
Abstract
The involvement of miR-335 in csolorectal cancer (CRC) development remains controversial. Here, we found that miR-335 was highly up-regulated in CRC specimens relative to normal mucosa, and high miR-335 expression level was markedly associated with the tumour size and differentiation of CRC. The overexpression of miR-335 in CRC cells facilitated cell proliferation in vitro and tumour growth in vivo. RASA1 was validated as a target of miR-335 that was downregulation in CRC. Forced expression of miR-335 silenced RASA1 and triggered Ras/ERK cascade in CRC. Together, miR-335-RASA1 contributes to cell growth in CRC, and elucidation of downstream pathway will provide new insights into the molecular mechanisms of CRC progression.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal carcinoma is the third most common cancer worldwide entity with multistep biological processes of tumourigenesis including somatic mutations, disruption of proliferation, apoptosis and differentiation [1–5]. Although the mortality rate of CRC is decreasing as a result of surgical treatment combined with adjuvant therapy, finding novel biomarkers for its prognosis and therapeutic targets remains urgent.
MiRNAs are endogenous, non-coding and single-stranded RNAs found in many organisms. Generally, they pair with complementary nucleotide sequences in the 3′untranslated regions (UTRs) of target mRNAs for translational repression and/or mRNA degradation. In this manner, miRNAs can regulate a series of biological processes such as cell proliferation, apoptosis and differentiation.
Calin DA et al. first described the relationship between miRNAs and cancer [6]. Following this initial finding, accumulating evidences indicate that miRNAs function as potential tumour suppressors or oncogenes to control migration, invasion and proliferation in many cancers [7–10]. Human miR-335 which transcribed from the genomic region chromosome 7q32.2 was primarily discovered by Michel J. Weber [11], decreased in metastatic breast cancer subcell lines compared with their parental cell lines and suppressed breast cancer metastasis by targeting the progenitor cell transcription factor SOX4 and extracellular matrix component tenascin C [12]. In addition, miR-335 impeded metastasis by targeting SP1 directly and indirectly through the Bclw-induced phosphoinositide 3-kinase-Akt-Sp1 pathway in gastric cancer [13]. Recent studies show that miR-335 was significantly up-regulated in human meningioma tissues and its effect of cell proliferation was associated with Rb1 signalling pathway [14].
Taken together, these evidences indicate the versatility of miR-335 in tumour progression. In this study, we concentrated on the functional and clinical significance of miR-335 in CRC. We found that miR-335 overexpression enhanced cell proliferation in vitro and tumour growth in vivo in CRC. We show that to acquire such proliferation capacity, miR-335 targets the RAS p21 protein activator 1 (RASA1), which acting as a suppressor of Ras function. Our result could be the basis for the development of new targeted treatments focused on the inhibition of miR-335 in CRC.
Materials and methods
Tissue samples and cell cultures
Paired primary colorectal cancer and adjacent normal colorectal tissues were obtained from the Nanfang Hospital, Southern Medical University. Written informed consents were obtained before specimen collection. The use of clinical materials for research purposes was approved by the Southern Medical University Institutional Board. Surgically removed tissues were immediately frozen in liquid nitrogen and stored at −80 °C for further experiment. The cell lines SW480, SW620, HT29, HCT116 and FHC were obtained from the American Type Culture Collection (ATCC). The human embryonic kidney cell lines HEK293A and HEK293FT were available in our laboratory. All cells were cultured as recommendation by the supplier and incubated in a humidified incubator with 5 % CO2 at 37 °C.
Lentivirus generation and transfection
A 402 bp fragment covering the region coding for pri-miR-335 and its upstream and downstream regions from genomic DNA was amplified by PCR and then cloned into the lentivirus vectors pLVTHM (Addgene). For packaging of the miR-335 constructs in pseudoviral particles, the HEK293FT cells were co-transfected with the above vectors, psPAX2 and pMD2.G using Lipofectamine 2000 reagents (Invitrogen). After 48 h of transfection, the supernatants containing miR-335 lentiviruses were collected, filtered and used for transduction of SW620 and HT29 cells accompanied with of 8 μg/ml of polybrene (Sigma). Empty pLVTHM lentiviruses were used as a control for the experiments. The cells stably expressed miR-335 were FACS sorted for green fluorescent protein (GFP) three days after transduction.
The miR-335 mimics, miR-335 inhibitors, the sh-miR-335 lentiviruses, the sh-RASA1 lentiviruses and their negative control were purchased from GenePharma and transfected into CRC cells using Lipofectamine 2000 reagents (Invitrogen) according to the manufacturer’s instructions.
RNA extraction and quantitative real-time (qRT)-PCR
Total RNA was extracted from the cells and tissues using the TRIzol reagent (Takara) according to the established protocols. The All-in-One™ miRNA qRT-PCR Detection Kit (GeneCopoeia) was used to detect and quantify the expression of miR-335 in cells and tissues according to the manufacturer’s protocols. The expression of U6 was used as an endogenous control. For RASA1 mRNA detection, cDNA was prepared using the Reverse Transcriptase System (Takara) and then utilized as template to quantify RASA1 expression by a SYBR green qRT-PCR assay (Takara). The expression data were normalized to the housekeeping gene Glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Each sample was analysed in triplicate. The relative expression was calculated using the comparative cycle threshold (C t) method.
Western blotting
Total protein from tumoural, normal colorectal tissues and cell lines was isolated using Whole Cell Lysis Assay (KeyGEN) according to the manufacturer’s protocol and solubilized in SDS loading buffer (KeyGEN). Equal amounts of proteins were separated by electrophoresis on 8–10 % SDS–polyacrylamide gel, transferred to a polyvinylidene difluoride (PVDF) membrane (Roche Applied Sciences) and blocked in 0.1 % Tween 20 containing 5 % skim milk in Tris Buffer Saline. Membranes were incubated with the rabbit anti-RASA1 monoclonal antibody (1:800, Epitomics), rabbit anti-Ras monoclonal antibody (1:500, Epitomics), rabbit anti-ERK1/2, Phospho-ERK1/2 (p-ERK1/2) monoclonal antibody (1:1000, Bioworld) and rabbit anti-Tubulin antibody (1:2000, Epitomics) overnight at 4 °C. Antibody binding was revealed by incubation with the appropriate horseradish peroxidase-conjugated secondary antibody. The reactive bands were visualized using Pierce ECL Western Blotting Substrate (Thermo Scientific).
Cell proliferation and plate colony formation assays
A Cell Counting Kit-8 (CCK-8) (Dojindo Laboratories) assay was performed to determine cell proliferation. The treated cells were plated in 96-well plates at a density of 0.8–1 × 104 cells/ml in a volume of 100 μl per well. 10 μl of CCK-8 solution was added to each well; then the plate was incubated for 2 h at 37 °C. The absorbance was measured at 450 nm using a Vmax microplate spectrophotometer (PerkinElmer). Each well of a 6-well plate was seeded with 2–3 × 102 cells for colony formation assays. After 14 days, the colonies were fixed with 4 % paraformaldehyde for 30 min and stained with Giemsa solution for 20 min. Only the colonies containing at least 50 cells formed were counted.
Cell-cycle analysis
Cells were washed twice with PBS and fixed overnight at 4 °C in 70 % ethanol. RNAase was added for getting rid of total RNA. After the cells were incubated at 37 °C for 30 min, their DNA was stained with propidium iodide (PI) (KeyGEN, Nanjin, China) for 20 min at room temperature. The samples were quantified by flow cytometry (Becton–Dickinson).
Luciferase reporter experiments
For luciferase reporter experiments, the 3′UTR segments of RASA1 predicted to interact with miR-335 were amplified by PCR from human genomic DNA and then inserted into downstream of the Renilla luciferase open reading frame of the psi-CHECK2 vector (Promega). Mutations in miR-335 seeding sequence within RASA1 3′UTR were generated using a KOD Plus Mutagenesis Kit (Toyobo). The HEK293A or SW480 cells were co-transfected with the wild-type or mutational reporter plasmids, along with miR-335 mimics or negative control (NC) miRNAs. Firefly and Renilla luciferase activities were measured using Dual-luciferase assays (Promega) 48 h after transfection. The relative luciferase activity was formulated as luminescence from Renilla luciferase divided by luminescence from firefly luciferase.
In vivo tumour growth assays
All experimental procedures involving animals were performed in accordance with animal protocols approved by Animal Care and Use Committee of Southern Medical University. Animal models were conducted as previously described [15]. HCT116-NC, HCT116-sh-miR-335 or HCT116-sh-miR-335/sh-RASA1 cells were injected subcutaneously on the hindlimb of nude mice (n = 4 per group). The tumour size was measured by a slide caliper, and tumour volume was determined by the formula 1/2 × length × width2 every three days. All mice were scarified 21 days after inoculation, and the tumours were excised and then analysed with H&E staining. To assess the proliferative activity of xenograft tumours after miR-335 up-regulation, immunohistochemistry was performed to examine the expression of Ki-67 (rabbit anti-Ki-67 monoclonal antibody, Bioworld) in tumours tissues.
Statistical analysis
Data were analysed using the SPSS v13.0 statistical software and expressed as the mean ± standard deviation (SD) of at least three independent experiments. Student’s t test was used to analyse differences between two groups, and Fisher’s exact test was used to analyse the relationship between miR-335 expression and the clinicopathologic features of CRC. Differences were considered statistically significant at a p value <0.05 (*p < 0.05, **p < 0.01 and ***p < 0.001).
Results
MiR-335 is overexpressed in CRC cells and tissues
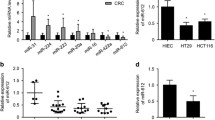
The expression of miR-335 was examined in 37 pairs of CRC biopsies and their matched adjacent normal tissues using qRT–PCR. An increase in miR-335 expression was seen in 89.19 % (33/37) of CRC tissues compared with their matched non-tumour tissues (p < 0.001; Fig. 1a, b). MiR-335 levels in CRC tissues were approximately 7.03-fold higher than those in matched non-tumour tissues. To further investigate the clinicopathologic significance of miR-335 expression in CRC patients, the median relative expression level of miR-335 in the 37 CRC samples was recommended as the cut-off point for dividing miR-335 level into a low expression group and a high expression group. Correlation analysis showed that the miR-335 expression level was significantly associated with tumour size and differentiation (p < 0.05; Table 1). However, there were no significant correlations between the miR-335 expression and serosal invasion, lymph metastasis or TNM classification. Moreover, the relative miR-335 expression was significantly higher in CRC cells compared with colon epithelium cell FHC (p < 0.05; Fig. 1c).

The expression of miR-335 in CRC tissues and cells. a qRT-PCR analysis of miR-335 expression in 37 paired human colorectal cancer tissues; the expression of miR-335 is expressed relative to the matched adjacent normal tissues. b Comparison of the miR-335 abundance in 37 paired primary CRC tissues (T) with matched adjacent normal tissues (N) shows its up-regulation in tumour tissues. c qRT-PCR analysis of miR-335 expression in CRC cell lines, including SW480, SW620, HCT116 and HT29, and the expression of miR-335 is expressed relative to colon epithelium cell FHC
RASA1 is direct target of miR-335
The target genes of miR-335 were predicted by three bioinformatic algorithms PicTar, TargetScan and microRNA.org. At least three of these databases predicted JAG1, MLLT3, JMJD2C, CD79B, WWP1, RASA1, SP1 and SNIP1 as the potential target genes of miR-335. All these genes are involved in the proliferation, metastasis or apoptosis during tumourigenesis, of which we focused on RASA1. Bioinformatic analysis indicated a putative miR-335 target site in the RASA1 3′UTR (Fig. 2a). We then experimentally confirmed the interactions of RASA1 with miR-335. A significant decrease in the wild-type RASA1 luciferase activity was observed upon overexpression of miR-335 in SW480. Moreover, miR-335 could not decrease the luciferase activity of mutant RASA1 in the miR-335 binding site (Fig. 2b), indicating miR-335 targets the specific region on RASA1 3′UTR. We further confirmed that miR-335 dramatically decreased the luciferase activity in the wild-type RASA1 rather than mutant RASA1 in the miR-335 binding site in HEK293A (Fig. 2b), thereby providing additional evidence of direct interaction between miR-335 and RASA1. Moreover, inhibition of miR-335 also block miR-335-induced downregulation of luciferase activity in cells transfected with RASA1 3′UTR in a dose-dependent manner (Fig. 2b). The RASA1 expression was significantly decreased in CRC specimens and cell lines compared to their normal counterparts (Fig. 2c, e). Spearman’s correlation analysis demonstrated that RASA1 and miR-335 were again inversely related in expression (p = 0.000; r = −0.940, Fig. 2d).

RASA1 is a direct target of miR-335. a Schematic illustration of base paring between miR-335 and the RASA1 3′UTR, as well as the mutated sequences used in this study. b Analysis of luciferase activity. HEK293A or SW480 cells were co-transfected with either miR-335 mimics or negative control miRNA and luciferase reporter plasmids comprising Luc-RASA1 3′UTR Wt or Luc-RASA1 3′UTR Mut. HCT116 or SW480 cells were co-transfected with Luc-RASA1 3′UTR and different dose of miR-335-inhibitors. c qRT-PCR analysis of RASA1 expression in 37 paired human colorectal cancer tissues. d Spearman correlation analysis showed a negative relationship between the miR-335 expression level and the RASA1 mRNA in 37 cases of CRC tissue samples. e Western blotting analysis of RASA1 expression in colorectal cancer cells and tissues
Overexpression of miR-335 promotes proliferation and tumourigenicity of CRC cells
To evaluate the possible function of miR-335 on proliferation in CRC, we transfected the CRC cells with miR-335 lentiviruses (p < 0.001; Fig. 3a) or sh-miR-335 lentiviruses (p < 0.001; Fig. 3b) as showed by RT-PCR analysis. The sh-RASA1 lentiviruses were transfected into the sh-miR-335-cells for the rescue experiments (Fig. 3b).The colony formation assays and CCK-8 cell proliferation assays demonstrated that the overexpression of miR-335 significantly increased the proliferation potentials of both SW620 and HT29 cells compared to their control cells (p < 0.01; Fig. 3c and p < 0.05; Fig. 3d). Concomitantly, the depletion of miR-335 evidently reduced cell viability of HCT116 and SW480 cells (p < 0.05) compared to negative control (NC) transfected cells. However, the proliferation potentials were restored in the sh-miR-335/sh-RASA1 cells (Fig. 3e). These evidences support the function of miR-335 on facilitating cell proliferation by targeting RASA1. Furthermore, HCT116-NC, HCT116-sh-miR-335 and HCT116-sh-miR-335/sh-RASA1 cells were implanted subcutaneously into nude mice, respectively. The subcutaneous tumours generated from HCT116-sh-miR-335 cells were smaller than those derived from HCT116-NC cells (p < 0.05). Tumours in mice injected with HCT116-NC cells showed no significant difference from HCT116-sh-miR-335/sh-RASA1 cells (Fig. 4a, b). Immunohistochemistry staining substantiated that the tumours from the HCT116-sh-miR-335 cells revealed much lower Ki-67 indexes than the tumours from the HCT116-NC cells, which was restored by downregulation of RASA1 (p < 0.001, Fig. 4c, d). qRT-PCR and Western blotting confirmed that RASA1 was negatively regulated by miR-335 in subdermal tumour specimens (Fig. 4e, f).

miR-335 promotes CRC cells proliferation in vitro. a SW620 and HT29 cells were infected with pLVTHM or miR-335 lentiviruses; miR-335 expression was tested by qRT-PCR. b The sh-miR-335 lentiviruses and sh-RASA1 lentiviruses were transfected in HCT116 and SW480 cells. c Colonies formed by pLVTHM or miR-335-infected cells and quantification of the number of Giemsa-stained cell colonies were shown 2 weeks after plating. d CCK-8 proliferation assays identified the effect of miR-335 on cell proliferation in the pLVTHM cells and the miR-335 cells. e The viability of HCT116 and SW480 cells after transfection with sh-miR-335 lentiviruses or sh-RASA1 lentiviruses was measured by CCK-8 proliferation assays

The downregulation of miR-335 inhibited tumour growth in vivo. a Tumour sizes were measured at different time points until day (d) 21 when mice were executed. Data points are presented as the mean tumour volume ± SD. b Effects of sh-miR-335 or sh-miR-335/sh-RASA1 infected HCT116 cells on subcutaneous tumor generation (n=4). c Representative photographs of H&E staining and immunohistochemistry staining of Ki-67 of the subcutaneous tumour sections are shown. d Quantitative analyses showing the intensity Ki-67 staining. e The expression of miR-335 was determined in the tumour xenografts by RT-PCR. f The expression of RASA1 was determined in the tumour xenografts by RT-PCR and western blotting
MiR-335 accelerates cell-cycle progression in CRC
We further evaluated the cell-cycle distribution by flow cytometry to explore the possible mechanism by which miR-335 regulates CRC cell proliferation. MiR-335 overexpression obviously increased the percentage of cells in the S phase in SW620 and HT29 (Fig. 5a). The percentage of S-phase cells in SW620-miR-335 (47.39 % ± 1.29) and HT29-miR-335 (47.61 % ± 1.86) were significantly higher than that in the control cells, whereas they were only 24.07 % ± 1.82 (p < 0.001) and 23.62 % ± 1.03 (p < 0.001), respectively. On the contrary, a significant decrease in the percentage of cells in the S phase was observed in sh-miR-335 cells. The percentage of S-phase cells in HCT116-sh-miR-335 (36.83 % ± 1.48) and SW480-sh-miR-335 (41.18 % ± 1.12) were significantly less than in HCT116-NC (51.65 % ± 0.92) (p < 0.01) and SW480-NC (53.25 % ± 0.66) (p < 0.01) cells, respectively, whereas knockdown of RASA1 increased the percentage of S-phase cells in HCT116-sh-miR-335 (51.15 % ± 1.58) and SW480-sh-miR-335 (53.65 % ± 1.03)and blocked the inhibitory effects of miR-335 (Fig. 5a). The expression levels of critical cell-cycle regulators were also detected. As shown in Fig. 5b, Cyclin D1 was significantly up-regulated, whereas p21 were strikingly downregulated in miR-335-overexpressing cells. In contrast, significant increases in the expression of p21 and decreases in Cyclin D1 were shown in miR-335-inhibiting cells. However, knockdown of RASA1 rescued those effects.

miR-335 accelerates cell-cycle progression. a Representative histograms depicting cell-cycle profiles of indicated cells. Cells were stained with PI and analysed by flow cytometry. b Western blotting analysis of the expression of p21 and Cyclin D1 proteins in the indicated cell
MiR-335 suppresses expression of RASA1 and activated the Ras/ERK pathway in CRC
RASA1, belonging to the GAP1 subfamily of Ras GTPase-activating proteins, restrains Ras activity in GDP-bound forms [16, 17]. The Ras signalling pathways are key regulators of cell growth and malignant transformation [18]. Thus, we investigate whether the modulation of Ras signalling activity by miR-335 was due to regulation of CRC proliferation. MiR-335 overexpression significantly downregulated RASA1 expression (Fig. 6a, c), up-regulated Ras expression and Ras-GTP activity, and promoted the phosphorylation of ERK1/2 (Fig. 6a). Conversely, miR-335 inhibition significantly increased RASA1 overexpression and reduced Ras expression, RAS-GTP activity and p-ERK1/2 protein levels (Fig. 6b).

miR-335 is responsive to the Ras/ERK pathway in CRC. a Western blotting analysis of RASA1, Ras-GTP, Ras, ERK1/2 and p-ERK1/2 in SW620-miR-335 and HT29-miR-335. Tubulin served as the loading control. b Western blotting analysis of RASA1, Ras-GTP, Ras, ERK1/2 and p-ERK1/2 expression in HCT116 and SW480 transfected with miR-335 inhibitors. Tubulin served as the loading control. c qRT-PCR analysis of RASA1 expression in indicated cells transfected with miR-335 expression lentiviruses or inhibitors. d The proposed model summarized the molecular mechanisms of CRC cell proliferation by the miR-335-RASA1-Ras/ERK cascade
Discussion
Characterization of microRNA expression profiles based on genome-wide microarrays has been widely employed in cancer studies [19–21]. Dysexpressed miRNAs in human cancers differentiated from corresponding counterpart indeed revealed an involvement of miRNAs in cancer developments and progressions. The abnormal expression and functional significance of miR-335 has been reported in some tumour types. Some studies have defined miR-335 as a tumour suppressor in malignant astrocytoma, ovarian, breast, prostate gastric and pancreatic cancer [12, 13, 22–24], as it suppresses cell proliferation, migration and invasion. On the contrary, miR-335 promoted cell growth and inhibited cell-cycle arrest in the G0/G1 phase in meningiomas [14]. Despite the confirmed cellular function, the role of miR-335 in various kinds of cancers remains controversial.
It is reported that the expression levels of miR-335 have been up- or downregulated in CRC. Michael et al. reported that miR-335 levels were significantly elevated in CRC primary tumours with a metastatic capacity [16]. A study based on a Genome-Wide Profiling and Validation Study confirmed that miR-335 was significantly up-regulated in patients with CRC [25]. While, Sun et al. reported that miR-335 was downregulated in human primary CRC tissues with lymph node metastases [26]. These contrary findings may be due in part to the different source organization and empirical method models. To the best of our knowledge, few studies have corrected miR-335 function with proliferation in colorectal cancer. In this study, we found that miR-335 was highly expressed in CRC tissues, and high miR-335 expression levels were found to be markedly associated with tumour size and differentiation in CRC, implicating a direct role of miR-335 in the growth of colorectal cancer. Increased expression levels of miR-335 in CRC encourage us to reason that it would be feasible for potential therapeutic target. Therefore, we further investigated its pathogenic effect. The overexpression of miR-335 enforced cell proliferation in vitro and tumour growth in vivo.
Our results confirmed that RASA1 was a direct target of miR-335. RASA1 known as p120 RasGAP contains a catalytic domain of Ras-GTP that is necessary for interaction with Ras and accountable for negative regulation of the Ras activity [27]. Accordingly, ectopic expression of miR-335 in CRC cells increased the Ras activity. A family of Ras proteins that representing the central players in the signal transduction pathway are the cellular vital mechanisms for controlling cell survival, differentiation, proliferation, metabolism, and motility in response to extracellular cues [28–31]. ERK encoded MAP kinases functions as the downstream effector of the Ras oncoprotein. In this pathway, Ras activation stimulates a signaling cascade by phosphorylation of MAPK which successively phosphorylates and activates the downstream proteins ERK1/2. ERK1/2 can also phosphorylate a series of nuclear transcription factors and kinasesa to affect the cell biological processes such as proliferation [28, 29, 32–34]. On the basis of these models, we examine the protein levels of Ras, ERK1/2 and p-ERK1/2. Ras and p-ERK1/2 proteins were elevated in the miR-335 up-regulation cells. Thus, we draw a conclusion that the effects of miR-335 on the proliferation of CRC cells may be mediated via downregulation of RASA1 and activation of Ras/ERK signal pathway (Fig. 6d). Similarly, RASA1 was a target of miR-31 in CRC. MiR-31 targets RASA1 to stimulate CRC cell proliferation and tumourigenesis [35].
In summary, the acting of miR-335 on RASA1 has valuable ramifications on tumour growth. This outcome helps us to further comprehend the potential molecular mechanisms of CRC proliferation.
Abbreviations
- CRC:
-
Colorectal carcinoma
- miRNA:
-
microRNA
- UTR:
-
Untranslated regions
- RASA1:
-
RAS p21 protein activator 1
- SD:
-
Standard deviation
- CCK-8:
-
Cell counting kit-8
- GFP:
-
Green fluorescent protein
References
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S et al (2013) Signatures of mutational processes in human cancer. Nature 500:415–421
Zhang L, Ren X, Alt E, Bai X, Huang S et al (2010) Chemoprevention of colorectal cancer by targeting APC-deficient cells for apoptosis. Nature 464:1058–1061
Myant KB, Cammareri P, McGhee EJ, Ridgway RA, Huels DJ et al (2013) ROS production and NF-kappaB activation triggered by RAC1 facilitate WNT-driven intestinal stem cell proliferation and colorectal cancer initiation. Cell Stem Cell 12:761–773
van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I et al (2002) The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 111:241–250
Ashley N, Yeung T, Bodmer WF (2013) Stem cell differentiation and lumen formation in colorectal cancer cell lines and primary tumours. Cancer Res 73:5798–5809
Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S et al (2002) Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA 99:15524–15529
Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T et al (2011) The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med 17:211–215
Hayashita Y, Osada H, Tatematsu Y, Yamada H, Yanagisawa K et al (2005) A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res 65:9628–9632
Schimanski CC, Frerichs K, Rahman F, Berger M, Lang H et al (2009) High miR-196a levels promote the oncogenic phenotype of colorectal cancer cells. World J Gastroenterol 15:2089–2096
Huang Q, Gumireddy K, Schrier M, le Sage C, Nagel R et al (2008) The microRNAs miR-373 and miR-520c promote tumour invasion and metastasis. Nat Cell Biol 10:202–210
Weber MJ (2005) New human and mouse microRNA genes found by homology search. FEBS J 272:59–73
Tavazoie SF, Alarcon C, Oskarsson T, Padua D, Wang Q et al (2008) Endogenous human microRNAs that suppress breast cancer metastasis. Nature 451:147–152
Xu Y, Zhao F, Wang Z, Song Y, Luo Y et al (2012) MicroRNA-335 acts as a metastasis suppressor in gastric cancer by targeting Bcl-w and specificity protein 1. Oncogene 31:1398–1407
Shi L, Jiang D, Sun G, Wan Y, Zhang S et al (2012) miR-335 promotes cell proliferation by directly targeting Rb1 in meningiomas. J Neurooncol 110:155–162
Zhou C, Liu G, Wang L, Lu Y, Yuan L et al (2013) MiR-339-5p regulates the growth, colony formation and metastasis of colorectal cancer cells by targeting PRL-1. PLoS ONE 8:e63142
Vickers MM, Bar J, Gorn-Hondermann I, Yarom N, Daneshmand M et al (2012) Stage-dependent differential expression of microRNAs in colorectal cancer: potential role as markers of metastatic disease. Clin Exp Metastasis 29:123–132
McCormick F (1989) GTPase activating protein Signal transmitter and signal terminator. Cell 56:5–8
Downward J (2003) Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 3:11–22
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J et al (2005) MicroRNA expression profiles classify human cancers. Nature 435:834–838
van Schooneveld E, Wouters MC, Van der Auwera I, Peeters DJ, Wildiers H et al (2012) Expression profiling of cancerous and normal breast tissues identifies microRNAs that are differentially expressed in serum from patients with (metastatic) breast cancer and healthy volunteers. Breast Cancer Res 14:R34
Murakami Y, Yasuda T, Saigo K, Urashima T, Toyoda H et al (2006) Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non-tumorous tissues. Oncogene 25:2537–2545
Shu M, Zheng X, Wu S, Lu H, Leng T et al (2011) Targeting oncogenic miR-335 inhibits growth and invasion of malignant astrocytoma cells. Mol Cancer 10:59
Xiong SW, Lin TX, Xu KW, Dong W, Ling XH et al (2013) MicroRNA-335 acts as a candidate tumor suppressor in prostate cancer. Pathol Oncol Res 19:529–537
Gao L, Yang Y, Xu H, Liu R, Li D et al (2014) miR-335 functions as a tumor suppressor in pancreatic cancer by targeting OCT4. Tumour Biol 35:8309–8318
Giraldez MD, Lozano JJ, Ramirez G, Hijona E, Bujanda L et al (2013) Circulating microRNAs as biomarkers of colorectal cancer: results from a genome-wide profiling and validation study. Clin Gastroenterol Hepatol 11(681–688):e683
Sun Z, Zhang Z, Liu Z, Qiu B, Liu K et al (2014) MicroRNA-335 inhibits invasion and metastasis of colorectal cancer by targeting ZEB2. Med Oncol 31:1–10
Tocque B, Delumeau I, Parker F, Maurier F, Multon MC et al (1997) Ras-GTPase activating protein (GAP): a putative effector for Ras. Cell Signal 9:153–158
Mendoza MC, Er EE, Blenis J (2011) The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci 36:320–328
Choi C, Helfman DM (2013) The Ras-ERK pathway modulates cytoskeleton organization, cell motility and lung metastasis signature genes in MDA-MB-231 LM2. Oncogene 33:3668–3676
Sheppard KE, Cullinane C, Hannan KM, Wall M, Chan J et al (2013) Synergistic inhibition of ovarian cancer cell growth by combining selective PI3K/mTOR and RAS/ERK pathway inhibitors. Eur J Cancer 49:3936–3944
Renshaw J, Taylor KR, Bishop R, Valenti M, De Haven Brandon A et al (2013) Dual blockade of the PI3K/AKT/mTOR (AZD8055) and RAS/MEK/ERK (AZD6244) pathways synergistically inhibits rhabdomyosarcoma cell growth in vitro and in vivo. Clin Cancer Res 19:5940–5951
Molina JR, Adjei AA (2006) The Ras/Raf/MAPK pathway. J Thorac Oncol 1:7–9
Zhang Z, Miao L, Lv C, Sun H, Wei S et al (2013) Wentilactone B induces G2/M phase arrest and apoptosis via the Ras/Raf/MAPK signaling pathway in human hepatoma SMMC-7721 cells. Cell Death Dis 4:e657
Fritsch R, de Krijger I, Fritsch K, George R, Reason B et al (2013) RAS and RHO families of GTPases directly regulate distinct phosphoinositide 3-kinase isoforms. Cell 153:1050–1063
Sun D, Yu F, Ma Y, Zhao R, Chen X et al (2013) MicroRNA-31 activates the RAS pathway and functions as an oncogenic MicroRNA in human colorectal cancer by repressing RAS p21 GTPase activating protein 1 (RASA1). J Biol Chem 288:9508–9518
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos 81272758 and 81302158), and the Natural Science Foundation of Guangdong Province (S2012010009351).
Author information
Authors and Affiliations
Corresponding author
Additional information
Yanxia Lu, Hui Yang, Li Yuan, Guobing Liu have contributed equally to this work.
Rights and permissions
About this article

Cite this article
Lu, Y., Yang, H., Yuan, L. et al. Overexpression of miR-335 confers cell proliferation and tumour growth to colorectal carcinoma cells. Mol Cell Biochem 412, 235–245 (2016). https://doi.org/10.1007/s11010-015-2630-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-015-2630-9