NCT02652780). Curiously, the three teams have reported improvement in best corrected visual acuity in both treated and sham/untreated eyes in some subjects. These results were interpreted as possible transfer of the AAV-ND4 across the optic chiasm (Wan et al. 2016; Feuer et al. 2016; Vignal et al. 2018; Yang et al. 2016).
MNGIE
MNGIE (mithochondrial neuro-gastro-intestinal encephalomyopathy) is an autosomal recessive mitochondrial disorder characterized by severe gastrointestinal dysmotility, cachexia, progressive external ophthalmoplegia, myopathy, and peripheral demyelinating neuropathy (Hirano 1993). It is caused by autosomal recessive mutations in TYMP gene, encoding thymidine phosphorylase (TP). TP is a cytosolic enzyme that catalizes the first step of thymidine and deoxyuridine catabolism. When the enzyme is deficient, thymidine and deoxyuridine accumulates and become toxic, leading to mtDNA instability. Two trials at Columbia University, USA, are currently recruiting to define natural history of the disease (NCT01694953) and to assess the safety of hematopoetic stem cell transplant (HSCT), as a mean to replace TP enzyme, for MNGIE patients (NCT01694953) (see also Sect. 11.4). Orthoptic liver transplantation has been performed on three MNGIE patients with early promising results and possible greater safety than HSCT) (D’Angelo et al. 2017; De Giorgio et al. 2016).
Primary Mitochondrial Myopathies
Primary mitochondrial myopathies, defined as genetically confirmed disorders of oxidative phosphorylation affecting predominantly skeletal muscle (Mancuso et al. 2017), have emerged as targets for novel potential therapies (Madsen et al. 2020; Karaa et al. 2020). Oral omaveloxone, a semi-synthetic oleanoic triterpenoid activator of Nrf2, up to 160mg daily was well-tolerated and appeared safe. Primary (peak cycling exercise workload) and secondary (6-minute walk test) did not achieve statistical significance, but improvements in exploratory endpoints (lowering heart rate and lactate production during submaximal exercise) were reported (Madsen et al. 2020). A phase 3 trial of MTP-131 (elampretide), a small peptide stabilizer of cardiolipin, has been completed but publication of results are pending (Karaa et al. 2020).
Heterogeneous Cohort of Patients with MDs
Many of the therapeutic interventions acting on common pathogenic pathways of MDs have been tested in non-homogenous cohort of patients or in different disorders. One example is EPI-743, a para-benzoquinone analog modified to exert a higher antioxidant effect compared with CoQ10 and idebenone (Enns et al. 2012). This molecule is supposed to enhance the biosynthesis of glutathione (GSH), which is an important cellular antioxidant. Two open label studies conducted independently in North America and in Italy showed promising results in a cohort of patients with various mitochondrial diseases and with Leigh syndrome, respectively (Blankenberg et al. 2012; Martinelli et al. 2012). As a result, a randomized clinical trial has started in patients with Leigh syndrome (ClinicalTrials.gov NCT01721733) as well as in patients with FA (NCT01728064), and in acutely ill patients (90 days of end-of-life care) (NCT01370447). A clinical trial on Pearson Syndrome has been terminated because results of other studies have not supported continuation (NCT02104336). EPI-743 has also been studied in an open-label trial of patients with LHON with favorable outcome (Sadun et al. 2012).
11.4 Emerging Therapies
In the past few years, many potential treatments have been proposed for mitochondrial disorders. These approaches act on different mechanisms and can be broadly divided in “non-tailored strategies”, acting on common pathways thus in theory relevant to different MDs, and “disease-tailored” strategies (Viscomi 2016). Examples of these strategies are summarized in Table 11.2. Components of the first group are, for instance, strategies aiming at: (1) activation of mitochondrial biogenesis; (2) regulation of mitophagy and mitochondrial dynamics; (3) bypass of OXPHOS defects; (4) mitochondrial replacement therapy (MRT). Part of the second group includes: (5) scavenging of specific toxic compounds; (6) supplementation of nucleosides; (7) cell replacement therapies; (8) gene therapy; (9) shifting heteroplasmy; and (10) stabilizing mutant tRNAs. Some of these approaches have been proven effective only in preclinical models while others have already been successfully applied in anecdotic patients with MDs (Table 11.2).
11.4.1 Activation of Mitochondrial Biogenesis
Energy failure is a hallmark of mitochondrial diseases and various therapeutic interventions have been used to stimulate mitochondrial biogenesis. Although these interventions do not fix the underlying cause of the disease, increasing the mitochondrial mass might increase energy production, thus ameliorating the phenotype. There is increasing evidence in in vitro studies and animal models that increased mitochondrial biogenesis might be beneficial in many mitochondrial diseases. Interestingly, a recent observation suggested that increased mitochondrial content influences incomplete penetrance in LHON patients and that mitochondrial biogenesis could be used as a therapeutic strategy in this group of patients (Giordano et al. 2014). The biological pathway that controls mitochondrial biogenesis is complex and relies mostly on the peroxisome proliferator-activated receptor gamma (PPAR γ) coactivator 1α (PGC1α). PGC1α interacts with several transcription factors, including nuclear respiratory factors 1 and 2 (NRF1 and NRF2) and the peroxisome proliferator-activated receptors α, β, and γ. Once activated, NRFs increases the transcription of OXPHOS genes and PPARs increase the expression of genes related to fatty acid oxidation (FAO) (Scarpulla 2008). Besides, PGC1α activity is increased by deacetylation and phosphorylation. Importantly, two enzymes responsible for these modifications, deacetylation by Sirt1 and phosphorylation by AMPK, can be modulated by drugs (Puigserver and Spiegelman 2003) and have been tested in preclinical models. Different agents used with this purpose are listed below.
Sirt1 is a nuclear deacetylase that utilizes NAD+ to deacetylate residues of acetyl-lysine of proteins. Notably, Sirt1 is activated by increased cellular levels of NAD+. This increase can be achieved by providing NAD precursor, such as nicotinamide riboside, or inhibiting NAD consuming enzymes, such as poly(ADP) ribosylpolymerase1 (PARP1). These approaches have been tested in animal models of mitochondrial myopathies with beneficial effects (Cerutti et al. 2014; Khan et al. 2014).
5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), an adenosine monophosphate analog, is an agonist of AMPK that has been used to increase the respiratory chain complex activities in three mouse models of COX deficiency (Surf−/−, Sco2KOKI, and ACTA-Cox15−/−) with striking improvement of motor performances only in the Sco2 model (Viscomi et al. 2011).
Bezafibrate, a pan-PPAR activator, was tested in fibroblasts of patients with different MDs and was able to stimulate PGC1α and improve the mitochondrial respiratory chain defects (Bastin et al. 2008). These findings were subsequently confirmed by in vivo studies in mouse models of COX-deficiency (Noe et al. 2013; Hofer et al. 2014). However, studies on other mouse models did not show induction of mitochondrial biogenesis or increased mitochondrial respiratory chain enzyme activities (Viscomi et al. 2011; Yatsuga and Suomalainen 2012; Dillon et al. 2012). PPAR-δ agonists have also been proposed as potential therapeutic agents for primary mitochondrial myopathies.
Resveratrol has also been described as an activator of mitochondrial biogenesis in animal models and in human fibroblasts (Lopes Costa et al. 2014; Mizuguchi et al. 2017), although its mechanism of action is still unknown and it did not appear to increase OXPHOS activities in another study on human fibroblasts (De Paepe et al. 2014).
Retinoic acid has been used to stimulate the retinoid X receptor-alfa (RXRalfa) in cybrid containing the m.3243A>G mutation, ameliorating the respiratory chain defect (Chae et al. 2013).
Endurance training has also been used as an activator of mitochondrial biogenesis and has been reported to be beneficial and safe in the mtDNA mutator mice (Safdar et al. 2011), and in patients with MDs (Jeppesen et al. 2009; Zeviani 2008). Endurance training seems able to regulate not only PGC1α but also PGC1β, AMPK, and the hypoxia inducible factors (HIFs) (Rowe et al. 2012).
11.4.2 Regulating Mitophagy and Mitochondrial Dynamics
Mitophagy is the selective elimination of dysfunctional mitochondria, a physiological process fundamental for maintaining normal mitochondrial function (Kim et al. 2007; Ashrafi and Schwarz 2013). This process is under the regulation of various pathways. One way of targeting mitophagy is via mTOR inhibition, which can be achieved by rapamycin. This approach has been investigated in a mouse model of Leigh syndrome (Ndufs4−/−) (Johnson et al. 2013) and in a knock-in mouse model of mtDNA depletion syndrome, (Siegmund et al. 2017) and appeared to ameliorate the clinical phenotype and life-span of the treated mice, even though the biochemical defect was not rescued.
The balance between mitochondrial fusion and fission also contributes to the maintenance of mitochondrial function and can theoretically be regulated by specific modulators. An inhibitor of the mitochondrial fission protein dynamin-related protein1 (DRP1), for example the selective inhibitor P110, could potentially decrease pathological hyper-fragmentation observed in some MDs (Qi et al. 2013).
11.4.3 Bypassing OXPHOS Blocks
The use of single-peptide enzymes derived from yeast or low eukaryotes to bypass mitochondrial respiratory chain defects has been tested in in vitro and in vivo models. In particular, Ndi1 substitutes complex I in yeast and transfers electron to CoQ, without pumping protons across the membrane. AOX is present in lower eukaryotes and bypasses complex III and IV transferring electrons from CoQ. Expression of these enzymes has been used to bypass complex I deficiency (Perales-Clemente et al. 2008; Sanz et al. 2010) and Complex III-IV deficiencies in human cells and drosophila (Dassa et al. 2009; Fernandez-Ayala et al. 2009), but not in mammals in vivo.
11.4.4 Mitochondrial Replacement Therapy
Mitochondrial DNA mutations are transmitted maternally and can cause fatal or severe disorders in children (Schon et al. 2012). Moreover, these mutations are relatively common with an estimated 12,423 women at risk for transmitting mtDNA pathogenic mutations and 778 affected children per year in the United States (Gorman et al. 2015). Prenatal and preimplantation diagnoses are currently the only options available to women carrying mtDNA mutations who want to give birth to healthy children genetically related to them (Richardson et al. 2015). These techniques can accurately predict the risk of the embryo of carrying a high mtDNA mutation load. However, they cannot be applied to women with homoplasmic or nearly-homoplasmic mutations. Mitochondrial replacement therapy is a promising new reproductive technique that prevents the transmission of mtDNA mutations. It consists in combining nuclear DNA (nDNA) from a woman with a mtDNA mutation with the mitochondria of a healthy donor. This can be achieved either transferring nuclear genetic material between the oocyte of the woman carrying the mutation and the oocyte of a healthy donor or between embryos (pronuclear transfer). These techniques have been first performed in non-human primates (Tachibana et al. 2009), and then in human embryos and oocytes of healthy individuals (Craven et al. 2010; Paull et al. 2013; Tachibana et al. 2013) and women with mtDNA mutations (Kang et al. 2016) with success. Optimization of these techniques is ongoing and long-term efficacy and safety still under examination. A reversion to the original mtDNA after MRT, for example, has been recently reported from different groups (Yamada et al. 2016; Hyslop et al. 2016; Kang et al. 2016) pointing towards the necessity of additional studies to evaluate the compatibility of donor mtDNA haplogroups. Moreover, there is a fervent debate on the ethical issues concerning manipulating oocytes. Nonetheless, 2 years after the approval of the UK parliament, the UK Human Fertilisation and Embryology Authority (HFEA) authorized the use of mitochondrial replacement on a case-by-case basis in 2016. The authorities have not yet approved mitochondrial replacement techniques in the United States, even if there is evidence that women carrying mtDNA mutations and oocyte donors would support the development of these procedures (Engelstad et al. 2016). The first successful mitochondrial replacement therapy via oocyte chromosomal spindle transfer has been reported; a woman carrier of the m.8993T>G mutation with a prior history of four miscarriages and 2 children who died of Leigh syndrome gave birth to a healthy boy with neonatal mtDNA mutation loads 2.36–9.23% in tested tissues (Zhang et al. 2017).
11.4.5 Scavenging of Specific Toxic Compounds
Ethylmalonic encephalopathy (EE) is a devastating disorder of infancy due to ETHE1 mutations. ETHE1 encodes a mitochondrial sulfur dioxygenase (SDO) involved in the elimination of H2S. Accumulation of H2S, produced by the catabolism of amino acids and by the anaerobic flora of the intestine, is toxic and leads to inhibition of COX activity and to endothelial damage. N-acetyl cysteine is a precursor of glutathione and can be used to buffer intracellular H2S. Metronidazole is an intestinal antibiotic active against anaerobic bacteria that produce H2S.The use of metronidazole and N-acetyl cysteine in a mouse model of ethylmalonic encephalopathy (Ethe1−/−) prolonged the lifespan and ameliorated the clinical phenotype of this model. Moreover, the administration of these compounds in a cohort of patients with EE was able to improve some of the clinical features of the disease (Viscomi et al. 2010). This treatment has not been tested in clinical trials yet.
In MNGIE, hemodialysis has been used to remove these toxins but was not effective in decreasing thymidine or deoxyuridine levels (Spinazzola et al. 2002).
11.4.6 Supplementation of Nucleotides/Nucleosides
Supplementation of deoxyribonucleotides and deoxyribonucleosides has been exploited in in vitro and in vivo models of mitochondrial deoxynucleotide triphosphate (dNTP) pool unbalance. Mitochondrial dNTP pool unbalance causes mtDNA instability and consequent mtDNA depletion, multiple deletions, and point mutations. Different enzymes are involved in the maintenance of dNTP pools, such as thymidine kinase 2 (TK2), deoxyguanosine kinase (dGK), and thymidine phosphorylase (TP).
Thymidine kinase 2 (TK2) is a mitochondrial matrix protein that phosphorylates thymidine and deoxycytidine nycleosides to generate deoxythymidine and deoxycytidine monophosphate (dTMP, dCMP), which are then converted to dNTPs, fundamental for mtDNA synthesis. Recessive mutations in TK2 gene cause dNTP pool unbalance and mtDNA instability. The consequent clinical phenotypes range from a severe infantile neuromuscular form to adult-onset chronic progressive external ophthalmoplegia. Promising results were obtained in a Tk2 knockin mouse model (Tk2H126N/ H126N) with oral administration of deoxycytidine and deoxythymidine monophosphates and subsequently deoxycytidine and deoxythymidine; both treatments increased mtDNA levels and mitochondrial respiratory chain enzyme activities, and prolonged the lifespan of the homozygous mutant mice (Garone et al. 2014; Lopez-Gomez et al. 2017). In 2019, a compassionate use (expanded access) study of 16 patients demonstrated safety and improved survival in early onset TK2 deficiency patient (onset <2 years-old) as well as well as motor functions in all forms of TK2 deficiency relative to the natural history studies (Dominguez-Gonzalez et al. 2019).
Depletion of mtDNA has been corrected in vivo in a Tymp knockout mouse model of MNGIE disease by administrating deoxycytidine or tetrahydrouridine (Camara et al. 2014). In the same study, the addition of deoxycytidine and tetrahydrouridine to a cell model of MNGIE disease (dThd-induced mtDNA depleted fibroblasts) was also able to prevent mtDNA depletion. mtDNA depletion was also corrected in dGK deficient human fibroblasts by adding deoxyguanosine (Camara et al. 2014).
11.4.7 Cell Replacement Therapies
Cell replacement has been explored in different mitochondrial disorders as a method to deliver a specific protein when deficient. For instance, platelets (Lara et al. 2006) or erythrocyte-encapsulated thymidine phsphorylase (Bax et al. 2013) have been transfused in patients with MNGIE disease with temporary reduction of thymidine and deoxyuridine levels, but currently there is no evidence of the sustained effect of these approaches.
Allogenic hematopoietic stem cell transplantation (HSCT) has shown potential long-term effects in treating MNGIE patients (Hirano et al. 2006) being able to restore TP function and improve biochemical and clinical manifestation of the disease. This procedure albeit has been associated with high mortality. A retrospective study (Halter et al. 2015) evaluated the experience of HSCT in MNGIE patients and underlined the effectiveness of this treatment and the importance of balancing risks and benefits of this procedure. A consensus statement for future transplants in MNGIE patients has been published (Halter et al. 2011) and HSCT should be recommended in selected patients with optimized transplant conditions (Halter et al. 2015).
Liver transplantation has been recently performed in an infant with EE due to ETHE1 mutations (Dionisi-Vici et al. 2016). The patient showed progressive improvement of the neurological function and normalization of the biochemical abnormalities. Liver transplantation can replace the deficient enzyme and clear the toxic compounds that accumulate in this disorder, constituting a feasible therapeutic option in patients with EE.
11.4.8 Gene Therapy
Nuclear DNA Defects
Adeno-associated viruses (AAV) are ideal candidates as viral vectors for gene therapy, given their low risk of insertional mutagenesis, and are currently the most widely used. AAV-mediated gene therapy has been performed in different mouse models of nuclear-encoded MDs. The first animal model was a Ant1−/− treated with muscle injection of AAV2 (Flierl et al. 2005). AAV2 vector targeted to retina was used to express AIF in the eye of the Harlequin mouse and restore complex I deficiency (Bouaita et al. 2012). A liver specific AAV2,8 serotype was used in a mouse model of EE and was able to dramatically improve the clinical course and the biochemical abnormalities of mutant mice (Di Meo et al. 2012). This study demonstrated that restoring ETHE1 activity selectively in the liver was sufficient to correct the enzymatic defect and led to the hypothesis that liver transplant could be used in patients with EE. Similarly, the same hepatotropic vector was used in a mouse model of MNGIE disease and was proven successful (Torres-Torronteras et al. 2014), suggesting that liver transplantation could be an option also in MNGIE patients (Boschetti et al. 2014). The first transplanted patient was described in 2016 (De Giorgio et al. 2016). The biochemical abnormalities rapidly normalized after the transplant, and clinical conditions remained stable after 400 days. AAV2,8 vector was also used to express the wild-type MPV17 protein in a mpv17 knockout mouse model of mtDNA depletion and hepato-cerebral syndrome (Bottani et al. 2014). The vector was able to rescue the mtDNA depletion and prevent liver steatosis induced by ketogenic diet in this model.
mtDNA Defects
Even more challenging is delivering gene therapy into mitochondria. One attempted approach is to allotopically express recombinant mtDNA encoded proteins containing a mitochondrial targeting sequence (MTS) in the nucleus. This approach has been tried in fibroblasts carrying mutations in ND1, ND4, and ATP6 genes (Bonnet et al. 2007, 2008; Kaltimbacher et al. 2006) and in an animal model of LHON (Ellouze et al. 2008). Despite the controversial preclinical results, clinical trials have started in patients with LHON (NCT01267422, NCT02064569 NCT02161380) and one recently completed clinical trials has shown promising results (Wan et al. 2016) (See Sect. 11.3).
11.4.9 Shifting mtDNA Mutation Heteroplasmy
Pathogenic mtDNA mutations are usually heteroplasmic, requiring a minimum critical mutation load to cause mitochondrial dysfunction. Shifting heteroplasmic levels in order to reduce the amount of mutated DNA below this threshold, therefore, has been used as a therapeutic approach. This can be achieved with different techniques: mitochondrial-targeted restriction endonucleases (Srivastava and Moraes 2001), zinc finger endonucleases (ZFNs) (Minczuk et al. 2008), transcription activator-like effectors nucleases (TALENs) (Bacman et al. 2013), and CRISPR (clustered regularly interspaced palindromic repeat)/Cas9. Restriction endonuclease SmaI has been used in cybrids carrying the m.8399T>G mutation and was able to reduce the mutation load and increase ATP levels (Tanaka et al. 2002; Alexeyev et al. 2008). Restriction endonucleases have been exploited also in heteroplasmic mouse models, using AAV vectors, with promising results (Bayona-Bafaluy et al. 2005; Bacman et al. 2007, 2010, 2012). The main limitation of this approach is that requires the generation of a suitable restriction site by the mtDNA mutation. The introduction of ZFNs and TALENs overcomes this limitation. ZFNs are engineered mitochondrially targeted heterodimeric zinc finger nucleases conjugated to the restriction enzyme FokI. Each zinc finger domain recognizes three nucleotides, so arranging zinc finger modules appropriately allow for recognition of virtually any DNA sequence. Expression of mtZFNs in cybrids was able to reduce the mutant mtDNA and restore mitochondrial function (Gammage et al. 2014). TALENs also work as heterodimers, requiring two monomers to bind close DNA sequence in order to allow the FokI nuclease to dimerize and cleave DNA, as for the mtZFNs. Reengineered TALENs targeted to different mtDNA point mutations and deletion (MitoTALENs) were able to permanently reduce the mutation load in patient-derived cells (Bacman et al. 2013). The CRISPR/Cas9 system, another endonuclease-based system, has been reported to be more effective than TALENs and has been demonstrated to rescue mitochondrial and skeletal muscle impairment in an iPS cell model of CoQ10 deficiency due to a mutation in the COQ4 gene (Doudna and Charpentier 2014; Romero-Moya et al. 2017). A possible limitation to the use of ZNFs and MitoTALENs in clinical practice is that AAV vectors usually are able to fit smaller constructs. Moreover, the risk of a rapid reduction in mtDNA copy numbers of inducing a potential mtDNA depletion syndrome remains a limitation for the potential clinical application of these approaches. Lastly, mitochondria-targeted restriction endonucleases and TALENs have also been used in the selective elimination of mtDNA mutations in the germline of the heteroplasmic mouse model and artificial mammalian oocytes as a potential approach for preventing transmission of mtDNA mutations (Reddy et al. 2015).
11.4.10 Stabilizing Mutant tRNAs
The majority of the mtDNA mutations are localized to tRNA genes. It is not surprising therefore that several therapies have been targeting mt tRNAs. In particular, tRNA synthetases are enzymes that catalyze the attachment of amino acids to their cognate tRNA during protein synthesis. Many studies indicated that overexpressing cognate and non-cognate aminoacyl mt-tRNA synthetase can stabilize mt-tRNAs and attenuate the detrimental effect of the mutation in yeast and human cell lines (De Luca et al. 2006, 2009; Li and Guan 2010; Rorbach et al. 2008; Perli et al. 2014; Hornig-Do et al. 2014).
11.5 Conclusions
Remarkable progress has been made in the past years in mitochondrial medicine. Many potential therapeutic approaches for MDs have been proposed and are now at different stages of development. Translating preclinical studies to bedside remains challenging and well-controlled trials of high quality are necessary to define the efficacy of potential therapies already in use and novel drugs (Pfeffer et al. 2013). Based on the knowledge acquired with the previous studies, these future trials may overcome the challenges posed by this heterogeneous group of disorders in the context of multicenter collaborations, by selecting numerous subgroups of homogeneous patients and by selecting outcome measures that are objective and relevant to patient care and quality of life. Undoubtedly, there is a need for evidenced-based guidelines in the treatment of mitochondrial patients and the development of more effective therapies is an exciting perspective for the near future.
Notes
- 1.
This chapter is based upon our published review article (Hirano et al. 2018) available via Open Access (https://portlandpress.com/essaysbiochem/article/62/3/467/78638/Emerging-therapies-for-mitochondrial-diseases) CCBY license ELVJWX.
References
Alexeyev MF, Venediktova N, Pastukh V, Shokolenko I, Bonilla G, Wilson GL (2008) Selective elimination of mutant mitochondrial genomes as therapeutic strategy for the treatment of NARP and MILS syndromes. Gene Ther 15(7):516–523. https://doi.org/10.1038/sj.gt.2008.11
Ashrafi G, Schwarz TL (2013) The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 20(1):31–42. https://doi.org/10.1038/cdd.2012.81
Bacman SR, Williams SL, Hernandez D, Moraes CT (2007) Modulating mtDNA heteroplasmy by mitochondria-targeted restriction endonucleases in a ‘differential multiple cleavage-site’ model. Gene Ther 14(18):1309–1318. https://doi.org/10.1038/sj.gt.3302981
Bacman SR, Williams SL, Garcia S, Moraes CT (2010) Organ-specific shifts in mtDNA heteroplasmy following systemic delivery of a mitochondria-targeted restriction endonuclease. Gene Ther 17(6):713–720. https://doi.org/10.1038/gt.2010.25
Bacman SR, Williams SL, Duan D, Moraes CT (2012) Manipulation of mtDNA heteroplasmy in all striated muscles of newborn mice by AAV9-mediated delivery of a mitochondria-targeted restriction endonuclease. Gene Ther 19(11):1101–1106. https://doi.org/10.1038/gt.2011.196
Bacman SR, Williams SL, Pinto M, Peralta S, Moraes CT (2013) Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nat Med 19(9):1111–1113. https://doi.org/10.1038/nm.3261
Bastin J, Aubey F, Rotig A, Munnich A, Djouadi F (2008) Activation of peroxisome proliferator-activated receptor pathway stimulates the mitochondrial respiratory chain and can correct deficiencies in patients’ cells lacking its components. J Clin Endocrinol Metab 93(4):1433–1441. https://doi.org/10.1210/jc.2007-1701
Bax BE, Bain MD, Scarpelli M, Filosto M, Tonin P, Moran N (2013) Clinical and biochemical improvements in a patient with MNGIE following enzyme replacement. Neurology 81(14):1269–1271. https://doi.org/10.1212/WNL.0b013e3182a6cb4b
Bayona-Bafaluy MP, Blits B, Battersby BJ, Shoubridge EA, Moraes CT (2005) Rapid directional shift of mitochondrial DNA heteroplasmy in animal tissues by a mitochondrially targeted restriction endonuclease. Proc Natl Acad Sci U S A 102(40):14392–14397. https://doi.org/10.1073/pnas.0502896102
Blanchet L, Smeitink JA, van Emst-de Vries SE, Vogels C, Pellegrini M, Jonckheere AI, Rodenburg RJ, Buydens LM, Beyrath J, Willems PH, Koopman WJ (2015) Quantifying small molecule phenotypic effects using mitochondrial morpho-functional fingerprinting and machine learning. Sci Rep 5:8035. https://doi.org/10.1038/srep08035
Blankenberg FG, Kinsman SL, Cohen BH, Goris ML, Spicer KM, Perlman SL, Krane EJ, Kheifets V, Thoolen M, Miller G, Enns GM (2012) Brain uptake of Tc99m-HMPAO correlates with clinical response to the novel redox modulating agent EPI-743 in patients with mitochondrial disease. Mol Genet Metab 107(4):690–699. https://doi.org/10.1016/j.ymgme.2012.09.023
Bonnet C, Kaltimbacher V, Ellouze S, Augustin S, Benit P, Forster V, Rustin P, Sahel JA, Corral-Debrinski M (2007) Allotopic mRNA localization to the mitochondrial surface rescues respiratory chain defects in fibroblasts harboring mitochondrial DNA mutations affecting complex I or v subunits. Rejuvenation Res 10(2):127–144. https://doi.org/10.1089/rej.2006.0526
Bonnet C, Augustin S, Ellouze S, Benit P, Bouaita A, Rustin P, Sahel JA, Corral-Debrinski M (2008) The optimized allotopic expression of ND1 or ND4 genes restores respiratory chain complex I activity in fibroblasts harboring mutations in these genes. Biochim Biophys Acta 1783(10):1707–1717. https://doi.org/10.1016/j.bbamcr.2008.04.018
Boschetti E, D’lessandro R, Bianco F, Carelli V, Cenacchi G, Pinna AD, Del Gaudio M, Rinaldi R, Stanghellini V, Pironi L, Rhoden K, Tugnoli V, Casali C, De Giorgio R (2014) Liver as a source for thymidine phosphorylase replacement in mitochondrial neurogastrointestinal encephalomyopathy. PLoS One 9(5):e96692. https://doi.org/10.1371/journal.pone.0096692
Bottani E, Giordano C, Civiletto G, Di Meo I, Auricchio A, Ciusani E, Marchet S, Lamperti C, d’Amati G, Viscomi C, Zeviani M (2014) AAV-mediated liver-specific MPV17 expression restores mtDNA levels and prevents diet-induced liver failure. Mol Ther 22(1):10–17. https://doi.org/10.1038/mt.2013.230
Bouaita A, Augustin S, Lechauve C, Cwerman-Thibault H, Benit P, Simonutti M, Paques M, Rustin P, Sahel JA, Corral-Debrinski M (2012) Downregulation of apoptosis-inducing factor in Harlequin mice induces progressive and severe optic atrophy which is durably prevented by AAV2-AIF1 gene therapy. Brain 135(Pt 1):35–52. https://doi.org/10.1093/brain/awr290
Camara Y, Gonzalez-Vioque E, Scarpelli M, Torres-Torronteras J, Caballero A, Hirano M, Marti R (2014) Administration of deoxyribonucleosides or inhibition of their catabolism as a pharmacological approach for mitochondrial DNA depletion syndrome. Hum Mol Genet 23(9):2459–2467. https://doi.org/10.1093/hmg/ddt641
Cerutti R, Pirinen E, Lamperti C, Marchet S, Sauve AA, Li W, Leoni V, Schon EA, Dantzer F, Auwerx J, Viscomi C, Zeviani M (2014) NAD(+)-dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab 19(6):1042–1049. https://doi.org/10.1016/j.cmet.2014.04.001
Chae S, Ahn BY, Byun K, Cho YM, Yu MH, Lee B, Hwang D, Park KS (2013) A systems approach for decoding mitochondrial retrograde signaling pathways. Sci Signal 6(264):rs4. https://doi.org/10.1126/scisignal.2003266
Craven L, Tuppen HA, Greggains GD, Harbottle SJ, Murphy JL, Cree LM, Murdoch AP, Chinnery PF, Taylor RW, Lightowlers RN, Herbert M, Turnbull DM (2010) Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature 465(7294):82–85. https://doi.org/10.1038/nature08958
D’Angelo R, Rinaldi R, Pironi L, Dotti MT, Pinna AD, Boschetti E, Capristo M, Mohamed S, Contin M, Caporali L, Carelli V, De Giorgio R (2017) Liver transplant reverses biochemical imbalance in mitochondrial neurogastrointestinal encephalomyopathy. Mitochondrion 34:101–102. https://doi.org/10.1016/j.mito.2017.02.006
Dassa EP, Dufour E, Goncalves S, Paupe V, Hakkaart GA, Jacobs HT, Rustin P (2009) Expression of the alternative oxidase complements cytochrome c oxidase deficiency in human cells. EMBO Mol Med 1(1):30–36. https://doi.org/10.1002/emmm.200900001
De Giorgio R, Pironi L, Rinaldi R, Boschetti E, Caporali L, Capristo M, Casali C, Cenacchi G, Contin M, D’Angelo R, D’Errico A, Gramegna LL, Lodi R, Maresca A, Mohamed S, Morelli MC, Papa V, Tonon C, Tugnoli V, Carelli V, D’Alessandro R, Pinna AD (2016) Liver transplantation for mitochondrial neurogastrointestinal encephalomyopathy. Ann Neurol 80(3):448–455. https://doi.org/10.1002/ana.24724
De Luca C, Besagni C, Frontali L, Bolotin-Fukuhara M, Francisci S (2006) Mutations in yeast mt tRNAs: specific and general suppression by nuclear encoded tRNA interactors. Gene 377:169–176. https://doi.org/10.1016/j.gene.2006.04.003
De Luca C, Zhou Y, Montanari A, Morea V, Oliva R, Besagni C, Bolotin-Fukuhara M, Frontali L, Francisci S (2009) Can yeast be used to study mitochondrial diseases? Biolistic tRNA mutants for the analysis of mechanisms and suppressors. Mitochondrion 9(6):408–417. https://doi.org/10.1016/j.mito.2009.07.004
De Paepe B, Vandemeulebroecke K, Smet J, Vanlander A, Seneca S, Lissens W, Van Hove JL, Deschepper E, Briones P, Van Coster R (2014) Effect of resveratrol on cultured skin fibroblasts from patients with oxidative phosphorylation defects. Phytother Res 28(2):312–316. https://doi.org/10.1002/ptr.4988
Di Meo I, Auricchio A, Lamperti C, Burlina A, Viscomi C, Zeviani M (2012) Effective AAV-mediated gene therapy in a mouse model of ethylmalonic encephalopathy. EMBO Mol Med 4(9):1008–1014. https://doi.org/10.1002/emmm.201201433
Di Prospero NA, Baker A, Jeffries N, Fischbeck KH (2007a) Neurological effects of high-dose idebenone in patients with Friedreich’s ataxia: a randomised, placebo-controlled trial. Lancet Neurol 6(10):878–886. https://doi.org/10.1016/S1474-4422(07)70220-X
Di Prospero NA, Sumner CJ, Penzak SR, Ravina B, Fischbeck KH, Taylor JP (2007b) Safety, tolerability, and pharmacokinetics of high-dose idebenone in patients with Friedreich ataxia. Arch Neurol 64(6):803–808. https://doi.org/10.1001/archneur.64.6.803
Dillon LM, Hida A, Garcia S, Prolla TA, Moraes CT (2012) Long-term bezafibrate treatment improves skin and spleen phenotypes of the mtDNA mutator mouse. PLoS One 7(9):e44335. https://doi.org/10.1371/journal.pone.0044335
DiMauro S, Hirano M (1993) Merrf. In: Pagon RA, Adam MP, Ardinger HH et al (eds) GeneReviews(R). University of Washington, Seattle
DiMauro S, Schon EA, Carelli V, Hirano M (2013) The clinical maze of mitochondrial neurology. Nat Rev Neurol 9(8):429–444. https://doi.org/10.1038/nrneurol.2013.126
Dionisi-Vici C, Diodato D, Torre G, Picca S, Pariante R, Giuseppe Picardo S, Di Meo I, Rizzo C, Tiranti V, Zeviani M, De Ville De Goyet J (2016) Liver transplant in ethylmalonic encephalopathy: a new treatment for an otherwise fatal disease. Brain 139(Pt 4):1045–1051. https://doi.org/10.1093/brain/aww013
Dominguez-Gonzalez C, Madruga-Garrido M, Mavillard F, Garone C, Aguirre-Rodriguez FJ, Donati MA, Kleinsteuber K, Marti I, Martin-Hernandez E, Morealejo-Aycinena JP, Munell F, Nascimento A, Kalko SG, Sardina MD, Alvarez Del Vayo C, Serrano O, Long Y, Tu Y, Levin B, Thompson JLP, Engelstad K, Uddin J, Torres-Torronteras J, Jimenez-Mallebrera C, Marti R, Paradas C, Hirano M (2019) Deoxynucleoside therapy for thymidine kinase 2-deficient myopathy. Ann Neurol 86(2):293–303. https://doi.org/10.1002/ana.25506
Doudna JA, Charpentier E (2014) Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346(6213):1258096. https://doi.org/10.1126/science.1258096
El-Hattab AW, Scaglia F (2016) Mitochondrial cardiomyopathies. Front Cardiovasc Med 3:25. https://doi.org/10.3389/fcvm.2016.00025
El-Hattab AW, Emrick LT, Hsu JW, Chanprasert S, Almannai M, Craigen WJ, Jahoor F, Scaglia F (2016) Impaired nitric oxide production in children with MELAS syndrome and the effect of arginine and citrulline supplementation. Mol Genet Metab 117(4):407–412. https://doi.org/10.1016/j.ymgme.2016.01.010
Elliott HR, Samuels DC, Eden JA, Relton CL, Chinnery PF (2008) Pathogenic mitochondrial DNA mutations are common in the general population. Am J Hum Genet 83(2):254–260. https://doi.org/10.1016/j.ajhg.2008.07.004
Ellouze S, Augustin S, Bouaita A, Bonnet C, Simonutti M, Forster V, Picaud S, Sahel JA, Corral-Debrinski M (2008) Optimized allotopic expression of the human mitochondrial ND4 prevents blindness in a rat model of mitochondrial dysfunction. Am J Hum Genet 83(3):373–387. https://doi.org/10.1016/j.ajhg.2008.08.013
Engelstad K, Sklerov M, Kriger J, Sanford A, Grier J, Ash D, Egli D, DiMauro S, Thompson JL, Sauer MV, Hirano M (2016) Attitudes toward prevention of mtDNA-related diseases through oocyte mitochondrial replacement therapy. Hum Reprod 31(5):1058–1065. https://doi.org/10.1093/humrep/dew033
Enns GM, Kinsman SL, Perlman SL, Spicer KM, Abdenur JE, Cohen BH, Amagata A, Barnes A, Kheifets V, Shrader WD, Thoolen M, Blankenberg F, Miller G (2012) Initial experience in the treatment of inherited mitochondrial disease with EPI-743. Mol Genet Metab 105(1):91–102. https://doi.org/10.1016/j.ymgme.2011.10.009
Fernandez-Ayala DJ, Sanz A, Vartiainen S, Kemppainen KK, Babusiak M, Mustalahti E, Costa R, Tuomela T, Zeviani M, Chung J, O’Dell KM, Rustin P, Jacobs HT (2009) Expression of the Ciona intestinalis alternative oxidase (AOX) in Drosophila complements defects in mitochondrial oxidative phosphorylation. Cell Metab 9(5):449–460. https://doi.org/10.1016/j.cmet.2009.03.004
Feuer WJ, Schiffman JC, Davis JL, Porciatti V, Gonzalez P, Koilkonda RD, Yuan H, Lalwani A, Lam BL, Guy J (2016) Gene therapy for Leber hereditary optic neuropathy: initial results. Ophthalmology 123(3):558–570. https://doi.org/10.1016/j.ophtha.2015.10.025
Finsterer J, Frank M (2017) Gastrointestinal manifestations of mitochondrial disorders: a systematic review. Ther Adv Gastroenterol 10(1):142–154. https://doi.org/10.1177/1756283X16666806
Finsterer J, Kothari S (2014) Cardiac manifestations of primary mitochondrial disorders. Int J Cardiol 177(3):754–763. https://doi.org/10.1016/j.ijcard.2014.11.014
Flierl A, Chen Y, Coskun PE, Samulski RJ, Wallace DC (2005) Adeno-associated virus-mediated gene transfer of the heart/muscle adenine nucleotide translocator (ANT) in mouse. Gene Ther 12(7):570–578. https://doi.org/10.1038/sj.gt.3302443
Gammage PA, Rorbach J, Vincent AI, Rebar EJ, Minczuk M (2014) Mitochondrially targeted ZFNs for selective degradation of pathogenic mitochondrial genomes bearing large-scale deletions or point mutations. EMBO Mol Med 6(4):458–466. https://doi.org/10.1002/emmm.201303672
Garone C, Tadesse S, Hirano M (2011) Clinical and genetic spectrum of mitochondrial neurogastrointestinal encephalomyopathy. Brain 134(Pt 11):3326–3332. https://doi.org/10.1093/brain/awr245
Garone C, Garcia-Diaz B, Emmanuele V, Lopez LC, Tadesse S, Akman HO, Tanji K, Quinzii CM, Hirano M (2014) Deoxypyrimidine monophosphate bypass therapy for thymidine kinase 2 deficiency. EMBO Mol Med 6(8):1016–1027. https://doi.org/10.15252/emmm.201404092
Giordano C, Iommarini L, Giordano L, Maresca A, Pisano A, Valentino ML, Caporali L, Liguori R, Deceglie S, Roberti M, Fanelli F, Fracasso F, Ross-Cisneros FN, D’Adamo P, Hudson G, Pyle A, Yu-Wai-Man P, Chinnery PF, Zeviani M, Salomao SR, Berezovsky A, Belfort R Jr, Ventura DF, Moraes M, Moraes Filho M, Barboni P, Sadun F, De Negri A, Sadun AA, Tancredi A, Mancini M, d’Amati G, Loguercio Polosa P, Cantatore P, Carelli V (2014) Efficient mitochondrial biogenesis drives incomplete penetrance in Leber’s hereditary optic neuropathy. Brain 137(Pt 2):335–353. https://doi.org/10.1093/brain/awt343
Gorman GS, Grady JP, Ng Y, Schaefer AM, McNally RJ, Chinnery PF, Yu-Wai-Man P, Herbert M, Taylor RW, McFarland R, Turnbull DM (2015) Mitochondrial donation – how many women could benefit? N Engl J Med 372(9):885–887. https://doi.org/10.1056/NEJMc1500960
Halter J, Schupbach WM, Casali C, Elhasid R, Fay K, Hammans S, Illa I, Kappeler L, Krahenbuhl S, Lehmann T, Mandel H, Marti R, Mattle H, Orchard K, Savage D, Sue CM, Valcarcel D, Gratwohl A, Hirano M (2011) Allogeneic hematopoietic SCT as treatment option for patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): a consensus conference proposal for a standardized approach. Bone Marrow Transplant 46(3):330–337. https://doi.org/10.1038/bmt.2010.100
Halter JP, Michael W, Schupbach M, Mandel H, Casali C, Orchard K, Collin M, Valcarcel D, Rovelli A, Filosto M, Dotti MT, Marotta G, Pintos G, Barba P, Accarino A, Ferra C, Illa I, Beguin Y, Bakker JA, Boelens JJ, de Coo IF, Fay K, Sue CM, Nachbaur D, Zoller H, Sobreira C, Pinto Simoes B, Hammans SR, Savage D, Marti R, Chinnery PF, Elhasid R, Gratwohl A, Hirano M (2015) Allogeneic haematopoietic stem cell transplantation for mitochondrial neurogastrointestinal encephalomyopathy. Brain 138(Pt 10):2847–2858. https://doi.org/10.1093/brain/awv226
Hirano M (1993) Mitochondrial neurogastrointestinal encephalopathy disease. In: Pagon RA, Adam MP, Ardinger HH et al (eds) GeneReviews(R). University of Washington, Seattle
Hirano M, Marti R, Casali C, Tadesse S, Uldrick T, Fine B, Escolar DM, Valentino ML, Nishino I, Hesdorffer C, Schwartz J, Hawks RG, Martone DL, Cairo MS, DiMauro S, Stanzani M, Garvin JH Jr, Savage DG (2006) Allogeneic stem cell transplantation corrects biochemical derangements in MNGIE. Neurology 67(8):1458–1460. https://doi.org/10.1212/01.wnl.0000240853.97716.24
Hirano M, Emmanuele V, Quinzii CM (2018) Emerging therapies for mitochondrial diseases. Essays Biochem 62(3):467–481. https://doi.org/10.1042/EBC20170114
Hofer A, Noe N, Tischner C, Kladt N, Lellek V, Schauss A, Wenz T (2014) Defining the action spectrum of potential PGC-1alpha activators on a mitochondrial and cellular level in vivo. Hum Mol Genet 23(9):2400–2415. https://doi.org/10.1093/hmg/ddt631
Hornig-Do HT, Montanari A, Rozanska A, Tuppen HA, Almalki AA, Abg-Kamaludin DP, Frontali L, Francisci S, Lightowlers RN, Chrzanowska-Lightowlers ZM (2014) Human mitochondrial leucyl tRNA synthetase can suppress non cognate pathogenic mt-tRNA mutations. EMBO Mol Med 6(2):183–193. https://doi.org/10.1002/emmm.201303202
Hyslop LA, Blakeley P, Craven L, Richardson J, Fogarty NM, Fragouli E, Lamb M, Wamaitha SE, Prathalingam N, Zhang Q, O’Keefe H, Takeda Y, Arizzi L, Alfarawati S, Tuppen HA, Irving L, Kalleas D, Choudhary M, Wells D, Murdoch AP, Turnbull DM, Niakan KK, Herbert M (2016) Towards clinical application of pronuclear transfer to prevent mitochondrial DNA disease. Nature 534(7607):383–386. https://doi.org/10.1038/nature18303
Jeppesen TD, Duno M, Schwartz M, Krag T, Rafiq J, Wibrand F, Vissing J (2009) Short- and long-term effects of endurance training in patients with mitochondrial myopathy. Eur J Neurol 16(12):1336–1339. https://doi.org/10.1111/j.1468-1331.2009.02660.x
Johnson SC, Yanos ME, Kayser EB, Quintana A, Sangesland M, Castanza A, Uhde L, Hui J, Wall VZ, Gagnidze A, Oh K, Wasko BM, Ramos FJ, Palmiter RD, Rabinovitch PS, Morgan PG, Sedensky MM, Kaeberlein M (2013) mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 342(6165):1524–1528. https://doi.org/10.1126/science.1244360
Kaltimbacher V, Bonnet C, Lecoeuvre G, Forster V, Sahel JA, Corral-Debrinski M (2006) mRNA localization to the mitochondrial surface allows the efficient translocation inside the organelle of a nuclear recoded ATP6 protein. RNA 12(7):1408–1417. https://doi.org/10.1261/rna.18206
Kang E, Wu J, Gutierrez NM, Koski A, Tippner-Hedges R, Agaronyan K, Platero-Luengo A, Martinez-Redondo P, Ma H, Lee Y, Hayama T, Van Dyken C, Wang X, Luo S, Ahmed R, Li Y, Ji D, Kayali R, Cinnioglu C, Olson S, Jensen J, Battaglia D, Lee D, Wu D, Huang T, Wolf DP, Temiakov D, Belmonte JC, Amato P, Mitalipov S (2016) Mitochondrial replacement in human oocytes carrying pathogenic mitochondrial DNA mutations. Nature 540(7632):270–275. https://doi.org/10.1038/nature20592
Karaa A, Kriger J, Grier J, Holbert A, Thompson JL, Parikh S, Hirano M (2016) Mitochondrial disease patients’ perception of dietary supplements’ use. Mol Genet Metab 119(1-2):100–108. https://doi.org/10.1016/j.ymgme.2016.07.005
Karaa A, Haas R, Goldstein A, Vockley J, Cohen BH (2020) A randomized crossover trial of elamipretide in adults with primary mitochondrial myopathy. J Cachexia Sarcopenia Muscle 11(4):909–918. https://doi.org/10.1002/jcsm.12559
Kerr DS (2013) Review of clinical trials for mitochondrial disorders: 1997–2012. Neurotherapeutics 10(2):307–319. https://doi.org/10.1007/s13311-013-0176-7
Khan NA, Auranen M, Paetau I, Pirinen E, Euro L, Forsstrom S, Pasila L, Velagapudi V, Carroll CJ, Auwerx J, Suomalainen A (2014) Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol Med 6(6):721–731. https://doi.org/10.1002/emmm.201403943
Kim I, Rodriguez-Enriquez S, Lemasters JJ (2007) Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys 462(2):245–253. https://doi.org/10.1016/j.abb.2007.03.034
Klopstock T, Yu-Wai-Man P, Dimitriadis K, Rouleau J, Heck S, Bailie M, Atawan A, Chattopadhyay S, Schubert M, Garip A, Kernt M, Petraki D, Rummey C, Leinonen M, Metz G, Griffiths PG, Meier T, Chinnery PF (2011) A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy. Brain 134(Pt 9):2677–2686. https://doi.org/10.1093/brain/awr170
Koenig MK, Emrick L, Karaa A, Korson M, Scaglia F, Parikh S, Goldstein A (2016) Recommendations for the management of strokelike episodes in patients with mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes. JAMA Neurol 73(5):591–594. https://doi.org/10.1001/jamaneurol.2015.5072
Koga Y, Akita Y, Nishioka J, Yatsuga S, Povalko N, Tanabe Y, Fujimoto S, Matsuishi T (2005) L-arginine improves the symptoms of strokelike episodes in MELAS. Neurology 64(4):710–712. https://doi.org/10.1212/01.WNL.0000151976.60624.01
Koga Y, Akita Y, Junko N, Yatsuga S, Povalko N, Fukiyama R, Ishii M, Matsuishi T (2006) Endothelial dysfunction in MELAS improved by l-arginine supplementation. Neurology 66(11):1766–1769. https://doi.org/10.1212/01.wnl.0000220197.36849.1e
Koga Y, Akita Y, Nishioka J, Yatsuga S, Povalko N, Katayama K, Matsuishi T (2007) MELAS and L-arginine therapy. Mitochondrion 7(1-2):133–139. https://doi.org/10.1016/j.mito.2006.11.006
Lara MC, Weiss B, Illa I, Madoz P, Massuet L, Andreu AL, Valentino ML, Anikster Y, Hirano M, Marti R (2006) Infusion of platelets transiently reduces nucleoside overload in MNGIE. Neurology 67(8):1461–1463. https://doi.org/10.1212/01.wnl.0000239824.95411.52
Li R, Guan MX (2010) Human mitochondrial leucyl-tRNA synthetase corrects mitochondrial dysfunctions due to the tRNALeu(UUR) A3243G mutation, associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like symptoms and diabetes. Mol Cell Biol 30(9):2147–2154. https://doi.org/10.1128/MCB.01614-09
Lopes Costa A, Le Bachelier C, Mathieu L, Rotig A, Boneh A, De Lonlay P, Tarnopolsky MA, Thorburn DR, Bastin J, Djouadi F (2014) Beneficial effects of resveratrol on respiratory chain defects in patients’ fibroblasts involve estrogen receptor and estrogen-related receptor alpha signaling. Hum Mol Genet 23(8):2106–2119. https://doi.org/10.1093/hmg/ddt603
Lopez-Gomez C, Levy RJ, Sanchez-Quintero MJ, Juanola-Falgarona M, Barca E, Garcia-Diaz B, Tadesse S, Garone C, Hirano M (2017) Deoxycytidine and deoxythymidine treatment for thymidine kinase 2 deficiency. Ann Neurol 81(5):641–652. https://doi.org/10.1002/ana.24922
Lynch DR, Perlman SL, Meier T (2010) A phase 3, double-blind, placebo-controlled trial of idebenone in friedreich ataxia. Arch Neurol 67(8):941–947. https://doi.org/10.1001/archneurol.2010.168
Madsen KL, Buch AE, Cohen BH, Falk MJ, Goldsberry A, Goldstein A, Karaa A, Koenig MK, Muraresku CC, Meyer C, O’Grady M, Scaglia F, Shieh PB, Vockley J, Zolkipli-Cunningham Z, Haller RG, Vissing J (2020) Safety and efficacy of omaveloxolone in patients with mitochondrial myopathy: MOTOR trial. Neurology 94 (7):e687–e698. https://doi.org/10.1212/WNL.0000000000008861
Mariotti C, Solari A, Torta D, Marano L, Fiorentini C, Di Donato S (2003) Idebenone treatment in Friedreich patients: one-year-long randomized placebo-controlled trial. Neurology 60(10):1676–1679
Martinelli D, Catteruccia M, Piemonte F, Pastore A, Tozzi G, Dionisi-Vici C, Pontrelli G, Corsetti T, Livadiotti S, Kheifets V, Hinman A, Shrader WD, Thoolen M, Klein MB, Bertini E, Miller G (2012) EPI-743 reverses the progression of the pediatric mitochondrial disease – genetically defined Leigh Syndrome. Mol Genet Metab 107(3):383–388. https://doi.org/10.1016/j.ymgme.2012.09.007
Mancuso M, McFarland R, Klopstock T, Hirano M, consortium on Trial Readiness in Mitochondrial M (2017) International Workshop: Outcome measures and clinical trial readiness in primary mitochondrial myopathies in children and adults. Consensus recommendations. 16–18 November 2016, Rome, Italy. Neuromuscul Disord 27 (12):1126–1137. https://doi.org/10.1016/j.nmd.2017.08.006
Meier T, Perlman SL, Rummey C, Coppard NJ, Lynch DR (2012) Assessment of neurological efficacy of idebenone in pediatric patients with Friedreich’s ataxia: data from a 6-month controlled study followed by a 12-month open-label extension study. J Neurol 259(2):284–291. https://doi.org/10.1007/s00415-011-6174-y
Minczuk M, Papworth MA, Miller JC, Murphy MP, Klug A (2008) Development of a single-chain, quasi-dimeric zinc-finger nuclease for the selective degradation of mutated human mitochondrial DNA. Nucleic Acids Res 36(12):3926–3938. https://doi.org/10.1093/nar/gkn313
Mizuguchi Y, Hatakeyama H, Sueoka K, Tanaka M, Goto YI (2017) Low dose resveratrol ameliorates mitochondrial respiratory dysfunction and enhances cellular reprogramming. Mitochondrion. https://doi.org/10.1016/j.mito.2016.12.006
Murphy R, Turnbull DM, Walker M, Hattersley AT (2008) Clinical features, diagnosis and management of maternally inherited diabetes and deafness (MIDD) associated with the 3243A>G mitochondrial point mutation. Diabet Med 25(4):383–399. https://doi.org/10.1111/j.1464-5491.2008.02359.x
Noe N, Dillon L, Lellek V, Diaz F, Hida A, Moraes CT, Wenz T (2013) Bezafibrate improves mitochondrial function in the CNS of a mouse model of mitochondrial encephalopathy. Mitochondrion 13(5):417–426. https://doi.org/10.1016/j.mito.2012.12.003
O’Toole JF (2014) Renal manifestations of genetic mitochondrial disease. Int J Nephrol Renov Dis 7:57–67. https://doi.org/10.2147/IJNRD.S37887
Parikh S, Goldstein A, Koenig MK, Scaglia F, Enns GM, Saneto R, Anselm I, Cohen BH, Falk MJ, Greene C, Gropman AL, Haas R, Hirano M, Morgan P, Sims K, Tarnopolsky M, Van Hove JL, Wolfe L, DiMauro S (2015) Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med 17(9):689–701. https://doi.org/10.1038/gim.2014.177
Paull D, Emmanuele V, Weiss KA, Treff N, Stewart L, Hua H, Zimmer M, Kahler DJ, Goland RS, Noggle SA, Prosser R, Hirano M, Sauer MV, Egli D (2013) Nuclear genome transfer in human oocytes eliminates mitochondrial DNA variants. Nature 493(7434):632–637. https://doi.org/10.1038/nature11800
Perales-Clemente E, Bayona-Bafaluy MP, Perez-Martos A, Barrientos A, Fernandez-Silva P, Enriquez JA (2008) Restoration of electron transport without proton pumping in mammalian mitochondria. Proc Natl Acad Sci U S A 105(48):18735–18739. https://doi.org/10.1073/pnas.0810518105
Perli E, Giordano C, Pisano A, Montanari A, Campese AF, Reyes A, Ghezzi D, Nasca A, Tuppen HA, Orlandi M, Di Micco P, Poser E, Taylor RW, Colotti G, Francisci S, Morea V, Frontali L, Zeviani M, d’Amati G (2014) The isolated carboxy-terminal domain of human mitochondrial leucyl-tRNA synthetase rescues the pathological phenotype of mitochondrial tRNA mutations in human cells. EMBO Mol Med 6(2):169–182. https://doi.org/10.1002/emmm.201303198
Pfeffer G, Majamaa K, Turnbull DM, Thorburn D, Chinnery PF (2012) Treatment for mitochondrial disorders. Cochrane Database Syst Rev 4:CD004426. https://doi.org/10.1002/14651858.CD004426.pub3
Pfeffer G, Horvath R, Klopstock T, Mootha VK, Suomalainen A, Koene S, Hirano M, Zeviani M, Bindoff LA, Yu-Wai-Man P, Hanna M, Carelli V, McFarland R, Majamaa K, Turnbull DM, Smeitink J, Chinnery PF (2013) New treatments for mitochondrial disease-no time to drop our standards. Nat Rev Neurol 9(8):474–481. https://doi.org/10.1038/nrneurol.2013.129
Puigserver P, Spiegelman BM (2003) Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev 24(1):78–90. https://doi.org/10.1210/er.2002-0012
Qi X, Qvit N, Su YC, Mochly-Rosen D (2013) A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J Cell Sci 126(Pt 3):789–802. https://doi.org/10.1242/jcs.114439
Rahman S (2012) Mitochondrial disease and epilepsy. Dev Med Child Neurol 54(5):397–406. https://doi.org/10.1111/j.1469-8749.2011.04214.x
Reddy P, Ocampo A, Suzuki K, Luo J, Bacman SR, Williams SL, Sugawara A, Okamura D, Tsunekawa Y, Wu J, Lam D, Xiong X, Montserrat N, Esteban CR, Liu GH, Sancho-Martinez I, Manau D, Civico S, Cardellach F, Del Mar O’Callaghan M, Campistol J, Zhao H, Campistol JM, Moraes CT, Izpisua Belmonte JC (2015) Selective elimination of mitochondrial mutations in the germline by genome editing. Cell 161(3):459–469. https://doi.org/10.1016/j.cell.2015.03.051
Richardson J, Irving L, Hyslop LA, Choudhary M, Murdoch A, Turnbull DM, Herbert M (2015) Concise reviews: assisted reproductive technologies to prevent transmission of mitochondrial DNA disease. Stem Cells 33(3):639–645. https://doi.org/10.1002/stem.1887
Rodan LH, Wells GD, Banks L, Thompson S, Schneiderman JE, Tein I (2015) L-arginine affects aerobic capacity and muscle metabolism in MELAS (mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes) syndrome. PLoS One 10(5):e0127066. https://doi.org/10.1371/journal.pone.0127066
Romero-Moya D, Santos-Ocana C, Castano J, Garrabou G, Rodriguez-Gomez JA, Ruiz-Bonilla V, Bueno C, Gonzalez-Rodriguez P, Giorgetti A, Perdiguero E, Prieto C, Moren-Nunez C, Fernandez-Ayala DJ, Victoria Cascajo M, Velasco I, Canals JM, Montero R, Yubero D, Jou C, Lopez-Barneo J, Cardellach F, Munoz-Canoves P, Artuch R, Navas P, Menendez P (2017) Genetic rescue of mitochondrial and skeletal muscle impairment in an induced pluripotent stem cells model of coenzyme Q10 deficiency. Stem Cells 35(7):1687–1703. https://doi.org/10.1002/stem.2634
Rorbach J, Yusoff AA, Tuppen H, Abg-Kamaludin DP, Chrzanowska-Lightowlers ZM, Taylor RW, Turnbull DM, McFarland R, Lightowlers RN (2008) Overexpression of human mitochondrial valyl tRNA synthetase can partially restore levels of cognate mt-tRNAVal carrying the pathogenic C25U mutation. Nucleic Acids Res 36(9):3065–3074. https://doi.org/10.1093/nar/gkn147
Rowe GC, El-Khoury R, Patten IS, Rustin P, Arany Z (2012) PGC-1alpha is dispensable for exercise-induced mitochondrial biogenesis in skeletal muscle. PLoS One 7(7):e41817. https://doi.org/10.1371/journal.pone.0041817
Sadun AA, Chicani CF, Ross-Cisneros FN, Barboni P, Thoolen M, Shrader WD, Kubis K, Carelli V, Miller G (2012) Effect of EPI-743 on the clinical course of the mitochondrial disease Leber hereditary optic neuropathy. Arch Neurol 69(3):331–338. https://doi.org/10.1001/archneurol.2011.2972
Safdar A, Bourgeois JM, Ogborn DI, Little JP, Hettinga BP, Akhtar M, Thompson JE, Melov S, Mocellin NJ, Kujoth GC, Prolla TA, Tarnopolsky MA (2011) Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. Proc Natl Acad Sci U S A 108(10):4135–4140. https://doi.org/10.1073/pnas.1019581108
Sanz A, Soikkeli M, Portero-Otin M, Wilson A, Kemppainen E, McIlroy G, Ellila S, Kemppainen KK, Tuomela T, Lakanmaa M, Kiviranta E, Stefanatos R, Dufour E, Hutz B, Naudi A, Jove M, Zeb A, Vartiainen S, Matsuno-Yagi A, Yagi T, Rustin P, Pamplona R, Jacobs HT (2010) Expression of the yeast NADH dehydrogenase Ndi1 in Drosophila confers increased lifespan independently of dietary restriction. Proc Natl Acad Sci U S A 107(20):9105–9110. https://doi.org/10.1073/pnas.0911539107
Scaglia F, Towbin JA, Craigen WJ, Belmont JW, Smith EO, Neish SR, Ware SM, Hunter JV, Fernbach SD, Vladutiu GD, Wong LJ, Vogel H (2004) Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics 114(4):925–931. https://doi.org/10.1542/peds.2004-0718
Scarpulla RC (2008) Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev 88(2):611–638. https://doi.org/10.1152/physrev.00025.2007
Schaefer AM, McFarland R, Blakely EL, He L, Whittaker RG, Taylor RW, Chinnery PF, Turnbull DM (2008) Prevalence of mitochondrial DNA disease in adults. Ann Neurol 63(1):35–39. https://doi.org/10.1002/ana.21217
Schon EA, DiMauro S, Hirano M (2012) Human mitochondrial DNA: roles of inherited and somatic mutations. Nat Rev Genet 13(12):878–890. https://doi.org/10.1038/nrg3275
Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS (2005) Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 308(5730):1909–1911. https://doi.org/10.1126/science.1106653
Siegmund SE, Yang H, Sharma R, Javors M, Skinner O, Mootha V, Hirano M, Schon EA (2017) Low-dose rapamycin extends lifespan in a mouse model of mtDNA depletion syndrome. Hum Mol Genet 26(23):4588–4605. https://doi.org/10.1093/hmg/ddx341
Sinnathuray AR, Raut V, Awa A, Magee A, Toner JG (2003) A review of cochlear implantation in mitochondrial sensorineural hearing loss. Otol Neurotol 24(3):418–426
Spinazzola A, Marti R, Nishino I, Andreu AL, Naini A, Tadesse S, Pela I, Zammarchi E, Donati MA, Oliver JA, Hirano M (2002) Altered thymidine metabolism due to defects of thymidine phosphorylase. J Biol Chem 277(6):4128–4133. https://doi.org/10.1074/jbc.M111028200
Srivastava S, Moraes CT (2001) Manipulating mitochondrial DNA heteroplasmy by a mitochondrially targeted restriction endonuclease. Hum Mol Genet 10(26):3093–3099
Szeto HH (2014) First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol 171(8):2029–2050. https://doi.org/10.1111/bph.12461
Tachibana M, Sparman M, Sritanaudomchai H, Ma H, Clepper L, Woodward J, Li Y, Ramsey C, Kolotushkina O, Mitalipov S (2009) Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature 461(7262):367–372. https://doi.org/10.1038/nature08368
Tachibana M, Amato P, Sparman M, Woodward J, Sanchis DM, Ma H, Gutierrez NM, Tippner-Hedges R, Kang E, Lee HS, Ramsey C, Masterson K, Battaglia D, Lee D, Wu D, Jensen J, Patton P, Gokhale S, Stouffer R, Mitalipov S (2013) Towards germline gene therapy of inherited mitochondrial diseases. Nature 493(7434):627–631. https://doi.org/10.1038/nature11647
Taivassalo T, Shoubridge EA, Chen J, Kennaway NG, DiMauro S, Arnold DL, Haller RG (2001) Aerobic conditioning in patients with mitochondrial myopathies: physiological, biochemical, and genetic effects. Ann Neurol 50(2):133–141
Taivassalo T, Gardner JL, Taylor RW, Schaefer AM, Newman J, Barron MJ, Haller RG, Turnbull DM (2006) Endurance training and detraining in mitochondrial myopathies due to single large-scale mtDNA deletions. Brain 129(Pt 12):3391–3401. https://doi.org/10.1093/brain/awl282
Tanaka M, Borgeld HJ, Zhang J, Muramatsu S, Gong JS, Yoneda M, Maruyama W, Naoi M, Ibi T, Sahashi K, Shamoto M, Fuku N, Kurata M, Yamada Y, Nishizawa K, Akao Y, Ohishi N, Miyabayashi S, Umemoto H, Muramatsu T, Furukawa K, Kikuchi A, Nakano I, Ozawa K, Yagi K (2002) Gene therapy for mitochondrial disease by delivering restriction endonuclease SmaI into mitochondria. J Biomed Sci 9(6 Pt 1):534–541
Tarnopolsky MA (2014) Exercise as a therapeutic strategy for primary mitochondrial cytopathies. J Child Neurol 29(9):1225–1234. https://doi.org/10.1177/0883073814538512
Torres-Torronteras J, Viscomi C, Cabrera-Perez R, Camara Y, Di Meo I, Barquinero J, Auricchio A, Pizzorno G, Hirano M, Zeviani M, Marti R (2014) Gene therapy using a liver-targeted AAV vector restores nucleoside and nucleotide homeostasis in a murine model of MNGIE. Mol Ther 22(5):901–907. https://doi.org/10.1038/mt.2014.6
Vignal C, Uretsky S, Fitoussi S, Galy A, Blouin L, Girmens JF, Bidot S, Thomasson N, Bouquet C, Valero S, Meunier S, Combal JP, Gilly B, Katz B, Sahel JA (2018) Safety of rAAV2/2-ND4 gene therapy for Leber hereditary optic neuropathy. Ophthalmology 125(6):945–947. https://doi.org/10.1016/j.ophtha.2017.12.036
Viscomi C (2016) Toward a therapy for mitochondrial disease. Biochem Soc Trans 44(5):1483–1490. https://doi.org/10.1042/BST20160085
Viscomi C, Burlina AB, Dweikat I, Savoiardo M, Lamperti C, Hildebrandt T, Tiranti V, Zeviani M (2010) Combined treatment with oral metronidazole and N-acetylcysteine is effective in ethylmalonic encephalopathy. Nat Med 16(8):869–871. https://doi.org/10.1038/nm.2188
Viscomi C, Bottani E, Civiletto G, Cerutti R, Moggio M, Fagiolari G, Schon EA, Lamperti C, Zeviani M (2011) In vivo correction of COX deficiency by activation of the AMPK/PGC-1alpha axis. Cell Metab 14(1):80–90. https://doi.org/10.1016/j.cmet.2011.04.011
Voet NB, van der Kooi EL, Riphagen II, Lindeman E, van Engelen BG, Geurts AC (2013) Strength training and aerobic exercise training for muscle disease. Cochrane Database Syst Rev 7:CD003907. https://doi.org/10.1002/14651858.CD003907.pub4
Wan X, Pei H, Zhao MJ, Yang S, Hu WK, He H, Ma SQ, Zhang G, Dong XY, Chen C, Wang DW, Li B (2016) Efficacy and safety of rAAV2-ND4 treatment for Leber’s hereditary optic neuropathy. Sci Rep 6:21587. https://doi.org/10.1038/srep21587
Yamada M, Emmanuele V, Sanchez-Quintero MJ, Sun B, Lallos G, Paull D, Zimmer M, Pagett S, Prosser RW, Sauer MV, Hirano M, Egli D (2016) Genetic drift can compromise mitochondrial replacement by nuclear transfer in human oocytes. Cell Stem Cell 18(6):749–754. https://doi.org/10.1016/j.stem.2016.04.001
Yang S, Ma SQ, Wan X, He H, Pei H, Zhao MJ, Chen C, Wang DW, Dong XY, Yuan JJ, Li B (2016) Long-term outcomes of gene therapy for the treatment of Leber’s hereditary optic neuropathy. EBioMedicine 10:258–268. https://doi.org/10.1016/j.ebiom.2016.07.002
Yatsuga S, Suomalainen A (2012) Effect of bezafibrate treatment on late-onset mitochondrial myopathy in mice. Hum Mol Genet 21(3):526–535. https://doi.org/10.1093/hmg/ddr482
Yu-Wai-Man P, Chinnery PF (1993) Leber hereditary optic neuropathy. In: Pagon RA, Adam MP, Ardinger HH et al (eds) GeneReviews(R). University of Washington, Seattle
Zeviani M (2008) Train, train, train! No pain, just gain. Brain 131(Pt 11):2809–2811. https://doi.org/10.1093/brain/awn264
Zhang J, Liu H, Luo S, Lu Z, Chavez-Badiola A, Liu Z, Yang M, Merhi Z, Silber SJ, Munne S, Konstantinidis M, Wells D, Tang JJ, Huang T (2017) Live birth derived from oocyte spindle transfer to prevent mitochondrial disease. Reprod BioMed Online 34(4):361–368. https://doi.org/10.1016/j.rbmo.2017.01.013
Acknowledgements
V.E. has been supported by the North American Mitochondrial Diseases Consortium (NAMDC) fellowship program 2016–2017 (U54 NS NS078059) and has also been supported by L’Oréal- UNESCO for Women in Science 2016, Italy. NAMDC is part of the Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR), National Center for Advanced Translational Sciences (NCATS), National Institutes of Health (NIH). NAMDC is jointly funded through an NIH U54 grant mechanism by NCATS, the National Institute of Neurological Disorders and Stroke (NINDS), the Eunice Kennedy Shriver National Institute of Child Health and Development (NICHD), and the Office of Dietary Supplements (ODS). C.M.Q and M.H. are supported by NIH grant (P01 HD080642). M.H. is also supported by U54 NS NS078059, the Marriott Mitochondrial Disorders Clinical Research Network; the Nicholas Nunno Fund; the Mileti Fund; the Bernard and Anne Spitzer Fund; and the Finn Foundation.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Emmanuele, V., Quinzii, C.M., Hirano, M. (2021). Therapies Approaches in Mitochondrial Diseases. In: Navas, P., Salviati, L. (eds) Mitochondrial Diseases. Springer, Cham. https://doi.org/10.1007/978-3-030-70147-5_11
Download citation
DOI: https://doi.org/10.1007/978-3-030-70147-5_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-70146-8
Online ISBN: 978-3-030-70147-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)