
Abstract
The application of multiple radiochronometers in a forensic investigation of bulk uranium provides increased confidence for interpreting age-dating results. We have developed a streamlined method for the purification of Pa and Th from bulk uranium and have applied this method in analyzing uranium certified reference materials (CRMs) IRMM-1000 and CRM U100, using the 231Pa–235U and 230Th–234U radiochronometers. Our improved 233Pa spike calibration technique has replaced the time-intensive, conventional calibration using geologic rock standards. Paired Pa–U and Th–U age-dating analyses of CRM U100 produce concordant results. In contrast, analyses of IRMM-1000 demonstrate reproducibly-discordant Th–U and Pa–U age-dates.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Nuclear forensic investigations of known or unknown nuclear material commonly involve determining the production history of that material. The material’s production date can in many cases be determined empirically, with a ‘model age’ of that material (i.e., the time elapsed since last chemical purification) measurable by radiochronometry. Model age information is an important signature in reconstructing a material’s provenance and processing history, both of which are critical components in a nuclear forensic investigation. To determine the model age of a nuclear material, the relative amounts of a parent isotope and the decay product of that parent isotope, a daughter isotope, are measured by mass spectrometry methods. Calculating the model age of a nuclear material requires two primary assumptions: (1) the daughter isotope was completely purified from the parent isotope, and (2) the sample has not lost or gained either parent or daughter isotopes, except by radioactive decay (i.e., the sample is effectively a closed system). The most common radiochronometer used in forensic investigations of bulk uranium is the 230Th–234U daughter–parent decay system. Model ages of many uranium certified reference materials (CRMs) have been measured using this system [1,2,3,4,5,6,7,8]. In some cases, other daughter–parent and even granddaughter–daughter–parent radiochronometers may be applied to CRMs and other nuclear materials, [e.g., 2, 4]. Using multiple radiochronometers can lead to greater confidence in the interpretation of age-dating results and provide insight into processes that lead to concordant or discordant ages. For example, the 231Pa–235U (Pa–U) and 230Th–234U (Th–U) radiochronometers can be applied in tandem to better elucidate sample production histories. Importantly, discordant model ages for a given sample from multiple radiochronometers demonstrate that the two major assumptions described above can be violated.
In 2013, the mass spectrometry group at Lawrence Livermore National Laboratory (LLNL) published analysis methods for 231Pa–235U age-dating and presented results for CRMs (U005A, U030A, U630, CRM 125A) with concordant 230Th–234U and 231Pa–235U model ages [9]. Since that time, we have improved our 231Pa–235U age-dating capability by modifying our 233Pa spike preparation and calibration methods, improving our sample purification chemistry, and refining our data reduction algorithms. We have streamlined our chemical purification procedures to quantify both daughter isotopes, 230Th and 231Pa, from single aliquots of dissolved bulk uranium material. One advantage of determining 230Th and 231Pa assay from the same aliquot is for the scenario where one is sample-limited, e.g., samples are low concentration; young, low enriched or depleted uranium; or limited availability. Taken together, our improved process has resulted in increased throughput of 231Pa–235U model age determinations of CRMs, nuclear materials, and samples for international round robin exercises (i.e., 4, 9).
The most significant improvement to our radiochronometry methods is the use of an in-house 231Pa standard to calibrate 233Pa spikes, which are produced as-needed approximately 4–5 times per year. By utilizing an in-house 231Pa standard solution as a spike calibrant, we have eliminated the complex chemical separation procedures required to produce 231Pa secular equilibrium reference solutions from geologic matrices, as described in [9, 10]. This advance permits rapid spike calibration on the order of approximately 2 days compared with previous calibration methods which can take up to 2 weeks. This rapid spike calibration facilitates the application of multiple radiochronometers to nuclear forensic samples in a timely manner. In this contribution we present radiochronometry results for CRMs U100 and IRMM-1000, and compare the 230Th–234U model dates to 231Pa–235U model dates for each material.
Theory
To review, in applying daughter–parent radiochronometers to nuclear materials, the following two criteria must be met if a model age records the time elapsed since last chemical purification: (1) the material was completely purified of daughter isotopes, and (2) no loss or gain of daughter isotope has occurred (i.e., closed system). The daughter–parent isotope age-dating equation (Eq. 1) is used to calculate the age, t, of the material. The decay constants (λ) used for calculating model ages are from [11,12,13]. The 230Th half-life = 7.569 × 104 ± 115 years, and the 234U half-life = 2.4525 × 105 years. The 231Pa half-life = 32,713 ± 110 years, and the 235U half-life = 7.0381 × 108 ± 4.8 × 105.
Generalized daughter–parent isotope age-dating equation.
All model ages are calculated relative to a reference date, usually the date of Th and/or Pa chemical separation from bulk U in the laboratory during sample purification. If the purification process completely separates the daughter isotope, 230Th or 231Pa, from the parent isotope, 234U or 235U, respectively, then the measured model age subtracted from the reference date results in a ‘model date’. This model date could represent the complete purification date of the daughter from the parent. However, if the purification of daughter from parent is incomplete, the model date will be older than the incomplete purification date. This potential discordance can provide insight into the chemical processes used to purify nuclear material and their separation efficiencies.
Experimental
Age-dating of uranium reference materials by the Th–U and Pa–U radiochronometers requires accurate uranium (U) assay. Uranium isotope dilution by mass spectrometry (IDMS) methods are not given in detail in this study as they are described in other publications [e.g., 5, 8]. Daughter/parent ratio were determined using multi-collector-inductively coupled plasma-mass spectrometry (MC-ICP-MS) at LNLL.
Determination of 231Pa (and 230Th) assay by IDMS includes three steps: (1) production of a 233Pa spike material to use as a tracer in unknown samples; (2) calibration of that spike using either geologic materials, e.g., Table Mountain Latite or BCR-2 (USGS), or a 231Pa standard material, e.g., that can be milked from highly enriched uranium; and (3) spiking the sample with a calibrated 233Pa spike (and 229Th spike), separating and purifying the Pa and Th from the sample matrix, and measurement by MC-ICP-MS. Each of the chemical separation and purification steps outlined below utilize ultra-high purity reagents, i.e., Seastar Chemicals.
233Pa spike production
Due to the relatively short half-life of 233Pa (26.967 ± 0.004 days [14]), a 233Pa spike has a useful life of a few months, and therefore generally must be produced as-needed. Pa233 is sourced at LLNL by milking 237Np aliquots that are sufficiently old to be in secular equilibrium with 233Pa (Fig. 1). The Np stock used in this study has approximately 20–30 mg 237Np. The 237Np stock material is dried in 20 µL HClO4 to eliminate residual HF that may be present, as this inhibits Pa sorption to anion exchange resin used in Np–Pa separation. The Np stock is sequentially dried down with a few drops of concentrated HNO3, followed by a few drops of concentrated HCl, and finally 0.25 mL concentrated HCl and 0.25 mL 1 M hydroxylamine hydrochloride. These dry-down steps first oxidize and then reduce the oxidation state of Np, converting the 237Np stock to Np(IV) required to sorb Np to the anion exchange resin medium. The dried Np stock should be a bright emerald green color when adequately reduced; critically, Np–Pa separation efficiency is highly dependent on the oxidation state of Np. The Np stock material is then dissolved in (1 mL) 5:1 parts 10 M HCl: 1 M hydroxylamine hydrochloride; heated (capped) to dissolve the Np stock; ultrasonicated; and allowed to cool for at least 10 min.

233Pa ingrowth curve relative to secular equilibrium between 233Pa and 237Np
The first Np–Pa separation step is accomplished by anion exchange chemistry with AG-MP-1 resin (BioRad, 100-200 mesh). A 2-mL resin bed is loaded in a labeled BioRad PolyPrep column and conditioned with 8 mL 10 M HCl. A cleaned, labeled, Teflon vial is placed to catch the load and the vial rinses using 10 M HCl (0.5 mL; 1 mL) of the Np stock. Minor loss of Pa and Np(V) may occur during the initial load and rinse of the column bed with 10 M HCl. Np(IV) should be visible as a bright green band at the top of the resin bed and should remain throughout successive additions of rinse acid. Np sorption can also be monitored using a beta–gamma probe to track activity or measured directly by gamma spectrometry. If necessary, these initial rinses can be combined with the recovered Np later. Subsequent rinses (a total of 9 mL) with 10 M HCl should be activity-free, with Pa and Np fully sorbed to the anion resin. A clean, labeled vial is then placed to collect the Pa elution, accomplished using 8 mL 10 M HCl + 0.05 HF; the 233Pa elution is then dried down on a hotplate following addition of trace (~ 20 µL) HClO4. Additional 233Pa elution dry-downs are performed using a few drops of concentrated HNO3, concentrated HCl, and finally, concentrated HCl + 1 M hydroxylamine hydrochloride as described above. The Pa fraction is then ready for an additional Np–Pa separation. A Teflon vial is positioned under the column, and the Np eluted with 10 mL 1 M HCl + 0.5 M HF (combined with the load and rinse of the Np stock material). This can be dried down and used for future 233Pa spike production. The separation and recovery of Np and Pa can be monitored before proceeding to the next anion column. The second Np–Pa separation is done on a smaller volume, 1-mL AG-MP-1 (BioRad, 100-200 mesh) anion exchange resin bed using proportionately smaller load and rinse volumes (relative to the resin volume); again, the load and rinses are collected and combined with the Np recovery after Pa elution. The 233Pa spike can now be dried in a few drops of concentrated HNO3 + trace HClO4 to remove residual HF present before the final chemical purification step.
The final purification step for the 233Pa spike preparation is done using silica gel resin (Supelco, 75–200 µm particle size; see [10], for the suggested cleaning procedure). A 1.8-mL resin bed is constructed in a BioRad PolyPrep column and conditioned with 4 mL 5% HNO3. The dried 233Pa fraction is dissolved in 0.5 mL 5% HNO3 and 50 µL saturated H3BO3; heated (capped) on a hotplate; sonicated; and allowed to sit for 15 min. A waste beaker or Teflon vial is positioned under the column, and the sample is loaded onto the silica gel resin bed. All Pa should be sorbed to the silica gel, the efficiency of which can again be monitored with a beta–gamma probe. Following incremental rinsing (0.5 mL; 1 mL) of the 233Pa vial with 5% HNO3, the column is rinsed with 8 mL 5% HNO3. These rinses should continue to flush residual 237Np from the 233Pa spike. The tip of the column is then rinsed with ultrapure water to remove trace 237Np that may remain on the tip of the column. It is also important to note the date and time of final 233Pa separation from 237Np in the following elution steps, as this is an important factor when reconstructing the spike calibration after 233Pa has fully decayed to 233U after many months. The 233Pa is eluted into a cleaned, labeled, Teflon vial (i.e., 30-mL capacity) that can be used as a working spike vial with 6 mL 5% HNO3 + 0.1 M HF. A small aliquot can be taken from this solution and screened by MC-ICP-MS to determine the Np/Pa separation factor, as well as to approximate the 233Pa concentration from the known sensitivity of the instrument. If Np/Pa < 1000, the spike is sufficiently pure. If not, the silica gel purification step is repeated, again, noting the time of separation of 233Pa from 237Np. Once sufficiently pure, the spike solution can be diluted with 5% HNO3 to an appropriate volume and/or concentration for use as a working spike.
233Pa calibration
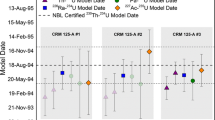
Currently, there is no 231Pa certified reference material that can be used to calibrate a 233Pa spike. Given this lack of a 231Pa reference material, laboratories have conventionally relied on using geologic 231Pa secular equilibrium materials, e.g., Table Mountain Latite (TML) and BCR-2 (USGS basalt standard), or, alternatively, 231Pa milked from highly enriched uranium, to calibrate 233Pa spike spikes. However, there are numerous complications in using geologic matrices to calibrate 233Pa spikes, including low and variable Pa recovery; incomplete dissolution of rock standards; and imprecise U assay of rock standards [9, 10]. Legacy 231Pa material that resided at LLNL has been purified and calibrated to use as an in-house standard solution for 233Pa spike calibration. Utilization of an in-house 231Pa standard solution to calibrate 233Pa spikes therefore represents a significant advance in 231Pa–235U radiochronometry. We note that while this 231Pa standard is not certified, it has been calibrated using a 233Pa spike that was itself calibrated using TML and BCR-2; therefore, the current 231Pa standard calibration is pinned to geologic secular equilibrium standards. Once calibrated, we have used this in-house 231Pa standard to calibrate a dozen spikes and compared this calibrant to rock matrix calibrations on the same 233Pa spikes. Results of this comparison demonstrate good correlation using multiple calibrants for 233Pa spikes (Fig. 3). Moreover, we have also demonstrated that use of the in-house 231Pa standard as the single calibrant for a 233Pa spike yields correct model ages for CRMs, including NBL U100, CRM 125A, and NBL U630. We have also calibrated our 233Pa spikes after they have decayed entirely to 233U to confirm original calibrations (see “Pa233 Spike calibration comparison: decayed spike (ingrown 233U) by IDMS” section). We are therefore confident in the current calibration and use of our in-house 231Pa standard for 231Pa–235U radiochronometry.
Pa spike calibration using rock standards
The procedure for 233Pa calibration using rock standards has been updated since publishing our methods in 2013 [9]. We sought to improve Pa recovery by adopting the first bulk matrix separation step described in [10]. Aliquots of fully dissolved TML and/or BCR-2 are weighed into Teflon vials and 233Pa spike was added to achieve 231Pa/233Pa ~ 0.5. Typically, multiple aliquots are made to facilitate a calibration check weeks after the initial calibration is made. Following bulk matrix separation with 4 M HF [10], samples are dried down with 20 µL HClO4. This dry down is followed by using a few drops of concentrated HNO3, and then a few drops of concentrated HCl. Repeated dry downs help to eliminate any fluorine that impedes Pa sorption on the following anion exchange column. Next, a 1.0-mL AG1X8 (BioRad, 100-200 mesh) anion exchange resin bed is prepared in a BioRad Poly-Prep column, then conditioned with 4 mL 9 M HCl. The Pa sample is dissolved in 1 mL 9 M HCl, trace (15 µL) concentrated HNO3, and trace (10 µL) saturated H3BO3; heated (capped) on a hotplate; cooled by sonication; and allowed to sit for 10 min. The sample is loaded on the conditioned anion resin bed and the sample vial rinsed with 0.5 mL and then 1 mL 9 M HCl. The column is rinsed with 2 mL 9 M HCl, with Pa remaining sorbed to the resin. The Pa is then eluted with 4 mL 9 M HCL + 0.05 M HF (note: U will be remain sorbed to the resin). The Pa elution is sequentially dried down with 20 µL HClO4, then concentrated HNO3. Now that the bulk of the rock matrix has been removed from the calibration fractions, the final silica gel purification step is described in the following section.
Pa spike calibration using 231Pa standard
The calibration technique using 231Pa is relatively straightforward and rapid. Multiple aliquots are prepared with the same amount of 231Pa and varying amount of prepared 233Pa spike; depending on the concentration of the 233Pa spike, the prepared aliquots target 231Pa/233Pa ranging from ~ 0.5 to 2. These same aliquots will be measured at a later date to confirm calibration. The mixtures are dried on a hotplate with 20 µL HClO4 and then concentrated HNO3. The sample is dissolved in 0.5 mL 5% HNO3 and trace (10 µL) saturated H3BO3; heated (capped) on a hotplate; cooled in a sonicator; and allowed sit for 15 min. A 1-mL silica gel resin bed is prepared in a BioRad column and conditioned with 4 mL 5% HNO3. The sample is loaded on the resin bed and the sample vial is rinsed with 0.5 mL and then 1 m 5% HNO3 followed by a column rinse using 4 mL 5% HNO3. Protactinium will sorb to the column, with ingrown 233U eluting during these load and rinse steps. Pa is then eluted using 4 mL 2% HNO3 + 0.05 M HF. Note the time and date of this final Pa from (ingrown) U purification for calculations. Mass spectrometry should be performed on these aliquots immediately following separation from ingrown U (see “U/Pa spike sensitivity” section below). Once the spike has been prepared and calibrated, it can be used to make IDMS measurements of 231Pa in unknown samples for approximately 3–6 months before nearly all 233Pa has decayed to 233U.
231Pa and 230Th assay by IDMS in U-bearing materials
Often age-dating efforts require that both radiochronometers, 230Th–234U and 231Pa–235U, are performed on the same material. As such, we have combined efforts to attain 230Th and 231Pa from a single aliquot, PaID + ThID (Fig. 2). The process for determining 230Th and 231Pa assay in U-bearing materials is described below. The first chemical separation step separates the bulk U from the ThID and PaID fractions. Each of those fractions is further processed for analysis by MC-ICP-MS.

Analytical flow diagram showing the path for U-bearing materials processing for 230Th and 231Pa assay
The following method assumes that a fully dissolved primary solution has been prepared. An important addition to this dissolved primary solution is HF. The presence of HF will keep the Th and Pa in solution and ideally inhibit these elements from adhering to the walls of the Teflon vial. First, a paired Pa isotope dilution (PaID) and Th isotope dilution (ThID) aliquot of primary solution is prepared and weighed so as to yield the desired amount of 231Pa and 230Th for measurement based on either a priori knowledge or an estimate about the sample, e.g., a certified reference material with a known purification age, U isotopic composition, or an unknown. Additional information, such as a pre-screening of the material by gamma or mass spectrometry may also be used to determine aliquot volumes. The process described here also assumes that the U assay has been determined by IDMS as a separate aliquot described in other publications [e.g., 1, 2, 3, 5, 8]. The desired amount of primary sample for 231Pa + 230Th assay is gravimetrically transferred to a cleaned, labeled Teflon vial, followed by gravimetric addition of the appropriate amounts of calibrated 229Th (or 232Th) spike and calibrated 233Pa spike so as to yield a sample/spike ratio of ~ 0.5–10. This mixture is then equilibrated by heating (capped) on a hotplate for several hours, followed by drying to completion with 20 µL HClO4 to ensure elimination of fluorine complexes. Chemical separations and purifications of ThID and PaID aliquots follow a series of chemistries described below.
The first step is to chemically separate bulk U from the desired 231Pa and 230Th. This is accomplished with a 1.0-mL AG1X8 (BioRad, 100-200 mesh) anion exchange resin bed in an Environmental Express® column. Thorium has been observed to sorb to the frit of BioRad Poly prep columns, whereas Environmental Express columns enable higher Th recovery, an important distinction for Th-limited samples. The dried sample is dissolved in 1 mL 9 M HCl + trace (25 µL) H3BO3 + trace (15 µL) concentrated HNO3, and the column conditioned with 3 mL 9 M HCl. A clean Teflon vial is placed to catch the ThID fraction, which will not sorb to the anion exchange resin in 9 M HCl. The vial is then rinsed with 0.5 mL and then 1 mL 9 M HCl, followed by a column rinse with 3 mL 9 M HCl. This ThID fraction is dried down, dried in concentrated HCl, and is ready for the second chemical purification of Th from U. The PaID fraction is then eluted into a cleaned Teflon vial with 3 mL 9 M HCl + 0.05 M HF and dried down with 30 µL HClO4. The dried PaID fraction is then dried with concentrated HNO3, followed by concentrated HCl. This fraction is ready for the second chemical separation of Pa from U matrix.
The next separation chemistry for the ThID fraction is performed using a 1-mL TEVA (Eichrom) resin bed. The ThID fraction is dissolved in 0.5 mL 4 M HNO3. The resin is conditioned with ultrapure 3 mL H2O followed by 4 mL 4 M HNO3. The sample is loaded on the resin bed, with both U and Th sorbing to TEVA resin. The vial is rinsed with 0.5 mL and then 1 mL 4 M HNO3, followed by a 4 mL 4 M HNO3 column rinse. A cleaned vial is placed to catch the Th elution, and the ThID fraction eluted, first, with 2 mL 9 M HCl, and then with 5 mL 0.1 M HCl + 0.005 HF. This elution is dried down, and then again with concentrated HCl. The last Th purification step for the ThID fraction is done using another 1-mL AG1x8 anion exchange resin (BioRad, 100-200 mesh) bed, conditioned with 3 mL 9 M HCl + 0.01 M HF. The sample is dissolved in 1 mL 9 M HCl + 0.01 M HF + trace (25 µL) concentrated HNO3. A new vial is placed to collect the sample load, and the vial and column rinsed with a total of 3.5 mL 9 M HCl + 0.01 HF. This aliquot is then dried down with concentrated HNO3 and dissolved in 2% HNO3 + 0.005 HF for mass spectrometry by MC-ICP-MS.
The next separation step for PaID involves further separating Pa from matrix elements. This second separation chemistry uses a 1-mL anion AG1x8 (BioRad, 100-200 mesh) resin bed. This second purification chemistry is the same chemistry as the first U matrix from ThID and PaID chemistry, omitting the elution of the ThID fraction. The PaID elution fraction is dried down with 30 µL HClO4 for the final silica gel resin purification. This final chemistry step is the silica gel chemistry that is described above in the “Pa spike calibration using 231Pa” section. The final elution in 2 M HNO3 + 0.05 M HF is ready for mass spectrometry by MC-ICP-MS. Empirical study in our lab has shown removal of > 99.8% of U matrix using silica gel resin procedure as described above.
Mass spectrometry methods
Both Pa and Th IDMS measurements were performed using a Nu Plasma HR MC-ICP-MS. Protactinium IDMS fractions were eluted in 2 M HNO3 + 0.05 M HF and Th samples dissolved in 2% HNO3 + 0.005 HF. Protactinium IDMS aliquots were analyzed using a one-cycle, static multicollection routine in which 231Pa and 233Pa are simultaneously measured on separate ion counters (ICs). Comparatively, thorium IDMS aliquots were analyzed using a two-cycle, dynamic multicollection routine in which 232Th is measured continuously on a Faraday collector and 229Th and 230Th are peak-hopped onto the same IC. Peak-hopping 229Th and 230Th with simultaneous 232Th Faraday measurement permits calculation of more precise 229Th/230Th by crossing 229Th/232Th (cycle #1) and 230Th/232Th (cycle #2), as uncertainties associated with Faraday/IC gain are obviated by this approach. Instrumental mass-dependent fractionation (mass bias) and Faraday/IC gain corrections for Pa and Th measurements were made using CRM U010 in the same analytical batch run by standard–sample–standard bracketing. Time-interpolation of mass bias and Faraday/IC gain factors for samples analyzed between bracketing CRM U010 measurements, in addition to a conservative expansion of uncertainties affiliated with interpolated correction factors, cumulatively yield more accurate and rigorous Pa and Th radiochronometric results. For complementary analytical details, see also [1, 2, 8, 9].
Pa233 Spike calibration comparison: decayed spike (ingrown 233U) by IDMS
To assess the relative accuracy and precision of various materials (including the 231Pa standard) used for calibrating 233Pa spikes at LLNL, we have measured decayed 233Pa spikes (as 233U) using a natural U assay standard (NBS 960). The half-life of 233Pa is 26.967 ± 0.004 days [14], which through beta decay creates 233U. Therefore, after approximately 6 months, 233Pa has near-fully decayed to 233U. To check the original calibration of our 233Pa spikes, we looked at previously produced spikes using this decayed spike IDMS method. Five independently prepared and calibrated 233Pa spikes are shown in Table 2 (Fig. 3).

233Pa spike prepared in April 2017 using multiple calibrants: TML, BCR-2, 231Pa, and 238U (decayed spike method)
Aliquots of decayed 233Pa spike were gravimetrically transferred to clean, dry Teflon vials. Aliquot sizes were determined to achieve 235U/233U of ~ 1. The same amount of natural U (NBS 960) of known concentration was added to each spike aliquot. When possible, multiple aliquots of each spike were analyzed. The decayed spike crosses were dried down, dried down again with concentrated HNO3, and dissolved in 2% HNO3 for analysis by MC-ICP-MS. The multicollection analysis method is the same as that used for measuring U by IDMS, where 238U, 235U, and 233U are all measured on Faraday collectors (234U is measured on an IC) (Table 1).
Figure 4 shows the relative difference between the decayed spike calibration (233U by IDMS) as compared with the original 233Pa assay determined by the calibrants: TML, BCR-2, and 231Pa. The difference is calculated as the percent difference between the original calibration as compared to the decayed spike calibration. The calculated uncertainty on the % difference is the combined uncertainty from both the decayed spike and original spike calibration. The uncertainty on the Faraday measurement of the 238U/233U ratio used to calculate the 233U assay is shown (Fig. 4) as a ± 0.25%, expanded uncertainty, k = 2.

Decayed 233Pa spike (measured as 233U) by IDMS using natural U (NBS 960)
U/Pa sensitivity
Another consideration for accurate PaID analysis is accounting for relative U/Pa sensitivity during mass spectrometry. Differing element-to-element instrumental sensitivity within an analytical session is a long-recognized and ubiquitous phenomenon. Elemental sensitivity tests demonstrate ~ 10% greater U sensitivity relative to concurrent Pa sensitivity on LLNL’s Nu Plasma HR MC-ICP-MS. This observation is crucial due to the relatively rapid ingrowth of isobaric 233U from decay of 233Pa spike (t1/2 ~ 27 days) in PaID aliquots between final silica gel purification chemistry and analysis by mass spectrometry. The magnitude of bias induced in 231Pa/233Pa increases with the time elapsed between PaID sample purification and analysis, resulting in increasingly-underestimated 231Pa/233Pa (sample/spike) due to the greater contribution of 233U at mass-233. Use of this low-biased 231Pa/233Pa for 231Pa–235U age-dating calculations will yield a model age that is too young (Fig. 5a), with the absolute magnitude of bias depending on (1) the time elapsed between chemistry and analysis, and (2) the absolute model age of the sample (Fig. 5b). It is therefore essential that mass spectrometry be performed as soon as PaID aliquots are purified to minimize the 233U isobar effect. Alternatively, U/Pa sensitivity would need to be determined empirically during the same batch run as sample analyses to make a “live” correction if significant time (> 2 days) passed between purification and mass spectrometry.

231Pa–235U model age bias effect from U233 ingrowth and contribution to mass-233 during PaID mass spectrometric analysis, assuming a relative U/Pa sensitivity of + 10%. a Relative (%) model age bias as a function of 233U ingrowth time (days). b Absolute (years) model age bias as a function of a sample’s true model age (years). Lines indicate a given 233U ingrowth time (i.e., time elapsed between final PaID purification and mass spectrometric analysis)
Results and discussion
Measured 230Th/234U and 231Pa/235U and corresponding expanded uncertainties (k = 2), and calculated model ages, model dates and expanded uncertainties (k = 2), for radiochronometry standards IRMM-1000 and CRM U100 are given in Table 2. Results for U100 230Th–234U and 231Pa–235U were processed as paired analyses as described above, where the Th and Pa assay originated from a single aliquot. Additionally, each radiochronometer aliquot has a unique U assay. IRMM-1000 results are a combination of analyses of REIMEP-22 from an interlaboratory exchange material [7] and an independently prepared reference unit of IRMM-1000, purchased from IRMM (Institute of Materials and Measurement) in 2015.
The 230Th–234U model dates calculated for CRM U100 span February–June 1958, with corresponding 231Pa–235U model dates ranging from October 1958–April 1959 (Fig. 6). The known production date of CRM U100 is January 8, 1959 [15]. Both radiochronometery daughter–parent pairs agree with one another within uncertainty, and are therefore concordant, in addition to preserving the material’s production date. This CRM can therefore act as a useful quality control standard when processing unknown U materials for age-dating as both chronometers yield similar results, suggesting near-complete purification of 230Th and 231Pa daughters during production, as well as being consistent with the production history of this material.

U100 results for paired ThID and PaID analyses, U100-1, U100-2, and U100-3. The horizontal line represents the number of aliquots of U100. Each pair of results has an individual U concentration by IDMS from the same aliquot. All uncertainties reported are expanded uncertainties, k = 2
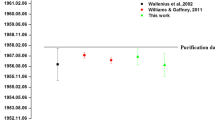
The reference material IRMM-1000 is a radiochronometry standard certified with a 230Th–234U production date of July 9th, 2012 ± 13 days [7]. The 230Th–234U model dates for this material range from late June–July 2012, with an average model date (n = 13) of July 2nd, 2012 ± 28 days, in agreement with the known production history of IRMM-1000. However, 231Pa–235U model dates range span April–early June 2012, with an average model date (n = 10) of April 26th, 2012 ± 56 days (Fig. 7). Long-term average IRMM-1000 Th–U and Pa–U model dates and uncertainties (k = 2) were calculated by propagating uncertainties from individual analyses. The average 231Pa–235U model date is therefore ~ 70 days older than the average 230Th–234U model date, with the two radiochronometers being clearly discordant at the k = 2 uncertainty level. This observable difference has been reproduced over time using two different IRMM-1000 stock solutions, one being from the REIMEP-22 interlaboratory exchange, as well as using two different 233U spikes, two different 229Th spikes, and six independently-prepared and -calibrated 233Pa spikes. As this material ages, the discordance becomes more difficult to observe because the absolute precision is a function of the age, thus older samples have larger absolute uncertainties on calculated model ages. This observed discordance between Th–U and Pa–U radiochronometers could be due to incomplete separation of Pa during the production of IRMM-1000 as a radiochronometry standard. Despite the observed model date discordance, we note that IRMM-1000 is still a valuable quality control material in nuclear forensics.

IRMM-1000 230Th–234U and 231Pa–235U model dates with expanded uncertainties shown relative to reference date. All analyses listed in Table 2 are in a. The certified 230Th–234U production date and combined uncertainty are in grey. The weighted average model dates for both radiochronometers are given in yellow (Th–U) and blue (Pa–U) (errors are stated in text below). Panel B inset shows our most recent data in finer detail as three separate paired ThID and PaID. U-assay for the paired analyses in b were determined using a newly prepared and calibrated in-house high purity 233U spike. All uncertainties reported are expanded uncertainties, k = 2. (Color figure online)
Conclusions
The 230Th–234U and 231Pa–235U model dates determined with our updated Pa–U age-dating methods for U100 are concordant and retain the production date of this material. By comparison, 230Th–234U and 231Pa–235U model ages for IRMM-1000 are discordant within uncertainties attainable using the improved Pa–U methodology. These methods can be applied to both relatively old (i.e., U100) and young (i.e., IRMM-1000) reference materials and unknowns and yield accurate results. Our improved methods are robust over time, using multiple distinct reference units, independently-prepared and -calibrated spikes, and newly available spikes, i.e., NFRM 229Th spike [16]. This study confirms the utility of calibrating 233Pa spikes with a 231Pa solution and the need for such a reference material for the age-dating community.
References
Gaffney AM, Hubert A, Kinman WS, Magara M, Okubo A, Pointurier F, Schorzman KS, Steiner RE, Williams RW (2016) Round-robin 230Th–234U age dating of bulk uranium for nuclear forensics. J Radioanal Nucl Chem 307:2055–2060
Kayzar TM, Williams RW (2015) Developing 226Ra and 227Ac age-dating techniques for nuclear forensics to gain insight from concordant and non-concordant radiochronometers. J Radioanal Chem 307:2061–2068
Mayer K, Wallenius M, Varga Z (2013) Nuclear forensic science: correlating measurable material parameters to the history of nuclear material. Chem Rev 113:884–900
Rolison JM, Treinen KC, McHugh KC, Gaffney AM, Williams RW (2017) Application of the 226Ra–230Th–234U and 227Ac–231Pa–235U radiochronometers to uranium certified reference materials. J Radioanal Nucl Chem 314:2459–2467
Treinen KC, Kinman WS, Yen C, Liuchao Z, Cardon AMR, Steiner RE, Kayzar-Boggs TM, Williams RW, Yong-Gang Z (2017) US-DOE and CIAE international cooperation in age-dating uranium standards. J Radioanal Nucl Chem 314:2469–2474
Varga Z, Wallenius M, Klaus M (2010) Age determination of uranium samples by inductively coupled plasma mass spectrometry using direct measurement and spectral deconvolution. J Anal Spectrom 25:1958–1962
Varga Z, Nicholl A, Wallenius M, Mayer K (2012) Development and validation of a methodology for uranium radiochronometry reference material preparation. Anal Chim Acta 718:25–31
Williams RW, Gaffney AM (2011) 230Th–234U model ages of some uranium standard reference materials. Radiochim Acta 1:31–35
Eppich GR, Williams RW, Gaffney AM, Schorzman KC (2013) 235U–231Pa age dating of uranium materials for nuclear forensic investigations. J Anal At Spectrom. https://doi.org/10.1039/c3ja50041a
Regelous M, Turner SP, Elliot TR, Rostami K, Hawkesworth CJ (2004) Measurement of femtogram quantities of protactinium in silicate rock samples by multicollector inductively coupled plasma mass spectrometry. Anal Chem 76:3584–3589
Cheng H, Edwards RL, Hoff J, Gallup CD, Richards DA, Asmerom Y (2000) The half-lives of uranium-234 and thorium-230. Chem Geol 169:17
Robert PJ, Miranda CF, Muxart R (1968) Mesure de la periode du protactinium 231 par microcalorimetrie. Radiochim Acta 11(2):104–108
Jaffey AH, Flynn KF, Glendenin LE, Bentley WC, Essling AM (1971) Precision measurement of half-lives and specific activities of 235U and 238U. Phys Rev C 4:1889–1906
Jones RT, Merritt JS, Okazaki A (1986) A measurement of the thermal neutron capture cross-section of 232Th. Nucl Sci Eng 93:171–180
Petit GS (1960) Preparation of uranium isotopic standards for the National Bureau of Standards. Union Carbide Nuclear Company, Oak Ridge, Tennessee. Oak Ridge National Laboratory, Report #KL-8, Department of Energy/K25 Archives
Essex RM, Mann JL, Williams RW, Kinman WS, Hubert A, Bennet ME, Gourgiotis A (2018) A new thorium-229 reference material. Appl Radiat Isotopes 134:23–31
Acknowledgements
This work was performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344. LLNL-CONF-749097.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Treinen, K.C., Gaffney, A.M., Rolison, J.M. et al. Improved protactinium spike calibration method applied to 231Pa–235U age-dating of certified reference materials for nuclear forensics. J Radioanal Nucl Chem 318, 209–219 (2018). https://doi.org/10.1007/s10967-018-6149-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-018-6149-x