
Abstract
Octamer-binding transcription factor 4 (OCT4) was closely related to pancreatic cancer progression, but its regulation in pancreatic cancer by microRNA (miRNA) is not fully clear. OCT4-positive and OCT4-negative pancreatic cells were isolated by flow cytometry, and it was found that OCT4-positive cells are enriched in transplanted pancreatic cancer cells compared with the primary ones and showed increasing proliferation and sphere formation. The data of miRNA array assay showed that miR-335 in OCT4-positive pancreatic cancer cells was lower than that in the negative ones. The results were confirmed in pancreatic cancer tissue and cell lines. Through expression analysis, it was found that miR-335 was underexpressed in OCT4(+) pancreatic cancer cells purified from primary tumors. Enforced expression of miR-335 in OCT4(+) pancreatic cancer cells inhibited clonogenic expansion and tumor development. miR-335 re-expression in OCT4(+) pancreatic cancer cells was blocked. Systemically delivered miR-335 inhibited pancreatic cancer metastasis and extended animal survival. Of significance, OCT4 was identified and validated as a direct and functional target of miR-335. Taken together, our results provide evidence that miR-335 might inhibit progression and stem cell properties of pancreatic cancer targeting OCT4.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pancreatic cancer is one of the poorest prognoses among all cancers. Aside from its silent nature and tendency for late discovery, pancreatic cancer also shows unusual resistance to chemotherapy and radiation [1]. Only 20 % of pancreatic cancer patients are eligible for surgical resection. The operations are very complex, and unless performed by surgeons specially trained and experienced in this procedure, they can be associated with very high rates of operative morbidity and mortality [2]. Unfortunately, many pancreatic cancers are not resectable at the time of diagnosis [2, 3]. Furthermore, there are limited treatment options available for the patients with pancreatic cancer because chemo- and radiotherapies are largely ineffective, and metastatic disease frequently redevelops even after surgery. Therefore, there is a need to discover novel and effective approaches for the prevention and/or treatment of pancreatic cancer.
Octamer-binding transcription factor 4 (OCT4) is always considered as a stem cell marker for pancreatic cancer [4]. OCT4 plays important roles in cancer progression such as proliferation, cell cycle, stem cell properties, metastasis, and so on [4–8]. Cancer stem cells (CSCs) have been proposed to be the cause of therapy failure, cancer relapse, and drug resistance [9]. CSCs and epithelial-mesenchymal transition (EMT)-type cells, which share molecular characteristics with CSCs, have been proposed to play critical roles in drug resistance and early cancer metastasis as demonstrated in several human malignancies including pancreatic cancer [10–13]. Thus, the discovery of molecular knowledge related to stem cell characteristics and EMT in pancreatic cancer is becoming an important area of research, and such knowledge is likely to be helpful in the discovery of novel molecular targets and strategies for the prevention of pancreatic cancer by targeting CSCs.
Emerging evidence indicates the critical roles of microRNAs (miRNAs) in the regulation of various biological and pathologic processes in cancer [14]. These small, noncoding molecules elicit their regulatory effects by imperfectly binding to the 3′ untranslated region of target mRNA, causing either degradation of mRNA or inhibition of their translation to functional proteins [15]. The expression of miRNAs has been recognized as integral components of many normal biological processes involving cell proliferation, differentiation, apoptosis, and stress resistance [16–18]. More importantly, it has been recently suggested that aberrant upregulation or downregulation of specific miRNAs and their targets in various types of cancer is associated with the development and progression of cancer [16–18]. The low expression of miR-335 has a close correlation with cancer development [19–25]. miR-335 has been identified as a metastasis-suppressor miRNA in cancers. The expression of miR-335 is lost in the majority of primary tumors from patients who relapse, and the loss of miR-335 expression is associated with poor distal metastasis-free survival. It regulates a set of genes, which are SOX4, PTPRN2, MERTK, and possible TNC, the collective expression of which in a large cohort of human tumors is associated with the risk of distal metastasis [26–30].
Our previous data of microarray showed that several stem cell markers such as OCT4, CD133, and C-myc were upregulated in transplanted pancreatic cancers. In this study, OCT4, CD133, and C-myc expression were verified by reverse transcriptase polymerase chain reaction (RT-PCR). OCT4 was increased much more than the other genes. We indentified that miR-335, directly targeting OCT4, was able to inhibit pancreatic cancer progression.
Materials and methods
Cell culture
Primary tumor cells were maintained in the Dulbecco’s modified Eagle’s medium (DMEM, Gibco, USA) supplemented with 10 % fatal bovine serum in a humidified incubator at 37 °C and 5 % CO2. Human pancreatic cancer cells SW1990, PANC-1, BxPC3, and CFPAC were obtained from the American Type Culture Collection (ATCC) and were cultured in DMEM supplemented with 10 % fetal bovine serum, 100 U/mL of penicillin, and 100 μg/mL of streptomycin. Cells were cultured at 37 °C in a humidified atmosphere of 5 % CO2. Human pancreatic normal cells HPDE6c7 were purchased from GuangZhou Jennio Biotech company and cultured according to the protocol.
Clinical specimens
Primary pancreatic cancer biopsy specimens and normal biopsies were obtained from the First Affiliated Hospital (Soochow University, China). Both tumors and their normal tissues were histologically confirmed by hematoxylin and eosin (H&E) staining. Informed consent was obtained from each patient, and the research protocols were approved by the Ethics Committee of the First Affiliated Hospital of Soochow University.
miRNA precursor expression profiling
Total RNA was isolated from the cell lines or tissues in 1 mL of Trizol (Invitrogen, USA). RNA concentration was determined by analyzing 1 μL of solution using the ND-1000 micro-spectrophotometer (NanoDrop Technologies, Wilmington, DE). RNA integrity was evaluated using the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA). RNA with a RIN ≥ 4 was included in the study. RNA was briefly treated with RNase-free DNAase I, and cDNA was synthesized from 1 μg of total RNA by using gene-specific primers to 222 miRNA precursors plus 18S rRNA. The expression of 222 miRNA precursors was profiled using a real-time quantitative PCR assay. Duplicate PCRs were performed for each miRNA precursor gene in each sample of cDNA. The mean CT was determined from the duplicate PCRs. Relative gene expression was calculated as 2 − (CTmiRNA − CT18S rRNA).
Oligonucleotide construction and lentivirus transduction
The oligonucleotide of mature miR-335 was chemosynthesized, amplified, and cloned into GV232-Puro Vectors by Genechem Co., Ltd. (Shanghai, China). The correct sequences and insertions were confirmed by DNA sequencing. Cells were lentivirally transfected with either the GV232-Puro-miR-335 recombined vector (LV-miR-335) or empty GV232-Puro vector (negative control, miR control). Oligonucleotide transfection or lentivirus construction was performed using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instructions. The transduced cells with a cell density of over 40 % confluency were exposed to puromycin dihydrochloride (1 mg/mL, Sigma, St. Louis, MO) for resistance selection. When all of the cells in the non-transfected control culture were killed, puromycin-resistant cell clones were picked and passaged in a medium containing a half concentration of puromycin (0.5 mg/mL) in the first round of selection. Lentivirus-mediated silencing of miR-335 was verified by qRT-PCR.
RNA isolation and real-time RT-PCR
Total RNA was isolated using the Trizol system (Invitrogen) according to the manufacturer’s guidelines. Oligo(dT)18 primer and M-MLV reverse transcriptase were used for the first-strand synthesis. Real-time RT-PCR was performed with Real-time RT-PCR Master Mix containing SYB Green I and Hot Start Taq DNA polymerase (Toyobo). The threshold cycle (Ct) values were determined by using an iCycler thermal cycler (Bio-Rad, Hercules, CA) performed according to the manufacturer’s instructions. OCT4, CD133, c-myc, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) primers were purchased from Invitrogen. The relative expression levels were calculated by comparing Ct values of the samples with those of the reference, all data normalized to the internal control GAPDH.
Fluorescence-activated cell sorting
Cells of 104 were suspended in fluorescence-activated cell sorting (FACS) buffer (phosphate-buffered saline (PBS) with 0.1 % BSA and 0.1 % Triton X100) followed by incubation with FITC-conjugated anti-OCT4 for 15 mins at room temperature. Cells were then washed with PBS and resuspended in PBS for FACS analysis.
MTT assay
3-[4,5-Dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT) assay was employed to detect the growth of pancreatic cancer cells, and the growth curve was delineated. Logarithmic-phase cells were collected, and the concentration of the cell suspension was adjusted to 5,000 cells per well (the edge wells of the plate are filled with aseptic PBS buffer). The cells were incubated at 37 °C and 5 % CO2 until cells cover the bottom of the well (a flat-bottom 96-well plate), and then the cells were cultured. Twenty microliters of the MTT solution was added to each well (5 mg/mL, 0.5 % MTT), and the cells were continued to culture for 4 h. After the incubation, the supernatant was discarded, and 150 μL dimethyl sulfoxide was added to each well, and the culture plate was shaken at a low speed for 10 min until crystal dissolved completely. The ELISA reader was used to measure the absorbance at 570 nm.
Colony-forming assay
Cells in logarithmic growth phase were digested in 0.5 % trypsin/0.04 % EDTA, and a single cell suspension was prepared. Then, these cells were added to six-well plates (200 cells/well) followed by incubation at 37 °C in an environment with saturated humidity and 5 % CO2 for 24 h. Nonadherent cells were removed. After culture for 10–14 days, colonies were present. The colony-forming efficiency and the morphology of colonies were observed.
Tumor spheroid assay
Spheroid-forming assays were performed as described. In brief, cells were plated in six-well ultralow attachment plates (Corning Inc., Corning, NY) at a density of 1,000 cells/mL in DMEM supplemented with 1 % N2 Supplement (Invitrogen), 2 % B27 Supplement (Invitrogen), 20 ng/mL human platelet growth factor (Sigma-Aldrich), 100 ng/mL epidermal growth factor (Invitrogen), and 1 % antibiotic-antimycotic (Invitrogen) at 37 °C in a humidified atmosphere of 95 % air and 5 % CO2. Spheroids were collected after 7 days and dissociated with Accutase (Innovative Cell Technologies, Inc.). The cells obtained from dissociation were sieved through a 40-μm filter and counted by a Coulter counter using trypan blue dye.
Western blot analysis
Western blots were performed as we described elsewhere. In brief, cells were lysed in RIPA buffer containing 1× protease inhibitor cocktail, and protein concentrations were determined using the Bradford assay (Bio-Rad, Philadelphia, PA). Proteins were separated by 12.5 % SDS/PAGE and transferred to membranes (Millipore, Bedford, MA) at 55 V for 4 h at 4 °C. After blocking in 5 % nonfat dry milk in Tris-buffered saline (TBS), the membranes were incubated with primary antibodies at 1:1,000 dilution in TBS overnight at 4 °C, washed three times with TBS-Tween 20, and then incubated with secondary antibodies conjugated with horseradish peroxidase from Santa Cruz (TX, USA) at 1:5,000 dilution in TBS for 1 h at room temperature. Primary antibodies including OCT4, E-cadherin, Fibronectin, Vimentin, and GAPDH were ordered from Cell Signaling (USA). α-Smooth muscle actin (α-SMA) antibody was purchased from Epitomics (CA, USA). Membranes were washed again in TBS-Tween 20 for three times at room temperature. Protein bands were visualized on X-ray film using an enhanced chemiluminescence detection system.
Transwell migration assay
For transwell migration assays, 1 × 105 pancreatic cancer cells were plated in the top chamber onto the noncoated membrane (24-well insert; pore size, 8 μm; Corning Costar) and allowed to migrate toward a serum-containing medium in the lower chamber. Cells were fixed after 24 h of incubation with methanol and stained with 0.1 % crystal violet (2 mg/mL, Sigma-Aldrich). The number of cells invading through the membrane was counted under a light microscope (three random fields per well).
Transwell invasion assay
For invasion assay, 1 × 105 cells were plated in the top chamber onto the Matrigel-coated membrane (24-well insert; pore size, 8 μm; Corning Costar). Each well was coated freshly with Matrigel (60 μg, BD Bioscience) before the invasion assay. Pancreatic CSCs were plated in a medium without serum or growth factors, and a medium supplemented with serum was used as a chemoattractant in the lower chamber. The cells were incubated for 48 h, and cells that did invade through the pores were removed by a cotton swab. CSCs on the lower surface of the membrane were fixed with methanol and stained with crystal violet. The number of cells invading through the membrane was counted under a light microscope (×40, three random fields per well).
Statistical analysis
The mean and SD were calculated for each experimental group. Differences between groups were analyzed by one- or two-way ANOVA, followed by Bonferroni’s multiple comparison tests using PRISM statistical analysis software (GraphPad Software, Inc., San Diego, CA). Significant differences among groups were calculated at P < 0.05.
Results
OCT4 promotes progression of pancreatic cancer
To compare with difference of gene expression in the primary tumor cells and transplanted ones, microarray was used to screen out the overexpressed gene in transplanted cells, which showed the expression levels of OCT4, CD133, and C-myc. The overexpressed genes were verified using real-time RT-PCR (Fig. 1a). OCT4-positive cells (+) were selected by flow cytometry (Fig. 1b). After cell separation and identification by flow cytometry, it indicated that OCT4(+) cells took more portions in transplanted tumor cells than in primary tumor cells. In order to know the role of OCT4(+) cells, cell biological functions were analyzed. The proliferation of OCT4(+) pancreatic cancer cells was increased compared with that of OCT4(−) ones (Fig. 1c, d). The sphere formation of OCT4(+) cells increased than that of OCT4(−) ones (Fig. 1e). These data indicated that OCT4-positive pancreatic cancer cells showed stem-like cells.

OCT4 promotes proliferation and stem cell properties in pancreatic cancer cells. a OCT4, CD133, and C-myc expression were verified by real-time RT-PCR. Primary tumor cells and transplanted tumor cells were cultured, and total RNA was collected for real-time RT-PCR. b Primary tumor cells and transplanted tumor cells were cultured and collected for flow cytometry. c Cell proliferation was analyzed by MTT assay. OCT4(+) cells and OCT4(−) cells were seeded in a 96-well plate; added with MTT in days 0, 1, 2, 3, 4, and 5; and then recorded. d Cell proliferation was analyzed by colony formation assay. One hundred cells were seeded in a six-well plate, and colonies were counted 10 days later. e Stem cell sphere formation was performed in OCT4(+) and OCT4(−) cells
Loss of miR-335 in pancreatic cancer cells with OCT4 expression in human pancreatic cancer
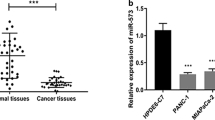
To compare the miRNA profile in the pancreatic cancer cells with OCT4(+) and (−), miRNA assay was used to analyze the different miRNAs. The data indicated that miR-335 in OCT4(+) pancreatic cancer cells was much lower than that in the OCT4(−) ones (Fig. 2a). Next, to investigate the possible role of miR-335 in pancreatic cancer, we examined the expression of miR-335 in human pancreatic specimens by real-time RT-PCR. We examined the expression of miR-335 in 16 tumor samples and 16 normal tissues. As shown in Fig. 2b, the expression levels of miR-335 in tumor samples were lower than those in normal ones. Similarly, miR-335 was lower in human pancreatic cancer cells compared with that in normal ones (Fig. 2c). These results provided us an initial evidence that miR-335 may play a suppressing miRNA in the development of human pancreatic cancer.

Loss of miR-335 in pancreatic cancer cells with OCT4 expression in human pancreatic cancer. a OCT4(+) and OCT4(−) cells were performed for microRNA array assay. b Average expression level of miR-335 in human pancreatic cancer specimens (n = 18) and normal tissues (n = 18). miRNA abundance was assessed by real-time RT-PCR and normalized to U6 RNA. c Real-time PCR analysis of miR-335 in SW1990, PANC-1, BxPC3, CFPAC pancreatic cancer cells, and normal pancreatic cells
OCT4 was a new target gene of miR-335 in pancreatic cancer cells
To clear the relationship between OCT4 and miR-335, TargetScan also showed that OCT4 may be directly suppressed by miR-335 (Fig. 3a). As shown in Fig. 3b, the luciferase activity of pGL4-OCT4-WT in OCT4 cells was much lower than that in control cells. The luciferase activity of pGL4-OCT4-Mut was rescued in cells. Compared with control, endogenous OCT4 mRNA levels were downregulated in the cells with miR-335 overexpression (Fig. 3c). We next examined whether miR-335 could regulate endogenous OCT4 expression in pancreatic cancer cells. When pancreatic cancer cells SW1990 and OCT4(+) with miR-335, OCT4 was decreased by Western blot (Fig. 3d). These data clearly indicated that OCT4 was a new target gene of miR-335, and miR-335 was a suppressor miRNA in pancreatic cancer.

OCT4 was a new target gene of miR-335 in pancreatic cancer cells. a The 3′- untranslated region (UTR) of the OCT4 gene contains binding sites for miR-335 according to bioinformatic analysis. b miR-335 suppressed the expression of a luciferase reporter gene harboring the 3′-UTR of OCT4. The pGL4 plasmid was modified by adding the human 3′-UTR or the 3′-UTR with mutations in regions complementary to miR-30a seed regions behind the firefly luciferase gene. Pancreatic cells were transiently co-transfected with negative control (mock) or miR-335 together with the indicated luciferase constructs, and luciferase activity was analyzed 48 h later. Data are presented as relative firefly luciferase activity normalized to Renilla luciferase activity from the same construct. The data presented are shown as means ± s.d. collected from three independent experiments. c miR-335 restoration downregulated OCT4 in OCT4-positive pancreatic cancer cells. Cells were transfected with miR-335 or miR control for 48 h and were then collected for Western blot analysis. miR-335 restoration downregulated OCT4 mRNA in OCT4-positive pancreatic cancer cells. Cells were transfected with miR-335 or miR control for 48 h and were then collected for real-time PCR. d miR-335 decreased OCT4 expression in SW1990 and OCT4(+) pancreatic cancer cells. Cells were transfected with miR-335 or miR- control for 48 h and were then collected for Western blot analysis
miR-335 inhibits progression and stem cell properties of pancreatic cancer cells by targeting OCT4
To explore the effect of miR-335 on cell growth, SW1990 cells were infected with LV-miR-335 or LV-miR-control, respectively. The results of colony formation assay displayed that miR-335 inhibited cell in pancreatic cancer cells (Fig. 4a). Because the miR control had no effect on cell growth, the data was not shown in the comparison of parent cells and the control ones, which indicated that the effect caused by miR-335 was highly specific. To further observe miR-335-mediated growth inhibition, cells were infected with LV-miR-335, and cell cycle distribution was examined. Compared with miR control, SW1990 cells infected with LV-miR-335 displayed an increased percentage of cells in G1 phase and fewer cells in S phase (Fig. 4b).

miR-335 inhibits proliferation and stem cell properties of pancreatic cancer cells by targeting OCT4. a Cell proliferation was analyzed by colony formation assay. SW1990 cells with LV-miR-335 or control or combined with OCT4 were seeded in a six-well plate, and colonies were counted after 10 days. b Cell cycle distribution of SW1990 cells was analyzed by flow cytometry. Cells with LV-miR-335 or control or combined with OCT4 were seeded in a six-well plate, and cell cycle was assayed. c Stem cell sphere formation was performed in SW1990 pancreatic cancer cells with LV-miR-335, control, or both of miR-335 and OCT4. d Comparison of the pancreatic cancer formation with OCT4(+) and OCT4(−) cells with LV-miR-335 or control on nude mice
Our above results have shown that miR-335 potently inhibits OCT4 expression. To investigate the potential role of miR-335 in pancreatic cancer stem cells, we examined whether miR-335 restoration could inhibit the cells and their self-renewal potential. Figure 4c shows that miR-335 restoration significantly decreased the OCT4(+) cells with 87 % reduction. This miR-335-induced reduction of the OCT4(+) cells was accompanied by reduced tumor sphere formation and smaller size of the tumor spheres. In order to explore the role of miR-335 in vivo of pancreatic cancer, models of pancreatic cancer were set up. Data of tumor growth curve showed that miR-335 could decrease the pancreatic cancer formation (Fig. 4d). miR-335 inhibits metastasis of pancreatic cancer cells by targeting OCT4.
To investigate the role of miR-335 in pancreatic cancer metastasis and EMT, OCT4 overexpressed cells were infected with LV-miR-335, and the results showed that upregulation of miR-335 could significantly decreased migration (Fig. 5a) and invasion (Fig. 5b) of pancreatic cancer cells. EMT markers were also detected by Western blot. The results showed that mesenchymal markers Fibronectin, Vimentin, α-SMA, and SNAIL1 decreased and epithelial marker, E-cadherin, increased in the pancreatic caner cells with LV-miR-335 (Fig. 5c). The results showed that miR-335 could inhibit metastasis and EMT of pancreatic cancer cells.

miR-335 inhibits metastasis of pancreatic cancer cells by targeting OCT4. a Migration of pancreatic cancer cells. Migration assay using the transwell chamber was performed for SW1990 cells with LV-miR-335 or control or combined with OCT4. b Invasion of pancreatic cancer cells. Invasion assay using the transwell chamber with matrigel in the upper chamber was performed for SW1990 cells with LV-miR-335 or control or combined with OCT4. c E-cadherin was detected by immunofluoresence, and EMT markers in pancreatic cancer cells were detected by Western blot. OCT4(+) cells with LV-miR-335 or control were seeded in the 12-well plate, fixed in the fixation solution, and cultured with E-cadherin antibody. OCT4(+) cells with LV-miR-335 or control were seeded in the six-well plate, and total protein was isolated from the cells and detected for E-cadherin, Fibronectin, Vimentin, α-SMA, and OCT4 protein
Discussion
Our data provide the first evidence that miR-335 is able to inhibit OCT4-positive tumor sphere-forming and tumor-initiating cancer stem cells in pancreatic cancer, implying that miR-335 might play a role in the self-renewal of pancreatic cancer stem cells. Similar results were also observed in pancreatic cells, where lentiviral miR-335 restoration significantly inhibited the clonogenic growth and tumor spheres.
Cancer stem cells have been defined as cells within a tumor that possess the capacity to self-renew and to cause the heterogeneous lineages of cancer cells that comprise the tumor [31]. These two definitive biological properties are what make the CSCs the prime candidate for initiation of relapse. These cells express stemness markers, are able to form floating spheres in a serum-free medium, a property associated with stem cells, and are also able to differentiate in an aberrant cell phenotype constituting tumor heterogeneity [32]. Experimentally, this population is identified by its ability to form new tumors through serial transplantations in immunodeficient hosts, re-establishing tumor heterogeneity [33]. There are three distinct and main methodologies to isolate CSCs from solid tumors: detection of side population (SP) phenotype by Hoechst 33342 exclusion [34], sphere formation by cultivation of a defined serum-free medium with growth factors that maintain the CSCs undifferentiated [35, 36], and isolation of CSCs by flow cytometry according to CSC-specific cell surface markers [37].
Cell surface markers of CSCs can help distinguish, isolate, and purify these tumor-initiating cells for further biological investigation. OCT4(+) cells comprised a small fraction of the total tumor population in all three samples studied, but represented an increased percentage of the sphere-forming cells. This suggests that OCT4(+) could act as a cell surface marker for CSCs in pancreatic cancer. We also investigated the use of this cell surface protein as a candidate marker to further identify the CSC phenotype in GBC. The self-renewal ability of OCT4(+) cells was tested using spheroid-forming assays in serum-free medium. OCT4(+) cells possessed higher clonogenicity than their antigen-negative counterparts. Subsequent in vivo tumorigenesis experiments demonstrated that OCT4(+) cells possessed higher tumorigenicity than the OCT4(−) subpopulation. Furthermore, the tumors generated in nude mice displayed the same phenotype as the primary pancreas tissue. Taken together, these results firmly suggest that CD133+ cells possess the potentials for self-renewal and high tumorigenicity, exhibiting cancer stem cell-like characteristics in human pancreatic cancer.
We used bioinformatics to search potential target genes and found that OCT4 was a target gene of miR-335. miR-335 could inhibit cell proliferation, stem cell properties, and EMT by downregulation of OCT4. EMT has been described as a cell biological program that is required for the remodeling of cells and tissues during embryogenesis, during certain types of wound healing, and during the acquisition of malignant traits by carcinoma cells. EMT is a key developmental program that is often activated during cancer invasion and metastasis and may promote resistance to chemotherapy. In a few years, evidence has mounted for EMT as the key means through which cancer cells acquire more highly mobile potentials to migrate and metastasize to distant sites during tumor progression [7]. E-cadherin, a classical cadherin from the cadherin superfamily, is required for maintaining epithelial cell plasticity. N-cadherin, known as an important member of the cadherin family that mediates calcium-dependent adhesion, is normally expressed in mesenchymal cells. Loss of E-cadherin and increased N-cadherin expression are now defined as a major hallmark of EMT [8, 9]. Snail, one member of the zinc finger family composed of a highly conserved COOH-terminal region, induces EMT and tumor invasion by binding the E-cadherin promoter through Ebox sequences. Over the past few years, accumulating data has demonstrated that EMT correlates closely with the acquisition of stem cell-like properties in cancer cells [10, 11]. Our results indicated that miR-335 could suppress EMT of OCT4-positive pancreatic cells. This is the first report that showed that miR-335 inhibited EMT of pancreatic cancer.
In summary, we identified miR-335 to be a tumor suppressor miRNA in pancreatic cancer, and low miR-335 expression was an unfavorable prognostic factor in patients with pancreatic cancer. miR-335 partially influences human pancreatic cancer through the regulation of OCT4. These results suggest that miR-335 is a potential target for treating pancreatic cancer, and the critical roles of miR-335 in pancreatic cancer tumorigenesis may aid patient prognosis and diagnosis. Our findings provide basic information to better understand the pathogenesis of pancreatic cancer and its possible therapeutic strategies.
References
Keane MG, Bramis K, Pereira SP, Fusai GK. Systematic review of novel ablative methods in locally advanced pancreatic cancer. World J Gastroenterol. 2014;20(9):2267–78.
He J, Page AJ, Weiss M, Wolfgang CL, Herman JM, Pawlik TM. Management of borderline and locally advanced pancreatic cancer: where do we stand? World J Gastroenterol. 2014;20(9):2255–66.
Herreros-Villanueva M, Bujanda L, Billadeau DD, Zhang JS. Embryonic stem cell factors and pancreatic cancer. World J Gastroenterol. 2014;20(9):2247–54.
Iki K, Pour PM. Expression of Oct4, a stem cell marker, in the hamster pancreatic cancer model. Pancreatology. 2006;6(4):406–13.
Wang D, Zhu H, Zhu Y, Liu Y, Shen H, Yin R, et al. Su Z.CD133(+)/CD44(+)/Oct4(+)/Nestin(+) stem-like cells isolated from Panc-1 cell line may contribute to multi-resistance and metastasis of pancreatic cancer. Acta Histochem. 2013;115(4):349–56.
Wang H, Wang S, Hu J, Kong Y, Chen S, Li L, et al. Oct4 is expressed in Nestin-positive cells as a marker for pancreatic endocrine progenitor. Histochem Cell Biol. 2009;131(5):553–63.
Wen J, Park JY, Park KH, Chung HW, Bang S, Park SW, et al. Oct4 and Nanog expression is associated with early stages of pancreatic carcinogenesis. Pancreas. 2010;39(5):622–6.
Chiou SH, Wang ML, Chou YT, Chen CJ, Hong CF, Hsieh WJ, et al. Coexpression of Oct4 and Nanog enhances malignancy in lung adenocarcinoma by inducing cancer stem cell-like properties and epithelial-mesenchymal transdifferentiation. Cancer Res. 2010;70(24):10433–44.
Park KS, Shin SW, Choi JW, Um SH. Specific protein markers for stem cell cross-talk with neighboring cells in the environment. Int J Stem Cells. 2013;6(2):75–86.
Bao B, Ahmad A, Li Y, Azmi AS, Ali S, Banerjee S, et al. Targeting CSCs within the tumor microenvironment for cancer therapy: a potential role of mesenchymal stem cells. Expert Opin Ther Targets. 2012;16(10):1041–54.
Nieto MA. Epithelial plasticity: a common theme in embryonic and cancer cells. Science. 2013;342(6159):1234850.
Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501(7467):328–37.
Espinoza I, Pochampally R, Xing F, Watabe K, Miele L. Notch signaling: targeting cancer stem cells and epithelial-to-mesenchymal transition. Onco Targets Ther. 2013;6:1249–59.
Palanichamy JK, Rao DS. miRNA dysregulation in cancer: towards a mechanistic understanding. Front Genet. 2014;5:54.
Tessitore A, Cicciarelli G, Del Vecchio F, Gaggiano A, Verzella D, Fischietti M, et al. MicroRNAs in the DNA damage/repair network and cancer. Int J Genomics. 2014;2014:820248.
Takahashi RU, Miyazaki H, Ochiya T. The role of microRNAs in the regulation of cancer stem cells. Front Genet. 2014;4:295.
Hauptman N, Glavac D. MicroRNAs and long non-coding RNAs: prospects in diagnostics and therapy of cancer. Radiol Oncol. 2013;47(4):311–8.
Cheng Q, Yi B, Wang A, Jiang X. Exploring and exploiting the fundamental role of microRNAs in tumor pathogenesis. Onco Targets Ther. 2013;6:1675–84.
Png KJ, Yoshida M, Zhang XH, Shu W, Lee H, Rimner A, et al. MicroRNA-335 inhibits tumor reinitiation and is silenced through genetic and epigenetic mechanisms in human breast cancer. Genes Dev. 2011;25(3):226–31.
Lynch J, Meehan MH, Crean J, Copeland J, Stallings RL, Bray IM. Metastasis suppressor microRNA-335 targets the formin family of actin nucleators. PLoS ONE. 2013;8(11):e78428.
Vimalraj S, Miranda PJ, Ramyakrishna B, Selvamurugan N. Regulation of breast cancer and bone metastasis by microRNAs. Dis Markers. 2013;35(5):369–87.
Wang Y, Zhao W, Fu Q. miR-335 suppresses migration and invasion by targeting ROCK1 in osteosarcoma cells. Mol Cell Biochem. 2013;384(1–2):105–11.
Gong M, Ma J, Guillemette R, Zhou M, Yang Y, Yang Y, et al. miR-335 inhibits small cell lung cancer bone metastases via IGF-IR and RANKL pathways. Mol Cancer Res. 2014;12(1):101–10.
Wang H, Li M, Zhang R, Wang Y, Zang W, Ma Y, et al. Effect of miR-335 upregulation on the apoptosis and invasion of lung cancer cell A549 and H1299. Tumour Biol. 2013;34(5):3101–9.
Cao J, Cai J, Huang D, Han Q, Yang Q, Li T, et al. miR-335 represents an invasion suppressor gene in ovarian cancer by targeting Bcl-w. Oncol Rep. 2013;30(2):701–6.
Dohi O, Yasui K, Gen Y, Takada H, Endo M, Tsuji K, et al. Epigenetic silencing of miR-335 and its host gene MEST in hepatocellular carcinoma. Int J Oncol. 2013;42(2):411–8.
Shi L, Jiang D, Sun G, Wan Y, Zhang S, Zeng Y, et al. miR-335 promotes cell proliferation by directly targeting Rb1 in meningiomas. J Neurooncol. 2012;110(2):155–62.
Lynch J, Fay J, Meehan M, Bryan K, Watters KM, Murphy DM, et al. MiRNA-335 suppresses neuroblastoma cell invasiveness by direct targeting of multiple genes from the non-canonical TGF-β signalling pathway. Carcinogenesis. 2012;33(5):976–85.
Shu M, Zhou Y, Zhu W, Zhang H, Wu S, Chen J, et al. MicroRNA 335 is required for differentiation of malignant glioma cells induced by activation of cAMP/protein kinase A pathway. Mol Pharmacol. 2012;81(3):292–8.
Xu Y, Zhao F, Wang Z, Song Y, Luo Y, Zhang X, et al. MicroRNA-335 acts as a metastasis suppressor in gastric cancer by targeting Bcl-w and specificity protein 1. Oncogene. 2012;31(11):1398–407.
Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–11.
Vermeulen L, Sprick MR, Kemper K, Stassi G, Medema JP. Cancer stem cells—old concepts, new insights. Cell Death Differ. 2008;15(6):947–58.
Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5(4):275–84.
Hirschmann-Jax C, Foster AE, Wulf GG, Goodell MA, Brenner MK. A distinct “side population” of cells in human tumor cells: implications for tumor biology and therapy. Cell Cycle. 2005;4(2):203–5.
Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–11.
Vermeulen L, Sprick MR, Kemper K, Stassi G, Medema JP. Cancer stem cells—old concepts, new insights. Cell Death Differ. 2008;15(6):947–58.
Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5(4):275–84.
Acknowledgments
The project was founded by the Post-Graduate Scientific Research Innovation Project of Education Department of Jiangsu Province (CXZZ12_0842), the Open fund of the Stem Cell and Biomedical Material Key Laboratory of Jiangsu Province (KJS1230), and Science and Technology Research Project of Science and Technology Bureau of Suzhou City (SYS201330), China.
Conflicts of interest
None
Author information
Authors and Affiliations
Corresponding author
Additional information
Ling Gao, Yijin Yang, Haiyan Xu and Ruqian Liu contributed equally to this work.
Rights and permissions
About this article

Cite this article
Gao, L., Yang, Y., Xu, H. et al. miR-335 functions as a tumor suppressor in pancreatic cancer by targeting OCT4. Tumor Biol. 35, 8309–8318 (2014). https://doi.org/10.1007/s13277-014-2092-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-014-2092-9