
Abstract
Diabetes is associated with an increase in skeletal fragility and risk of fracture. However, the underlying mechanism for the same is not well understood. Specifically, the results from osteoblast cell culture studies are ambiguous due to contradicting reports. The use of supraphysiological concentrations in these studies, unachievable in vivo, might be the reason for the same. Therefore, here, we studied the effect of physiologically relevant levels of high glucose during diabetes (11.1 mM) on MC3T3-E1 osteoblast cell functions. The results showed that high glucose exposure to osteoblast cells increases their differentiation and mineralization without any effect on the proliferation. However, high glucose decreases their migratory potential and chemotaxis with a decrease in the associated cell signaling. Notably, this decrease in cell migration in high glucose conditions was accompanied by aberrant localization of Dynamin 2 in osteoblast cells. Besides, high glucose also caused a shift in mitochondrial dynamics towards the appearance of more fused and lesser fragmented mitochondria, with a concomitant decrease in the expression of DRP1, suggesting decreased mitochondrial biogenesis. In conclusion, here we are reporting for the first time that hyperglycemia causes a reduction in osteoblast cell migration and chemotaxis. This decrease might lead to an inefficient movement of osteoblasts to the erosion site resulting in uneven mineralization and skeletal fragility found in type 2 diabetes patients, in spite of having normal bone mineral density (BMD).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diabetes is a chronic metabolic disorder primarily characterized by hyperglycemia. One of the complications strongly associated with diabetes is bone fragility and delayed fracture healing [1, 2]. In the case of type 1 diabetes, bone mass decreases due to a reduction in the release of insulin. However, in type 2 diabetes, fracture risk, and skeletal fragility develops even when the bone mineral density (BMD) either remains unchanged or paradoxically increases [3]. This contradiction is unexplained and might be due to some microarchitectural abnormalities. While osteoporosis characterized by low BMD is a well established complication in diabetes type 1, despite increase in fracture risk it doesn’t develop in type 2 diabetes.
Hyperglycemia is primary and the most common symptom associated with diabetes. The normal fasting blood glucose level for non-diabetics remains between 3.9 and 7.1 mM (70 to 130 mg/dL) with global mean plasma blood glucose level being ~ 5.5 mM (100 mg/dL) [4]. The optimum nonfasting blood glucose levels for the same should be below 6.9 mM (125 mg/dL). A value between 7.6 mM (139 mg/dL) and 11 mM (199 mg/dL) is considered as prediabetic, and the value equal to or above 11.1 mM (200 mg/dL) is regarded as diabetic.[5]. Glucose levels higher than 49 mM are life-threatening in vivo, even the sustained levels above 20 mM are not physiological and may lead to hyperglycemic hyperosmolar non-ketotic coma [6]. However, to date, most of the in vitro studies focussing on the effect of hyperglycemia on osteoblast functions were performed using very high concentrations of glucose ranging from 22 mM to 49.5 mM [7,8,9,10,11,12,13,14,15]. Results obtained at these concentrations showed a decrease in osteoblast cell proliferation, differentiation, mineralization, and expression of osteogenic genes [7, 9, 11,12,13, 16]. However, these results do not explain how patients with type 2 diabetes retain comparable or even higher BMD values compared to healthy individuals [3, 17].
In the case of diabetic patients, the glucose levels of 11–15 mM are physiologically relevant and can sustain for more extended periods [18]. Some of the studies performed in osteoblast cells at these concentrations have shown that high glucose exposure increases osteoblast proliferation, differentiation, and formation of mineralized nodules when compared with a healthy physiological concentration of 5.5 mM [18,19,20]. These studies, in part, explain the occurrence of higher BMD observed in people with type 2 diabetes. Still, they fail to substantiate the reason for the skeletal fragility observed in these patients.
Reports suggest that, in the process of bone remodeling, pre-osteoblasts derived from osteoblast precursors and resident stem cell pool, have to migrate to the site of bone formation within the bone [21]. Any alteration in the migratory behavior of the osteoblasts might lead to an uneven bone deposition and poor bone health seen in diabetics. Taking the lead from this information, we sought to understand the effect of physiologically relevant levels of glucose during diabetes on MC3T3-E1 pre-osteoblast cells. Specifically, the impact of high glucose on the migratory potential of osteoblast cells and the molecular mechanism leading to the modulation of osteoblast activity was also investigated.
Materials and methods
Reagents and chemicals
Cell culture medium and supplements were purchased from Sigma Aldrich unless otherwise indicated. All the cell culture flasks and plates were procured from Nunc, ThermoFisher scientific. Antibodies were purchased from Cell signaling technologies unless mentioned otherwise. FITC Phalloidin and DAPI stain were from Sigma Aldrich, and Mitotracker Green FM and Antifade mounting medium were bought from Invitrogen, ThermoFisher scientific.
Cell culture
MC3T3-E1 cells (subclone 4, obtained from ATCC) were cultured under standard conditions (5% CO2 and 37 ℃), with α-Minimum Essential Medium (α-MEM) containing 0.026 mM sodium bicarbonate, 10% fetal bovine serum (FBS), antibiotic/antimycotic solution, 1% non-essential amino acids, and 1 mM Sodium pyruvate, unless indicated otherwise. Cells were trypsinized and subcultured at 80% confluence. For osmotic control (OC), equimolar amount of cell culture grade mannitol was added to the α-MEM media.
BrdU cell proliferation assay
Bromodeoxyuridine (BrdU) cell proliferation assay was performed following a previously published method [22, 23]. Briefly, MC3T3-E1 cells at 70% to 80% confluence were trypsinized, and 4000 cells/well were seeded in 96‐well plate in six replicates. After 16 h, the media was changed to high glucose or OC media supplemented with 0.5% FBS. After 24 h of treatment, cell proliferation was measured using BrdU cell proliferation ELISA colorimetric kit (Roche) according to the manufacturer's instructions. Absorbance at 450 nm was measured with a microplate reader.
ALP assay
Alkaline phosphatase (ALP) activity was done as described earlier [24, 25]. Briefly, cells were seeded at the density of 4,000 cells/well in 96 well plates. After 16 h, the media was changed to high glucose or OC α‐MEM supplemented with 10% FBS, 10 mM β‐glycerophosphate, and 50 µg/mL of ascorbic acid. Treatment was given every alternate day for 3 days. To determine the ALP activity, media was removed, cells were freeze-thawed twice and incubated with pNPP buffer (0.2% 4-nitrophenyl phosphate, 50 mM glycine, 1 mM MgCl2, 100 mM TRIS, pH 10.5) for 5–15 min at 37 °C. The resulting formation of 4-nitrophenol (pNPP—yellow color) was determined photometrically at 405 nm. For ALP staining, 12,500 cells/well/500 µl of media were seeded in 24 well plates. After 16 h, media was changed to high glucose or OC Osteoblast differentiation media. Treatment was given every alternate day for 8–10 days. To stain ALP, Alkaline phosphatase detection staining kit (Sigma) was used according to the manufacturer's protocol.
Mineralization
25,000 cells/well were seeded in 12 well plates. After 16 h, media was changed to high glucose or OC Osteoblast differentiation media. Media was changed every alternate day for 18 days with OB differentiation media. Alizarin red staining was done as described earlier [26]. Briefly, cells were washed once, fixed with 3.7% formaldehyde at room temperature for 30 min, and mineralized nodules were stained with 2% Alizarin red S solution (pH 4.2) for 30 min at ambient temperature. The stain was removed, and wells were washed 5–6 times with tap water.
Migration studies
Wound healing assay: After growing the cells to full confluence in a 24 well cell culture dish, a vertical scratch was made in the center, using a 20 µL tip. The media was changed to high glucose or OC α‐MEM supplemented with 10% FBS. Phase-contrast images were taken at 4X and 10X magnification at 0, 6, and 18 h. Image J software was used to determine the distance traveled by the cells on the scratch area.
Circularity, Feret’s diameter, and polarity index of the cells were calculated following a previously published method [27]. Briefly, 1000 cells/well were grown in 4 well-chambered slides. After 16 h, high glucose or OC treatment was given for 24 h. After that, the media was removed, and cells were washed three times with PBS, fixed for 10 min using 3.7% formaldehyde. Then cells were washed twice with PBS and stained with phalloidin for 40 min at room temperature, after washing with PBS stained with DAPI for 1 min. Cells were again washed thrice with PBS and then mounted in Antifade mounting media. Images were taken using fluorescent microscopy, and Image J software was used to determine circularity (4π × area/perimeter2, where a value of 1.0 indicates a perfect circle), Feret’s diameter (The longest distance between any two points along the selection boundary), and polarity index (the length of the migratory axis divided by the length of the axis perpendicular to the migratory axis, that intersects the center of the cell nucleus) of the cells.
Chemotaxis assay
Transwell chamber assay was carried out using a translucent cell culture insert of diameter 6.5 mm and pore size 8.0 µm (Himedia), as described previously [28]. After 6 h of OC or High glucose treatment, cells migrated to the lower side of the insert were fixed and stained with crystal violet. Images were taken at × 4 and × 10. Total no. of cells migrated to the lower surface of the insert were counted using ImageJ.
Immunostaining
Immunostaining was performed as described earlier [29]. 1000 cells per well were seeded in 4 well-chambered slides. After 16 h, media was changed to high glucose or OC media. After 24 h, the media was removed, cells were washed with PBS twice and fixed in 4% formaldehyde at RT for 10 min. Cells were then permeabilized for 5 min with 0.25% Triton X 100, blocked for one hour with 1% BSA in PBST at RT and after overnight incubation at 4℃ in primary antibody, i.e., 1:1000 Dynamin 2 (Abcam-56555), washed thrice in PBS and incubated in 1:1000 secondary antibody (Abcam-150080) for one hour at RT. Cells were then incubated with FITC phalloidin for 40 min, washed thrice, stained with DAPI for 1 min, and then mounted in Antifade mounting media. Confocal images were taken at 20X.
Mitochondrial studies
1000 cells were seeded on a coverslip, and after high glucose or OC treatment, cells were washed twice with PBS and incubated in 100 µM Mitotracker green FM for 30 min at 37 ℃ and 5% CO2. Cells were then washed twice and mounted in PBS and immediately imaged in the fluorescent microscope at 60 ×. 60–100 images were taken for each sample. The length of the mitochondria per cell was measured using ImageJ software. Mitochondria were categorized based on their length into fragmented (< 2.1 µm), intermediate (2.1–8.9 µm) and fused (> 8.9 µm).
Western blot
Western blotting was performed using the previously published methods [29, 30]. Briefly, cells were lysed in ice-cold cell lysis buffer (cell signalling technologies) with protease and phosphatase inhibitors (Sigma). Protein concentration was determined by BCA kit (Thermofisher scientific) according to the manufacturer’s protocol. 30 µg of protein was separated by SDS-PAGE and transferred to PVDF membranes. After blocking and overnight incubation with primary antibodies at 4 °C, membranes were incubated with the corresponding HRP-conjugated secondary antibody for two hours at room temperature. Chemiluminescent signals were detected using a chemiluminescence kit, and images were taken on a Chemi-Doc.
Statistical analysis
The measurements are represented by the mean ± SEM of at least three independent experiments unless otherwise indicated. The student's t-test was used for comparisons between two groups using GraphPad Prism 3.02 software.
Results
Effect of high glucose on osteoblast cell functions
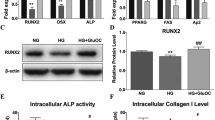
MC3T3-E1 cells were exposed to α-MEM media containing 11.1 mM, 16.6 mM, and 27.7 mM glucose or media containing equimolar mannitol as osmotic controls. BrdU cell proliferation ELISA results showed no change in the proliferation of MC3T3-E1 cells under 11.1 mM and 16.6 mM glucose when compared to their respective osmotic controls. However, at higher glucose concentration of 27.7 mM, there was a significant decrease in proliferation (Fig. 1a). On the contrary, osteoblast differentiation assessed by both ALP colorimetric assay and staining was found to be increased at all the concentrations (Fig. 1b, c). Similarly, alizarin red staining for mineralization in the cells also showed an increase in mineralized nodule formation at all the concentrations of glucose when compared to their respective osmotic controls (Fig. 1d).

High glucose increases MC3T3-E1 cell differentiation and mineralization. a Fold change in cell proliferation as compared to OC1 after 24 h of high glucose exposure by BrdU cell proliferation ELISA. b Fold change in ALP activity as compared to OC1 after 72 h of high glucose exposure, assessed by colorimetric method. c Representative image for ALP staining after 9 days of treatment. d Representative image for Alizarin red-stained cells after 18 days of treatment. OC1 (osmotic control 1) contains 5.55 mM of mannitol; OC2 contains 11.1 mM mannitol; OC3 contains 22.2 mM of mannitol. Data are presented as mean ± S.E.M. (*P < 0.05, **P < 0.01 as compared to the respective osmotic control)
Effect of high glucose on the migration potential of osteoblast cells
As the aim of this study was to determine the effect of physiologically relevant levels of hyperglycemia under diabetic conditions on osteoblast cells, further experiments were performed with 11.1 mM glucose. To investigate whether hyperglycemia modulates osteoblast migration, we carried out wound healing assay and measured the wound closure at 6 h and 18 h (Fig. 2a). Pre-osteoblasts treated with high glucose migrated lesser distances on the scratch area as compared to OC (Fig. 2b).

High glucose decreases osteoblast cell migration. a Representative images of wound healing assay for osmotic control (OC) and high glucose treated MC3T3-E1 cells at 0, 6, and 18 h. b Quantification of the distance covered by cells at 6 and 18 h. c Representative phalloidin-stained images of OC and high glucose exposed cells (24 h) to show their morphology. Quantification of the osteoblast cells d circularity, e Feret’s Diameter and f polarity index after 24 h of indicated treatments. Data are presented as mean ± S.E.M. (*P < 0.05 as compared to the respective osmotic control)
Migrating cells tend to have a lower circularity and higher Feret’s diameter and polarity index compared to the non-migrating ones. We observed that the cells exposed to high glucose media for 24 h were less elongated and had a significantly higher circularity as compared to the OC (Fig. 2c, d). In agreement, the Feret’s diameter and cell polarity index of the cells also decreased in the hyperglycemic condition when compared to the OC (Fig. 2e, f).
Effect of high glucose on osteoblast cells chemotaxis and associated signaling pathway
Chemotaxis is an essential characteristic of osteoblast cells due to which they migrate towards the growth factors and cytokines. As these are important components of serum, we carried out trans-well migration assays with serum supplemented (10% FBS) and serum depleted (0.5% FBS) conditions. MC3T3-E1 cells were plated with or without high glucose in the top well in 0.5% FBS media, and the bottom wells contained either 0.5% or 10% FBS media. As expected, in osmotic control, the number of cells migrated was higher in the case of 10% FBS media as compared to 0.5% FBS media. Notably, the number of cells migrated in through the pores decreased in both serum supplemented, and serum depleted conditions in high glucose media compared to the respective OC. However, the decrease was more prominent and significant in 10% FBS (Fig. 3a, b). Furthermore, there was no significant difference in the cells migrated in the presence of 0.5% FBS and 10% FBS in case of high glucose. These results suggest that, in case of high glucose, the serum components might not be able to induce/reinforce migratory signals as it does in OC.

High glucose inhibits osteoblast cells chemotaxis and activation of migration-related signaling. a Representative images showing the invasion of osteoblast cells in trans-well chambers carried out after 6 h of the indicated treatment and b quantification of cells migrated to the lower side of the chamber through 8 µm filter. c Representative images for western blots for p-Akt, Akt, p-Erk, Erk, and MMP2, and quantification of the fold change in the ratio of d p-Erk/Erk, e p-Akt/Akt, f MMP2/β-Actin as compared to the osmotic control (OC) at indicated time points. Data are presented as mean ± S.E.M. (*P < 0.05 and ***P < 0.001 as compared to the respective osmotic control)
Next, to check whether the decrease in cell migration is associated with the decline in the signaling pathways responsible, we performed western blotting. Akt and Erk are well-known regulators of cell motility [31,32,33,34]. High glucose exposure led to a decrease in Akt and Erk phosphorylation at least within 6 h post-treatment, which normalized at later time points (Fig. 3c–e). Also, it was observed that the expression of matrix metalloproteinase 2 (MMP2), a known promigratory protein, decreased with time under high glucose conditions (Fig. 3c, f).
Effect of high glucose on the expression and localization of Dynamin 2
Dynamin is a GTP hydrolase, whose inactivation is found to be associated with decreased cell migration and increased osteoblast differentiation and mineralization [35,36,37,38]. We observed a similar effect under high glucose conditions. Therefore to investigate further, we first compared the protein expression levels of dynamin 2 (Dyn-2) in high glucose exposed osteoblast cells as compared to OC. Western blotting results showed no change in the protein expression of Dyn-2 at all the time points studied (Fig. 4a, b).

High Glucose leads to the perinuclear localization of Dynamin. a Representative image for western blots for Dynamin 2 and β-actin at 6, 12, and 18 h and b the quantification of the same. c Confocal images for MC-3T3-E1 cells after 24 h treatment with OC and high glucose media showing DAPI staining (blue), Phalloidin FITC (green), Anti-Dynamin 2 (red), and merged images. (Color figure online)
Then we checked if the high glucose conditions have affected the localization of Dyn-2 in the cells. Immunocytometry results showed that, after 24 h of glucose treatment, Dyn-2 was mostly localized in the perinuclear region in case of high glucose, compared to control cells, where it showed strong enrichment in the membrane extensions towards the leading edge called lamellipodia (Fig. 4c). This differential localization of Dyn-2 indicated an aberration in its function when exposed to high glucose.
Effect of high glucose on mitochondrial biogenesis
Dynamin is also involved in mitochondrial biogenesis and fragmentation, which increases in the migrating cells. Therefore, next we evaluated the effect of high glucose on mitochondrial fragmentation in the osteoblast cells.
Mitochondrial staining by Mitotracker green FM showed a decrease in the fragmented mitochondrial filaments in high glucose treated cells at different time points as compared to OC (Fig. 5a). Further, we quantitated the number of fragmented (length less than 2.1 µm), intermediate (length between 2.1 and 8.9 µm), and fused (length more than 8.9 µm) mitochondria in both OC and high glucose exposed cells. The results showed a significant decrease in fragmented and increase in the intermediate and fused mitochondria in cells cultured in high glucose media as compared with OC (Fig. 5b).

High glucose results in a reduction of mitochondrial biogenesis. a Representative live-cell fluorescent images of mitochondria, stained with mitotracker green at 6, 12, and 24 h. b Quantification of the total number of fused, intermediate, and fragmented mitochondria in osmotic control (OC) and hyperglycemic condition, n ≥ 60. c Representative western blot images for Drp1 and Mfn2. Quantification of d Drp1/β-Actin and e Mfn2/ β-Actin. Data are presented as mean ± S.E.M. (*P < 0.05, **P < 0.01 and ***P < 0.001 as compared to the respective osmotic control)
To check whether this shift in the mitochondrial dynamics is due to increased fusion or decreased fission, we performed western blotting for Drp1 and Mfn2, which are responsible for fission and fusion, respectively. Results showed that the protein expression levels of Drp1 decreased after 6 and 12 h of high glucose treatment (Fig. 5c, d), while expression levels of Mfn2 did not change when compared to OC (Fig. 5c, e).
Discussion
Substantial evidence from human studies suggests that skeletal fragility is one of the severe comorbidities associated with diabetes [1, 2]. However, results from the cell culture and animal studies provide wrangling results regarding skeletal health in hyperglycemia [17]. Specifically, results obtained from osteoblast cell culture studies under high glucose conditions are very contradicting. Most of the research evaluating the effect of high glucose on osteoblasts reported a decrease in their proliferation, differentiation, and mineralization [7, 8, 12, 13, 16]. However, these studies were carried out at a physiologically very high concentration of glucose (> 22 mM). Moreover, they also contradicted the clinical studies where individuals with type 2 diabetes mellitus possess either normal or higher BMD as compared to healthy individuals [6]. Studies done at comparatively lower concentrations (11 mM to 16.5 mM) showed completely opposite results where differentiation and mineralization of osteoblast increased under high glucose conditions [18,19,20]. In our studies also, we found that high glucose at 11.1, 16.6, and 27.7 mM concentrations enhance osteoblast cell differentiation and mineralization, whereas only at 27.7 mM concentration proliferation of osteoblast cells decreases. Also, it is noteworthy, that at 27.7 mM glucose concentration, the increase in differentiation and mineralization is not as pronounced as it is at lower concenterations. This suggests that glucose increases the proliferation and differentiation of pre-osteoblasts at physiologically relevant glucose levels in diabetic conditions, however, at higher concentrations it has a negative effect. Nevertheless, these results don’t explain why skeletal fragility increases in physiologically relavent hyperglycemia.
To better understand the effect of high glucose on osteoblast cell functions and skeletal fragility in type 2 diabetes, we decided to investigate the effect of physiologically relevant levels of glucose in diabetic conditions (11.1 mM) on osteoblast cells. One of the essential functions of pre-osteoblast cells during bone remodeling is to migrate to the site of bone erosion, differentiate and mineralize to fill the resorption pit [39]. As high glucose was increasing the osteoblast cell differentiation and mineralization, which cannot be the reason for increased bone fragility, we next checked its effect on osteoblast cell migration. In the wound healing assay, there was no difference in the distance covered between OC and normal glucose control (without mannitol media; Supplementary Fig. 1). However, high glucose exposure decreased osteoblast cell migration, which is a novel observation not reported earlier. The establishment of cell polarity is an essential hallmark of migrating cells. Migrating cells tend to lose their circularity and elongate to adhere to the Extracellular matrix (ECM) [38]. Thus, cell shape descriptors significantly correlate to the cell migratory behavior [39]. In our results, we found that the cells exposed to hyperglycemic media were more circular and less polar than the cells in osmotic control media. These characteristics, which are the hallmark of non-migrating cells strengthened our observation of reduced migration of pre-osteoblasts under high glucose conditions. Moreover, most of the cells in high glucose condition had smaller lamellipodia unlike those observed in the OC. Interestingly, we also found that the difference in the invasion of cells cultured in high glucose versus control condition was more pronounced when they had to migrate towards chemoattractants present in the media supplemented with 10% FBS as compared to the serum depleted media. Moreover, the total number of cells migrated in serum-depleted control media was equivalent to the cells migrated in serum supplemented high glucose media. Serum is a source for growth factors, chemokines, hormones, and other nutrients which stimulates migration. This observation suggests that the cells cultured in hyperglycemic media were not able to respond to growth factors and chemokines required for migration. Overall, these results suggested that hyperglycemia results in aberrant migration and chemotaxis in osteoblast cells.
MAP kinase and PI3 kinase are two prominent signaling pathways activated during cell migration [40,41,42]. While MAP kinase activation results in the phosphorylation of Erk, P38, or JNK, PI3 kinase activation induces Akt phosphorylation [43]. Activation of these signaling pathways results in the reorganization of cytoskeleton and adhesion complexes, which enable cell movement [31, 32]. As compared to OC, we observed no change in the phosphorylation of P38 and JNK (Supplementary Fig. 2) in osteoblast treated with high glucose at various time points. However, there was a decrease in the phosphorylation of Erk when osteoblast cells were exposed to high glucose media. Similarly, Akt phosphorylation was also reduced under high glucose conditions. Further, MMP2, a known promigratory gene whose upregulation leads to increased cell migration and its inhibition is known to reduce migration and invasion [33, 34], also decreased over time in hyperglycemia.
Dynamin is a GTP hydrolase, which is involved in several cellular processes such as endocytosis [44, 45], actin remodeling [46], and migration [47]. Recently, a study from Pierre et al. showed that ablation of Dyn-2 levels and activity in osteoblast cells results in decrease in its migration and increase in its differentiation [38]. These results were similar to our results. Therefore, to evaluate whether there is any role of Dyn-2 in the reduced migration of osteoblast cells in hyperglycemia, we checked its protein levels in osteoblast cells. Western blotting results showed no difference in the protein levels of Dyn-2 between high glucose and OC osteoblasts. However, immunocytometry results revealed that while Dyn-2 in the control cells was localized both in lamellipodia as well as around nucleus, the cells cultured in high glucose had its localization only in the perinuclear region. Dyn-2 is a known regulator of clathrin-mediated endocytosis, and it also stabilizes actin assembly at the filopodia [48]. Hence, it might be affecting the endocytotic machinery and actin assembly in the osteoblast cells to reduce their migration. Also, Dyn-2 is involved in mitochondrial fission along with other accessory proteins [49]. Recent evidence suggests that mitochondrial fission is a prerequisite for cell migration [50,51,52], and faster-migrating cells contain higher number of fragmented mitochondria [51, 52]. Mitochondrial fission allows the cells to generate the energy needed for their movement. As Dyn-2 function was affected by high glucose, its effect on mitochondrial dynamics was evaluated. We found that OC osteoblast cells had more fragmented mitochondria, whereas cells cultured under a hyperglycemic environment had fused mitochondria. Importantly, mitochondrial DNA to nuclear DNA ratio also decreased in the cells exposed to high glucose (Supplementary Fig. 3), indicating a reduction in mitochondrial biogenesis. Drp1 is another dynamin family protein, which along with Mfn2 maintains mitochondrial dynamics in the cells. While Drp1 drives mitochondrial biogenesis/fission, Mfn2 induces fusion of mitochondria [36, 37]. Western blot analysis showed a decrease in Drp1 expression in high glucose treated cells when compared to OC. Notably, there was no change in the expression of Mfn2, indicating defective mitochondrial biogenesis without any change in the regulation of mitochondrial fusion. These results were in agreement with the previously published report where silencing of drp1 in cancer cells showed a reduction in their migratory potential [36]. Further research in this direction is required to shed more light on how high glucose changes Drp1 levels in the osteoblast cells. Moreover, coupling of Dyn-2 with Drp1 can also be studied in more detail.
In conclusion, here in this study, we have reported for the first time that high glucose induces a reduction in the migratory potential and chemotaxis of the pre-osteoblast cells. This reduction in migration is due to a decrease in mitochondrial biogenesis owing to the aberration in Dyn-2 localization and reduced Drp1 levels. These results unravel a new mechanism that could explain the mechanism behind lower rates of fracture healing in diabetic microenvironment. As hyperglycemia increases matrix mineralization but decreases osteoblast movement, it also explains the skeletal fragility observed in the type 2 diabetics even when their BMD is equivalent or higher than healthy subjects.
References
Cozen L (1972) Does diabetes delay fracture healing? Clin Orthop Relat Res 82:134–140
Jiao H, Xiao E, Graves DT (2015) Diabetes and its effect on bone and fracture healing. Curr Osteoporos Rep 13:327–335. https://doi.org/10.1007/s11914-015-0286-8
Oei L, Zillikens MC, Dehghan A, Buitendijk GH, Castano-Betancourt MC, Estrada K, Stolk L, Oei EH, van Meurs JB, Janssen JA, Hofman A, van Leeuwen JP, Witteman JC, Pols HA, Uitterlinden AG, Klaver CC, Franco OH, Rivadeneira F (2013) High bone mineral density and fracture risk in type 2 diabetes as skeletal complications of inadequate glucose control: the Rotterdam Study. Diabetes Care 36:1619–1628. https://doi.org/10.2337/dc12-1188
American Diabetes A (2006) Standards of medical care in diabetes—2006. Diabetes Care 29(Suppl 1):S4–42
Danaei G, Finucane MM, Lu Y, Singh GM, Cowan MJ, Paciorek CJ, Lin JK, Farzadfar F, Khang YH, Stevens GA, Rao M, Ali MK, Riley LM, Robinson CA, Ezzati M, Global Burden of Metabolic Risk Factors of Chronic Diseases Collaborating Group (2011) National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants. Lancet 378:31–40. https://doi.org/10.1016/S0140-6736(11)60679-X
Stoner GD (2017) Hyperosmolar hyperglycemic state. Am Fam Phys 96:729–736
Bartolome A, Lopez-Herradon A, Portal-Nunez S, Garcia-Aguilar A, Esbrit P, Benito M, Guillen C (2013) Autophagy impairment aggravates the inhibitory effects of high glucose on osteoblast viability and function. Biochem J 455:329–337. https://doi.org/10.1042/BJ20130562
Botolin S, McCabe LR (2006) Chronic hyperglycemia modulates osteoblast gene expression through osmotic and non-osmotic pathways. J Cell Biochem 99:411–424. https://doi.org/10.1002/jcb.20842
Cunha JS, Ferreira VM, Maquigussa E, Naves MA, Boim MA (2014) Effects of high glucose and high insulin concentrations on osteoblast function in vitro. Cell Tissue Res 358:249–256. https://doi.org/10.1007/s00441-014-1913-x
Liu Z, Jiang H, Dong K, Liu S, Zhou W, Zhang J, Meng L, Rausch-Fan X, Xu X (2015) Different concentrations of glucose regulate proliferation and osteogenic differentiation of osteoblasts via the PI3 kinase/Akt pathway. Implant Dent 24:83–91. https://doi.org/10.1097/ID.0000000000000196
Ma P, Gu B, Xiong W, Tan B, Geng W, Li J, Liu H (2014) Glimepiride promotes osteogenic differentiation in rat osteoblasts via the PI3K/Akt/eNOS pathway in a high glucose microenvironment. PLoS ONE 9:e112243. https://doi.org/10.1371/journal.pone.0112243
Shao X, Cao X, Song G, Zhao Y, Shi B (2014) Metformin rescues the MG63 osteoblasts against the effect of high glucose on proliferation. J Diabetes Res 2014:453940. https://doi.org/10.1155/2014/453940
Terada M, Inaba M, Yano Y, Hasuma T, Nishizawa Y, Morii H, Otani S (1998) Growth-inhibitory effect of a high glucose concentration on osteoblast-like cells. Bone 22:17–23. https://doi.org/10.1016/s8756-3282(97)00220-2
Wu YY, Yu T, Zhang XH, Liu YS, Li F, Wang YY, Wang YY, Gong P (2012) 1,25(OH)2D3 inhibits the deleterious effects induced by high glucose on osteoblasts through undercarboxylated osteocalcin and insulin signaling. J Steroid Biochem Mol Biol 132:112–119. https://doi.org/10.1016/j.jsbmb.2012.05.002
Zayzafoon M, Stell C, Irwin R, McCabe LR (2000) Extracellular glucose influences osteoblast differentiation and c-Jun expression. J Cell Biochem 79:301–310
Lopez-Herradon A, Portal-Nunez S, Garcia-Martin A, Lozano D, Perez-Martinez FC, Cena V, Esbrit P (2013) Inhibition of the canonical Wnt pathway by high glucose can be reversed by parathyroid hormone-related protein in osteoblastic cells. J Cell Biochem 114:1908–1916. https://doi.org/10.1002/jcb.24535
Patel VB (2017) Biomarkers in bone disease. Springer, Berlin Heidelberg, New York
Garcia-Hernandez A, Arzate H, Gil-Chavarria I, Rojo R, Moreno-Fierros L (2012) High glucose concentrations alter the biomineralization process in human osteoblastic cells. Bone 50:276–288. https://doi.org/10.1016/j.bone.2011.10.032
Balint E, Szabo P, Marshall CF, Sprague SM (2001) Glucose-induced inhibition of in vitro bone mineralization. Bone 28:21–28. https://doi.org/10.1016/s8756-3282(00)00426-9
Zhen D, Chen Y, Tang X (2010) Metformin reverses the deleterious effects of high glucose on osteoblast function. J Diabetes Complicat 24:334–344. https://doi.org/10.1016/j.jdiacomp.2009.05.002
Thiel A, Reumann MK, Boskey A, Wischmann J, von Eisenhart-Rothe R, Mayer-Kuckuk P (2018) Osteoblast migration in vertebrate bone. Biol Rev Camb Philos Soc 93:350–363. https://doi.org/10.1111/brv.12345
Ali SJ, Ellur G, Patel K, Sharan K (2019) Chlorpyrifos exposure induces parkinsonian symptoms and associated bone loss in adult swiss Albino mice. Neurotox Res 36:700–711. https://doi.org/10.1007/s12640-019-00092-0
Sharan K, Lewis K, Furukawa T, Yadav VK (2017) Regulation of bone mass through pineal-derived melatonin-MT2 receptor pathway. J Pineal Res. https://doi.org/10.1111/jpi.12423
Ali SJ, Ellur G, Khan MT, Sharan K (2019) Bone loss in MPTP mouse model of Parkinson's disease is triggered by decreased osteoblastogenesis and increased osteoclastogenesis. Toxicol Appl Pharmacol 363:154–163. https://doi.org/10.1016/j.taap.2018.12.003
Sharan K, Mishra JS, Swarnkar G, Siddiqui JA, Khan K, Kumari R, Rawat P, Maurya R, Sanyal S, Chattopadhyay N (2011) A novel quercetin analogue from a medicinal plant promotes peak bone mass achievement and bone healing after injury and exerts an anabolic effect on osteoporotic bone: the role of aryl hydrocarbon receptor as a mediator of osteogenic action. J Bone Miner Res 26:2096–2111. https://doi.org/10.1002/jbmr.434
Lewis KE, Sharan K, Takumi T, Yadav VK (2017) Skeletal site-specific changes in bone mass in a genetic mouse model for human 15q11-13 duplication seen in autism. Sci Rep 7:9902. https://doi.org/10.1038/s41598-017-09921-8
Lamers ML, Almeida ME, Vicente-Manzanares M, Horwitz AF, Santos MF (2011) High glucose-mediated oxidative stress impairs cell migration. PLoS ONE 6:e22865. https://doi.org/10.1371/journal.pone.0022865
Chen HC (2005) Boyden chamber assay. Methods Mol Biol 294:15–22. https://doi.org/10.1385/1-59259-860-9:015
Khan MP, Mishra JS, Sharan K, Yadav M, Singh AK, Srivastava A, Kumar S, Bhaduaria S, Maurya R, Sanyal S, Chattopadhyay N (2013) A novel flavonoid C-glucoside from Ulmus wallichiana preserves bone mineral density, microarchitecture and biomechanical properties in the presence of glucocorticoid by promoting osteoblast survival: a comparative study with human parathyroid hormone. Phytomedicine 20:1256–1266. https://doi.org/10.1016/j.phymed.2013.07.007
Bachagol D, Joseph GS, Ellur G, Patel K, Aruna P, Mittal M, China SP, Singh RP, Sharan K (2018) Stimulation of liver IGF-1 expression promotes peak bone mass achievement in growing rats: a study with pomegranate seed oil. J Nutr Biochem 52:18–26. https://doi.org/10.1016/j.jnutbio.2017.09.023
Klemke RL, Cai S, Giannini AL, Gallagher PJ, de Lanerolle P, Cheresh DA (1997) Regulation of cell motility by mitogen-activated protein kinase. J Cell Biol 137:481–492. https://doi.org/10.1083/jcb.137.2.481
Stahle M, Veit C, Bachfischer U, Schierling K, Skripczynski B, Hall A, Gierschik P, Giehl K (2003) Mechanisms in LPA-induced tumor cell migration: critical role of phosphorylated ERK. J Cell Sci 116:3835–3846. https://doi.org/10.1242/jcs.00679
Tanimura S, Takeda K (2017) ERK signalling as a regulator of cell motility. J Biochem 162:145–154. https://doi.org/10.1093/jb/mvx048
Xue G, Hemmings BA (2013) PKB/Akt-dependent regulation of cell motility. J Natl Cancer Inst 105:393–404. https://doi.org/10.1093/jnci/djs648
Kruchten AE, McNiven MA (2006) Dynamin as a mover and pincher during cell migration and invasion. J Cell Sci 119:1683–1690. https://doi.org/10.1242/jcs.02963
Razidlo GL, Wang Y, Chen J, Krueger EW, Billadeau DD, McNiven MA (2013) Dynamin 2 potentiates invasive migration of pancreatic tumor cells through stabilization of the Rac1 GEF Vav1. Dev Cell 24:573–585. https://doi.org/10.1016/j.devcel.2013.02.010
Singh M, Jadhav HR, Bhatt T (2017) Dynamin functions and ligands: classical mechanisms behind. Mol Pharmacol 91:123–134. https://doi.org/10.1124/mol.116.105064
Eleniste PP, Huang S, Wayakanon K, Largura HW, Bruzzaniti A (2014) Osteoblast differentiation and migration are regulated by dynamin GTPase activity. Int J Biochem Cell Biol 46:9–18. https://doi.org/10.1016/j.biocel.2013.10.008
Abdelgawad ME, Soe K, Andersen TL, Merrild DM, Christiansen P, Kjaersgaard-Andersen P, Delaisse JM (2014) Does collagen trigger the recruitment of osteoblasts into vacated bone resorption lacunae during bone remodeling? Bone 67:181–188. https://doi.org/10.1016/j.bone.2014.07.012
Kawabata T, Otsuka T, Fujita K, Sakai G, Kim W, Matsushima-Nishiwaki R, Kuroyanagi G, Kozawa O, Tokuda H (2018) HSP70 inhibitors reduce the osteoblast migration by epidermal growth factor. Curr Mol Med 18:486–495. https://doi.org/10.2174/1566524019666181213112847
Sun Y, Liu WZ, Liu T, Feng X, Yang N, Zhou HF (2015) Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J Recept Signal Transduct Res 35:600–604. https://doi.org/10.3109/10799893.2015.1030412
Aoki K, Kondo Y, Naoki H, Hiratsuka T, Itoh RE, Matsuda M (2017) Propagating wave of ERK activation orients collective cell migration. Dev Cell 43(305–317):e5. https://doi.org/10.1016/j.devcel.2017.10.016
Huang C, Jacobson K, Schaller MD (2004) MAP kinases and cell migration. J Cell Sci 117:4619–4628. https://doi.org/10.1242/jcs.01481
Fiuza M, Rostosky CM, Parkinson GT, Bygrave AM, Halemani N, Baptista M, Milosevic I, Hanley JG (2017) PICK1 regulates AMPA receptor endocytosis via direct interactions with AP2 alpha-appendage and dynamin. J Cell Biol 216:3323–3338. https://doi.org/10.1083/jcb.201701034
McMahon HT, Boucrot E (2011) Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol 12:517–533. https://doi.org/10.1038/nrm3151
Gonzalez-Jamett AM, Baez-Matus X, Olivares MJ, Hinostroza F, Guerra-Fernandez MJ, Vasquez-Navarrete J, Bui MT, Guicheney P, Romero NB, Bevilacqua JA, Bitoun M, Caviedes P, Cardenas AM (2017) Dynamin-2 mutations linked to centronuclear myopathy impair actin-dependent trafficking in muscle cells. Sci Rep 7:4580. https://doi.org/10.1038/s41598-017-04418-w
Yamada H, Takeda T, Michiue H, Abe T, Takei K (2016) Actin bundling by dynamin 2 and cortactin is implicated in cell migration by stabilizing filopodia in human non-small cell lung carcinoma cells. Int J Oncol 49:877–886. https://doi.org/10.3892/ijo.2016.3592
Maritzen T, Schachtner H, Legler DF (2015) On the move: endocytic trafficking in cell migration. Cell Mol Life Sci 72:2119–2134. https://doi.org/10.1007/s00018-015-1855-9
Lee JE, Westrate LM, Wu H, Page C, Voeltz GK (2016) Multiple dynamin family members collaborate to drive mitochondrial division. Nature 540:139–143. https://doi.org/10.1038/nature20555
Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, Abel PW, Tu Y (2013) Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 32:4814–4824. https://doi.org/10.1038/onc.2012.494
Ferreira-da-Silva A, Valacca C, Rios E, Populo H, Soares P, Sobrinho-Simoes M, Scorrano L, Maximo V, Campello S (2015) Mitochondrial dynamics protein Drp1 is overexpressed in oncocytic thyroid tumors and regulates cancer cell migration. PLoS ONE 10:e0122308. https://doi.org/10.1371/journal.pone.0122308
Che TF, Lin CW, Wu YY, Chen YJ, Han CL, Chang YL, Wu CT, Hsiao TH, Hong TM, Yang PC (2015) Mitochondrial translocation of EGFR regulates mitochondria dynamics and promotes metastasis in NSCLC. Oncotarget 6:37349–37366. https://doi.org/10.18632/oncotarget.5736
Acknowledgements
This study was supported by Science and Engineering Research Board (SERB), Government of India (K.S.), and the Department of Biotechnology, Government of India (K.S.). Funding from the CSIR-Central Food Technological Research Institute, Mysore, India, is acknowledged. Research fellowship grants from the DBT (H.P.), and Council of Scientific and Industrial Research (M.T.K.), Government of India, are also acknowledged.
Data Sharing
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Pahwa, H., Khan, M.T. & Sharan, K. Hyperglycemia impairs osteoblast cell migration and chemotaxis due to a decrease in mitochondrial biogenesis. Mol Cell Biochem 469, 109–118 (2020). https://doi.org/10.1007/s11010-020-03732-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-020-03732-8