
Abstract
Recent disruptions in the molybdenum-technetium generator supply chain prompted a review of non-reactor based production methods for both 99Mo and 99mTc. Small medical cyclotrons (E p ~ 16–24 MeV) are capable of producing Curie quantities of 99mTc from isotopically enriched 100Mo using the 100Mo(p,2n)99mTc reaction. Unlike most other metallic target materials for routine production of medical radioisotopes, molybdenum cannot be deposited by reductive electroplating from aqueous salt solutions. To overcome this issue, we developed a new process for solid molybdenum targets based on the electrophoretic deposition of fine 100Mo powder onto a tantalum plate, followed by high temperature sintering. The targets obtained were mechanically robust and thermally stable when irradiated with protons at high power density.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction and background
More than 80 % of all diagnostic nuclear medical procedures are based on radiopharmaceuticals labelled with the single photon emitter 99mTc (T 1/2 = 6.0 h, E γ = 141 keV). In clinical practice, 99mTc is eluted as pertechnetate from a radionuclide generator containing its parent isotope 99Mo (T 1/2 = 66.0 h). 99mTc-pertechnetate is then converted into radiopharmaceuticals by means of commercially available kits.
At present, the essential isotope 99Mo is exclusively produced in high flux reactors by fission of highly enriched uranium-235 targets. The 99Mo distributed universally is mainly generated in five nuclear facilities, the NRU reactor (Chalk River, Ontario, Canada; 40 %), HFR (Petten, The Netherlands; 30 %), BR-2 (Mol, Belgium; 12 %), Safari-1 (Palindaba, South Africa; 12 %) and OSIRIS (Saclay, France; 5 %). All of these reactors are between 45 and 55 years old. Proposals exist for a small number of newer plants to establish 99Mo production, such as the 20 MW Munich FRM-II reactor (commissioned in 2005) [1].
Starting in late 2007, several independent reactor outages significantly disrupted global 99Mo supplies. During the isotope crisis 2009–2010, triggered by an extended shutdown of the NRU reactor and a contemporaneous maintenance period at HFR, cancellations or delays in patient services ranged from 20 to 70 % of planned procedures.
In 2010, the Canadian Government announced its decision to discontinue support of the Chalk River reactor after 2016. At the same time, Natural Resources Canada (NRCan) funded a research initiative for a ‘non-reactor-based isotope supply contribution program’ (NISP) to investigate the feasibility of alternative approaches to the production of 99Mo and 99mTc.
TRIUMF’s Nuclear Medicine Division, in collaboration with partner institutions in British Columbia and Ontario, proposed the direct production of 99mTc instead of the 99Mo parent, using Canada’s existing medical cyclotrons. Local radiopharmacies would then deliver the purified 99mTc in major urban centres and to remote hospitals, similarly to the decentralised distribution of short-lived radiopharmaceuticals for Positron Emission Tomography, such as 18F-Fluorodeoxyglucose.
99mTc can be obtained on small cyclotrons by proton irradiation of isotopically enriched 100Mo targets via the 100Mo(p,2n)99mTc reaction. Although this method has been known since 1971 [2], it has never been practically applied to generate clinical quantities of 99mTc.
High yield production of 99mTc requires a 100Mo target capable of stopping protons in an energy range between approximately 20 and 10 MeV at beam currents of up to several hundred microamperes. 100Mo should preferably be applied in metal form to provide good thermal conductivity and mechanical stability.
Thick metallic targets for commercial scale radioisotope production are commonly prepared by reductive galvanic plating from aqueous salt solutions. Molybdenum cannot be electroplated in this manner because of its particular physical and chemical properties, which inhibit the formation of layers of more than ~10 μm thickness. Therefore a new procedure suitable for the manufacture of 100–500 μm thick deposits had to be developed.
We initially considered techniques such as plasma spraying [3], magnetron sputtering [4] as well as thermo-compression bonding of pressed and sintered molybdenum pellets [5]. Spraying and sputtering are well developed industrial processes but require comparatively large quantities of starting material, only a small fraction of which is actually deposited on the substrate. Most of the sprayed or sputtered material ends up on the walls of the deposition chamber, the fixtures that hold the substrates or in the vacuum system. While this is usually not an issue for many commonly used metals, such techniques are unsuitable for isotopically enriched materials due to their high cost and limited availability.
The pressed pellet approach, suggested by researchers at the University of Alberta, affords a very dense target that is rather difficult to dissolve after the irradiation, thus adding a long delay to the separation and purification process.
Eventually it was decided to proceed with a new multi-step process based on the electrophoretic deposition (EPD) of isotopically enriched molybdenum from an organic solvent, followed by high temperature sintering.
The EPD of metals and compounds was reported in 1962 [6]. Gutierrez et al. applied this versatile method to coat substrates with metals or compounds such as carbides. In summary, the process and setup is similar to a galvanic deposition, however, no chemical reaction takes place. The material to be deposited, in powder form, is suspended in a polar organic solvent and a small amount of protein binder is added. A voltage of several hundred volts DC is applied between the anode and the cathode while the suspension is stirred vigorously, allowing the powder particles to stay in suspension and to move between the electrodes. Within minutes, several hundred milligrams of powder can be deposited uniformly on the cathode. The thickness of the coating can be preselected by plating a suspension of known concentration to depletion. The deposition is considered complete when the liquid phase appears clear and transparent, with only a small number of particles visibly floating in the solvent. Figure 1 shows a schematic of the plating cell. In general, the wall material of the plating cell is not relevant as long as it does not react with or contaminate the organic solvent. In our case, a commercial polyethylene container of approximately 14 × 9 × 4.5 cm was used with good results.

Schematic of polyethylene plating cell for electrophoretic deposition (not to scale)
Materials and methods
Electrophoretic deposition
EPD yields smooth, homogeneous coatings if powders of small grain size (≤20 μm, ideally 1–6 μm) are used. Comminution of the starting material using a ball mill may be necessary prior to deposition; however, the use of a mechanical crushing device can introduce contaminants into the target material. EPD is remarkably affected by minute impurities in the powder and the solvent. For example, traces of water in the organic medium lead to undesired chemical reactions (e.g. oxidation of the suspended metal powder) that impede the deposition.
In our experience, isotopically enriched molybdenum powder (97.4–99.3 % 100Mo) obtained from suppliers (Trace Sciences International, Richmond Hill ON, Canada; Isoflex USA, San Francisco CA, USA) exhibited a highly non-uniform grain size distribution, which resulted in substantial variability in the target manufacturing process. To mitigate this risk, commercial 100Mo powders were oxidized by dissolution in hydrogen peroxide followed by reducing the dried trioxide back to molybdenum metal with hydrogen under carefully controlled conditions as described below (cf. ‘Recovery of enriched 100Mo’). Uniform deposits were achieved by using re-processed molybdenum powder of <10 μm grain size (Fig. 2).

SEM picture of pre-processed 100Mo powder
Particular attention had to be given to the selection and preparation of the substrates. In view of the eventual sintering of the targets, the backing material must be able to withstand temperatures in the order of 2,000 °C. It should have high thermal conductivity to facilitate cooling of the target plate during bombardment. In addition, it should show as little activation as possible when irradiated with 10 MeV protons. Tantalum was eventually chosen as it fulfils most of these requirements. In addition, tantalum is readily available commercially and can be machined by conventional methods.
In the scope of this project, we designed two different types of targets, a smaller circular one for irradiation on the GE PETtrace cyclotrons at the Lawson Health Research Institute and the Centre for Probe Development and Commercialisation (Fig. 3) and a large area target with cooling channels for the high current station on the ACSI TR-PET cyclotron at the British Columbia Cancer Agency (Fig. 4). Both types of target plates were manufactured from 1 mm thick tantalum sheet.

100Mo-targets for the GE PETtrace cyclotrons

100Mo Targets for the TR-PET cyclotron (front and back)
Before being subjected to EPD, the tantalum backings were prepared by roughing the surface to be deposited on, followed by degreasing, rinsing with deionised water and drying in air. EPD of ~600 and 1,000 mg 100Mo powder (for small and large area targets, respectively) was performed in dry nitroethane with addition of 1–2 ml of zein binder prepared as per [7]. Complete deposition was achieved in approximately 5–8 min at voltages between 450 and 1,500 VDC. The targets were removed from the plating bath and dried at ambient temperature.
Sintering
The plates obtained from the EPD process were placed in alumina boats and sintered in a tube furnace under UHP argon atmosphere. Optimum results were obtained by applying the following heating and cooling protocol. The temperature in the furnace was increased at a rate of 5 °C/min between room temperature and 1,300 °C, then at 2 °C/min between 1,300 and 1,700 °C. The furnace was held at 1,700 °C for 5 h and then allowed to cool to 1,300 °C at 2 °C/min. Between 1,300 °C and room temperature, the furnace temperature was decreased by 5 °C/min. The furnace was flushed with argon at 3 l/min during the entire cycle. A full sintering process took about 1 day.
Recovery of enriched 100Mo after separation of 99mTc
After separation of the desired 99mTc isotope using an automated purification module [8] and a decay period of 30 days, the fraction containing 100Mo-molybdate in aqueous sodium hydroxide was passed through a cation exchange column (Dowex 50W-X8, H+ form, 80 ml) to quantitatively remove sodium as well as trace cations from the solution. The slightly acidic eluate (pH ~ 6) contained various hydrates of molybdenum trioxide (‘molybdic acids’), which were isolated by evaporation to dryness. Upon further heating, the molybdic acids decomposed to water and 100MoO3.
100MoO3 was converted back to molybdenum metal in a two step reduction process similar to the methods described previously [9, 10] using a tube furnace and hydrogen gas. MoO3 was first reduced to MoO2 in an argon/hydrogen atmosphere (2 % H2, 3 l/min) at 700–800 °C (ramp rate 5 °C/min). After 30 min, the process gas was changed to 100 % H2 (3 l/min) and the temperature gradually increased at 5 °C/min to 1,100 °C and held for 30 min. The furnace was then allowed to cool down to room temperature at a rate of 5 °C/min.
Results and discussion
EPD proved to be an efficient, reliable and fast method for coating quantities of 600–1,200 mg of molybdenum powder onto tantalum backings. Losses of enriched isotope in the plating bath were negligible, typically in the order of a few milligrams, which could be recovered after evaporating the solvent. The deposits were uniform and adherent but not sufficiently dense for immediate irradiation.
Our sintering process for molybdenum follows standard industrial practices. Close control of process parameters was necessary to ensure reproducible results. The target plates manufactured according to this protocol were extremely robust. The molybdenum layer was scratch resistant and firmly bonded to the tantalum backing. A sinter density of ~70 % of the bulk material could be achieved. The sintered layer retained sufficient porosity to facilitate the dissolution of the molybdenum after irradiation, which reduced the time required for the separation and purification of the 99mTc considerably. Typically, a target thus produced could be completely dissolved in hydrogen peroxide in less than ten minutes.
We discovered that sintering in this temperature range also significantly increased both tensile strength and hardness of the tantalum backing. Hence the thickness of the backing plate could be further reduced, which consequently improved the heat transfer from the target to the cooling water during irradiation.
Targets were irradiated at the GE PETtrace cyclotron at the Lawson Health Research Institute with 16.5 MeV protons and beam currents of up to 80 μA (power density 1.1 kW/cm2). Visual inspection after irradiation proved that all targets were perfectly intact—data from irradiations of the large area targets at the BC Cancer Agency’s TR-PET cyclotron are not yet available.
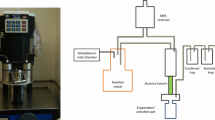
99mTc was separated from the molybdenum matrix and used successfully in labelling compounds such as 99mTc sestamibi (Cardiolite®), 99mTc exametazime (Ceretec®) and 99mTc-medronate (Draximage®). The formulated radiopharmaceuticals were analysed and found to meet or exceed USP purity requirements.
Recovery of the 100Mo metal was performed with >90 % efficiency. Gamma spectroscopy of the recovered powder revealed small traces of 95Nb (T 1/2 = 35 days), a contaminant produced from 98Mo impurities in the starting material via 98Mo(p,α)95Nb. While the actual quantities do not constitute a radiological hazard, repeated processing of the same powder would eventually lead to an accumulation of 95Nb in the target material. It is planned to modify the recovery procedure such that this contaminant is removed quantitatively before the eluate from the Dowex column is evaporated to dryness.
Conclusions
Our results demonstrate the feasibility of electrophoretic deposition, followed by high temperature sintering, as a reliable method for the manufacture of 100Mo targets for the direct cyclotron production of 99mTc. Furthermore, in the course of the NISP project, we established a complete production cycle including target preparation, proton irradiation at high power densities, rapid separation of high purity 99mTc, and efficient recovery of the enriched target material.
Future work will focus on process optimisation as well as establishing the cyclotron production of 99mTc at a large number of medical centres in Canada.
References
Technical University of Munich: Production of molybdenum-99 for medicine at the neutron source Heinz Maier-Leibnitz (FRM-II). http://cdn.frm2.tum.de/fileadmin/stuff/industry/Documents/moly99-flyer_en.pdf. Accessed 30 Oct 2012
Beaver JE, Hupf HB (1971) J Nucl Med 12:739–741
ASM International (1996) ASM Handbook vol. 5 surface engineering. ASM International, Materials Park, pp 944–973
Kelly PJ, Arnell RD (2000) Vacuum 56:159–172
Wilson J, Gagnon K, McQuarrie S (2012) Production of technetium from a molybdenum metal target. Patent Application WO2012139220
Gutierrez CP, Mosley JR, Wallace TC (1962) J Electrochem Soc 109:923–927
Reichard HF, Scheible, HG, Kibrick M, Katz H (1956) Report KLX-10029, Vitro Laboratories, West Orange NJ, May 1956
Morley TJ, Dodd M, Gagnon K et al (2012) Nucl Med Biol 39:551–559
Tuominen SM (1981) Powder Technol 30:73–76
Meyer HW, Baker JD, Ceckler WH (1977) US Patent 4,045,216
Acknowledgments
This project was funded by Natural Resources Canada under NRCan NISP.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hanemaayer, V., Benard, F., Buckley, K.R. et al. Solid targets for 99mTc production on medical cyclotrons. J Radioanal Nucl Chem 299, 1007–1011 (2014). https://doi.org/10.1007/s10967-013-2626-4
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-013-2626-4