
Abstract
Developing new bone pain palliation agents are a mandate in handling end-stage cancer patient’s around the world. 177Lu (1-hydroxy-2-imidazol-1-yl-phosphonoethyl)phosphonic acid (177Lu–ZLD) is a possible therapeutic agent which can be used in bone palliation therapy. In this study, 177Lu–ZLD complex was prepared successfully using commercial ZLD ligand and 177LuCl3 at 25 and 60 °C at various ligand:metal ratios for 60–360 min. 177Lu chloride was obtained by thermal neutron irradiation (4 × 1013 n cm−2s−1) of natural Lu2O3 samples. Radiochemical purity of 177Lu–ZLD was checked by ITLC and HPLC. Stability studies of final preparation in the presence of human serum were performed as well as protein binding studies and hydroxyapatite (HA) binding test. The biodistribution of 177Lu–ZLD and 177LuCl3 in mice were determined for 7 days. A comparative accumulation study for 177Lu–ZLD and 177Lu–EDTMP was performed for vital organs up to 7 days. The complex was obtained in high radiochemical purity ITLC (>97 %) and HPLC (>99.9 %) and satisfactory stability in presence of human serum and final formulations were obtained (≈90 % in 48 h). HA binding assay demonstrated >95 % binding from 5 to 20 mg of HA in 24 h at room temperature. The complex protein binding was about 55–58 %. The high bone uptake ratios at all time intervals was obtained (>9 % at day 7), bone:kidney and bone:liver uptake ratios were significantly high for ZLD at 7 day post injection but not superior to 177Lu–EDTMP. Due to longer physical half life of 177Lu compared to 153Sm and comparable ratios for 177Lu–ZLD compared to 177Lu–EDTMP, 177Lu–ZLD can be an interesting new candidate for clinical trials for bone pain palliation therapy.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Bone metastases are common in the progression of various tumors such as prostate, breast and lung carcinoma and they often entail an occurrence of progressive pain [1] and occur in many patients with solid malignant tumors [2]. Approximately 50 % of patients with breast carcinoma and 80 % of patients with prostate carcinoma develop metastatic bone diseases and nearly half of them experience bone pain [3]. In these patients who have progressive disease despite treatment, a systemic bone avid radiopharmaceutical for treatment of widespread bone metastases has potential benefits [4].
Lu-labeled ethylenediamine-N,N,N′,N′-tetrakis methylene phosphonic acid (177Lu–EDTMP), has been used to palliate metastatic bone pain as a new bone-seeking radiopharmaceutical in a phase II study, and has been assessed for its efficacy and safety for bone pain palliation in patients with breast cancer and hormone refractory prostate cancer with bone metastases.
Multidentate polyaminopolyphosphonic acid ligands are known to form stable chelates with many metals including lanthanides. (1-Hydroxy-2-imidazol-1-yl-phosphonoethyl)phosphonic acid; zoledronic acid (ZLD) can be envisaged as a possible carrier moiety, for the development of beta emitter-based radiopharmaceutical for bone palliation.
Considering the inhibitory binding affinity constant (Ki) of bisphosphonates used in clinics including etidronate (Ki: 91 μM), ibandronate (116 μM), pamidronate (83 μM), risedronate (85 μM) and zoledronate (81 μM), the zoledronate demonstrates the highest affinity for the osteoclast [5] as resulting in the highest possible bone affinity and when labeled with therapeutic radionuclides can be a potential compound for bone pain palliation therapy.
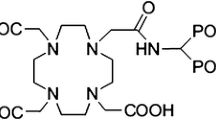
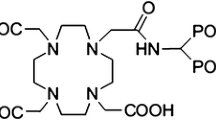
Zoledronic acid, a bisphosphonic acid which is an inhibitor of osteoclastic bone resorption, designated chemically as (1-hydroxy-2-imidazol-1-yl-phosphonoethyl)phosphonic acid monohydrate. The principal pharmacologic action of zoledronic acid (ZLD) is inhibition of bone resorption. Although the antiresorptive mechanism is not completely understood, several factors are thought to contribute to this action. The bone affinity of zoledronic acid among series of bisphosphonates is significantly high (Fig. 1).

Chemical structure for zoledronic acid monohydrate
Interesting characteristics ZLD suggest an ideal ligand for radiopharmaceutical development. For instance, ZLD does not undergo biotransformation in vivo. In animal studies, <3 % of the administered intravenous dose was found in the feces, with the balance either recovered in the urine or taken up by bone, indicating that the drug is eliminated intact via the kidney. Following an intravenous dose of 20 nCi 14C-zoledronic acid in a patient with cancer and bone metastases, only a single radioactive species with chromatographic properties identical to those of parent drug was recovered in urine, which suggests that zoledronic acid is not metabolized [6].
The drug and its analogs has been radiolabeled with technetium-99m for bone imaging [7] as well as labelling with 177Lu and 47Sc without reporting the evaluation and the biological data [8]. Also as mentioned 14C-labeled zoledronic acid has been used in the basic research studies for the drug binding as well as metabolic studies [9].
177Lu-radiopharmaceuticals have been developed and used in the bone palliation therapy [10]. However liver uptake is still a major problem in most of therapeutic bisphosphonate ligands and the search for an ultimate ligand with the highest binding activity to the bones with the least possible dissociation in the biological environment is still needed.
Owing to 177Lu suitable decay characteristics [t 1/2 = 6.73 days, Eβmax = 497 keV, Eγ = 112 keV (6.4 %), 208 keV (11 %)] as well as the feasibility of large-scale production in adequate specific activity and radionuclidic purity using a moderate flux reactor, 177Lu seems a suitable radionuclide in the development of a Lu–ZLD complex as a possible therapeutic radiopharmaceutical .
In this research, 177Lu (1-hydroxy-2-imidazol-1-yl-phosphonoethyl)phosphonic acid (177Lu–ZLD) complex was prepared followed by preclinical evaluation including in vitro/vivo stability, biodistribution studies by post-mortem studies.
Experimental
177Lu was produced with a specific activity of approximately 70–80 mCi mg−1 and radionuclidic purity of 99.98 % by irradiation of natural Lu2O3 targeted at a thermal neutron flux of approximately 4 × 1013 n cm−2 s−1 for 5 days at the Tehran Research Reactor (TRR). Whatman No. 3 paper was obtained from Whatman (UK). Zoledronic acid and sodium zoledronate were purchased from Sigma-Aldrich Co., UK. Radio-chromatography was performed by Whatman paper using a thin layer chromatography scanner, Bioscan AR2000, Paris, France. Analytical HPLC to determine the specific activity was performed by a Shimadzu LC-10AT, armed with two detector systems, flow scintillation analyzer (Packard-150 TR) and UV–Visible (Shimadzu) using Whatman Partisphere C-18 column 250 × 4.6 mm (Whatman Co., NJ, USA). A high purity germanium (HPGe) detector coupled with a Canberra™ (model GC1020-7500SL, Canberra Industries, Inc., CT, USA.) multichannel analyzer and a dose calibrator ISOMED 1010 (Elimpex-Medizintechnik, Austria) were used for counting distributed activity in mice organs. All other chemical reagents were purchased from Merck (Germany). Calculations were based on the 112 keV peak for 177Lu. All values were expressed as mean ± standard deviation and the data were compared using Student’s t test. Animal studies were performed in accordance with the United Kingdom Biological Council’s Guidelines on the Use of Living Animals in Scientific Investigations. 1987. The approval of NSTRI Ethical Committee was obtained for conducting this research. The wild-type mice were purchased from Pasteur Institute of Iran, Karaj, all weighing 20–25 g and were acclimatized at proper rodent diet and 12 h/12 h day/night light/darkness.
Production and quality control of 177LuCl3 solution
Lutetium-177 was produced by neutron irradiation of 1 mg of natural Lu2O3 (99.999 % from Aldrich Co., UK) according to reported procedures [11] at TRR, General Atomics, USA. The irradiated target was dissolved in 200 μL of 1.0 mol L−1 M HCl, to prepare 177LuCl3 and diluted to the appropriate volume with ultra pure water, to produce a stock solution of final volume of 5 mL (0.04 mol L−1). The mixture was filtered through a 0.22 μm filter for sterilization, Waters, USA. The radionuclidic purity of the solution was tested for the presence of other radionuclides using purity germanium (HPGe) spectroscopy for the detection of various interfering gamma emitting radionuclides. The radiochemical purity of the 177LuCl3 was checked using 2-solvent systems for instant thin layer chromatography (ITLC) [A: 10 mmol L−1 diethylene triamine pentaacetic acid (DTPA) pH 5 and B: 10 % ammonium acetate:methanol (1:1)].
Radiolabeling of ZLD with 177LuCl3
A stock solution of sodium zoledronate (MW. 334) was prepared by dissolution in double distilled ultra pure water, to produce a solution of 50 mg mL−1. For labeling, an appropriate amount of the 177LuCl3 solution containing the required activity (0.1 mL, 5 mCi) was added to the desired amount of NaZLD solution (0.3 mL, 1:5; 1:10; 1:15; 1:20; 1:40 and 1:50 ratios for Lu:ZLD). The complex solutions were kept at room temperature for 60–360 min. Also another set of experiment was performed at 60 °C warm bath for 60–360 min. The radiochemical purity was determined using ITLC and HPLC. The final solution was passed through a 0.22-μm membrane filter and pH was adjusted to 7–8.5 with 0.05 mol L−1 phosphate buffer (pH 5.5).
Radiochemical purity of 177Lu–ZLD
Instant thin layer chromatography
A 5 μL sample of the final fraction was spotted on a chromatography Whatman No. 3, paper, and developed in Whatman 3 MM chromatography paper or ITLC-SG eluted with NH4OH (56 %):MeOH (100 %):H2O (100 %) (0.2:2:4; v/v/v) as mobile phase mixture.
High performance liquid chromatography
HPLC was performed with a flow rate of 1 mL/min, pressure: 130 kgF/cm2 for 20 min. HPLC was performed on the final preparation using a mixture of water:acetonitrile 3:2(v/v) as the eluent by means of reversed phase column Whatman Partisphere C18 4.6 × 250 mm.
Sterility and apyrogenicity of the radiopharmaceutical
Sterility was controlled on a random sampling following decay of radioactivity. The limulus amoebocyte lysate (LAL) test was used for validation of radiopharmaceutical production according to the European protocol [12].
Stability of 177Lu–ZLD in final formulation
Stability of 177Lu–ZLD in final preparation was determined by storing the final solution at 25 °C for up to 48 h and performing frequent ITLC analysis to determine radiochemical purity using Whatman 3 MM chromatography paper or ITLC-SG eluted with NH4OH (56 %):MeOH (100 %):H2O (100 %) (0.2:2:4; v/v/v).
In vitro protein binding of 177Lu–ZLD in presence of human serum
In vitro protein binding of 177Lu–ZLD was carried out in human blood by protein precipitation according to published procedure [13]. To 3 mL fresh human plasma, 1 mL of the labeled complex was mixed and incubated for 1 h at 37 °C. Contents of the tube were centrifuged at 3,000 rpm for 10 min for separation of serum and blood cells. After mixing approximately equal volume of 10 % trichloroacetic acid (TCA), the mixture was centrifuged at 3,000 rpm for 10 min. Residue was separated from supernatant and both layers were counted for radioactivity in a well type gamma counter. Protein binding of the complex was expressed as the fraction of radioactivity bound to protein, in percentage of the total radioactivity.
In vitro stability of 177Lu–ZLD in presence of human serum
Final solution (200 μCi, 50 μL) was incubated in the presence of freshly prepared human serum (300 μL) (purchased from Iranian Blood Transfusion Organization, Tehran) and kept at 37 °C for 2 days. Every 30 min to a portion of the mixture (50 μL), TCA (10 %, 100 μL) was added and the mixture was centrifuged at 3,000 rpm for 5 min followed by decanting the supernatant from the debris. The stability was determined by performing frequent ITLC analysis of supernatant using above mentioned ITLC system.
Hydroxyapatite binding assay
The hydroxyapatite binding assay was performed according to the procedure described previously [14], with only a slight modification. In brief, to vials containing 1.0, 2.0, 5.0, 10.0, 20.0 and 50.0 mg of solid hydroxyapatite, 2 mL of saline solution of pH 7.4 were added and the mixtures were shaken for 1 h. Then, 50 mL of the radioactive preparation was added and the mixtures were shaken for 24 h at room temperature. The suspensions were centrifuged, and two aliquots of the supernatant liquid were taken from each vial and the radioactivity was measured with a well-type counter. Test experiments were performed using a similar procedure, but in the absence of hydroxyapatite. The percentage binding of 177Lu to hydroxyapatite (HA) was calculated according to HB = 1 − A/B × 100, where A is the mean radioactivity value of the supernatant sample under study and B is the mean total value of whole activity used.
Biodistribution studies
The biodistribution of free Lu3+ cation as well as of 177Lu–ZLD was determined in wild type mice. For each radiochemical compound, 100 μL (150 μCi) of radioactive solution was injected directly to normal mice through caudal vein. The animals (n = 3) were sacrificed at selected times after injection (2 h to 7 days) and percentage of injected dose in the tissues was determined with a γ-ray scintillation or a dose calibrator.
Discussion
Radionuclide production
The radionuclide was prepared at a range of specific activity of 3–5 MBq mg−1 for radiolabeling use, after counting the samples on an HPGe detector for 5 h, two major photons (6.4 % of 0.112 MeV and 11 % of 0.208 MeV) were observed.
The radiochemical purity of the 177Lu solution was checked in two solvents. In 10 mmol L−1 DTPA aq. solution (solvent 1), free Lu3+ is coordinated to more lipophilic moiety as Lu(DTPA)2− and migrates to higher Rf. Small radioactive fraction remains at the origin could be associated to colloids, since in presence of very strong complexing agent (i.e. DTPA), existence of other ionic species than Lu(DTPA)2− is impossible. On the other hand, 10 % ammonium acetate:methanol mixture (1:1) (solvent 2) was also used for the determination of radiochemical purity. The fast eluting species was possibly Lu3+ and other ionic forms of 177Lu such as LuCl4 − remained at the origin (Rf. 0) as well as colloids (Fig. 2). The differences in the impurity peaks in the two chromatograms could be in part related to the presence of a colloidal impurity which was insignificant. Also insignificant (about <1 %) amount of activity can be attributed to other ionic impurities.

ITLC chromatograms of 177LuCl3 solution in a DTPA solution (pH 5) (a), 10 % ammonium acetate:methanol (1:1; v/v) solution (b)
Radiolabeling
ITLC studies approved the production of a single radiolabeled compound, at the Rf. 0.8 while Lu3+ retains to the lower Rf due to the polarity (Fig. 3). The HPLC studies also demonstrated the existence of only one radiolabeled species using both UV and scintillation detectors.

ITLC radiochromatogram for 177Lu–ZLD eluted using NH4OH:MeOH:H2O (0.2:2:4) using Whatman 3 MM
A more fast-eluting compound at 3.06 min (scintillation detector) related to 3.22 min peak (UV detector 425 nm, not shown) demonstrated a hydrophilic species compared to lutetium cation. A single radiolabeled species was eluted at 10 min from the column which contained more than 99 % of the area under curve related to cold Lu–ZLD control peak (Figs. 4, 5). The effect of various factors on the labeling yield of 177Lu–ZLD including ligand concentration and temperature were also studied. The results are shown in Table 1.

HPLC chromatograms of 177LuCl3 on a reversed phase column using acetonitrile:water 40:60, up; using scintillation detector

HPLC chromatograms of 177Lu–ZLD (down) on a reversed phase column using acetonitrile:water 40:60, up; using scintillation detector
Labeling yield increased with increasing molar ratio Lu:ZLD (from 1:5 to 1:50) and reached more than 97 % in 60 min (Table 1). The stability of prepared 177Lu–ZLD complex was checked up to 48 h after preparation. The complex was stable in final pharmaceutical sample and its radiochemical purity was above 97 % even 48 h after preparation using Whatman 3 MM eluted with NH4OH:MeOH:H2O (0.2:2:4).
Stability test was developed for the complex in presence of human serum at 37 °C using ITLC as mentioned above and all data within 48 were above 89 % at all time intervals.
For determination of protein binding the data showed the 57 % protein binding using ITLC of the serum-radiopharmaceutical mixture, while 43 % is found in free form in the circulation. The protein binding for the ZLD ligand has been reported in different references from 60 to 77 % in free form [15], however no protein biding for metal ZLD complexes were available in the literature.
HA assay demonstrated high capacity binding for Lu–ZLD to hydroxyl apatite. Even at 2–3 mg amount of HA, more than 90 % binding was observed, while at 5 mg HA used >97 % of binding was obtained (Fig. 6).

Hydroxy apatite binding assay for 177Lu–ZLD at 37 °C in 24 h
Regarding the animal studies, the tissue uptakes were calculated as the percent of area under the curve of the related photo peak per gram of tissue (% ID/g) (Fig. 7). The biodistribution of 177Lu cation was determined in wild-type animals for 2 h to 7 days post injection. The liver radioactivity uptake of the cation is comparable to other radio-lanthanides such as Yb, Sm and Tb [16]. About 3 % of the cation radioactivity accumulates in the liver in 48 h. Low blood radioactivity content demonstrates the rapid removal of 177Lu from the circulation after injection. Lung, muscle and skin do not demonstrate significant 177Lu uptake while it is in accordance with other cations accumulation. A 4 % bone uptake is observed for 177Lu which remains almost constant after 7 h (5 %). Spleen also has significant 177Lu uptake possibly related to reticuloendothelial system.

Percentage of injected dose per gram of 177LuCl3 in wild type mice 2 h-7 days post injection (n = 3)
The radioactivity biodistribution of 177Lu–ZLD (200 μCi in 150 μL volume) in mice organs up to 7 days post injection was determined and it is clearly shown that the major portion of injected radioactivity of 177Lu–ZLD was transferred from blood circulation into bones (Fig. 8). After 7 days post injection, medium activity uptake was observed for soft tissues such as heart, lung, liver and stomach. The significant excretion of the radioactivity is observed for kidney as anticipated due to the major ZLD non-metabolized excretion through the kidneys already reported.

Percentage of injected dose per gram of 177Lu–ZLD in wild-type mice tissues at 2 h-7 days post injection (n = 3)
It can be suggested to use a mild diuretic agent would remove the un concentrated portion of the radiopharmaceutical into bones from the circulation through urinary tract, possibly enhance the bone:non target ratio for the therapeutic radiopharmaceutical leading to un-wanted irradiation doses to the patients (esp. gonads). The liver however plays insignificant role in metabolism (around 3 %) and also lower GI (intestines, colon) uptake is observed.
The target:non-target ratio of 177Lu–ZLD was determined in order to demonstrate the targeting property of radiopharmaceutical 2 h to 7 days post injection as shown in Table 2. As shown in the table, 7 days post injection (almost one physical half life) the best targeting can be observed regarding blood, kidney and liver. By plotting the ratio data against time an almost increasing trend can be observed especially in the case of liver and blood (Fig. 9).

Comparative target:non target ratios for 177Lu–ZLD for 7 days in mice
As a comparable 177Lu bone pain palliation agent, 177Lu–EDTMP has been proposed as a potential clinically used radiopharmaceutical and clinical trials have already been started according to the published data [17, 18]. A 3 % liver uptake has been reported for 177Lu–EDTMP which is considered a disadvantage as a bone pain palliative agent leading to unwanted liver dose in patients (Fig. 10).

Percentage of injected dose per gram of 177Lu–EDTMP in wild-type mice tissues at 2, 4, 24, 48 h and 7 days post injection [18]
Also, the target non-target ratio of 177Lu–EDTMP was determined in order to demonstrate the targeting property of radiopharmaceutical 2 h to 7 days post injection as shown in Table 3. As shown in the table, during 7 days the best targeting can be observed just for blood, while for the kidney and the liver (Fig. 11).

Comparative target:non target ratios for 177Lu–EDTMP for 7 days in mice
Conclusion
177Lu–ZLD was prepared (radiochemical purity >99 %) using optimization studies. 177Lu–ZLD and 177LuCl3 preparations were administered intravenously through the tail vein to wild-type rats and biodistribution data was checked 2 h to 7 days later showing at least 65 % accumulation of the drug in the bone tissues. A comparative accumulation study for the complex was obtained in high radiochemical purity ITLC (>97 %) and HPLC (>99.9 %) and satisfactory stability in presence of human serum and final formulations were obtained. HA binding assay demonstrated >95 % binding from 5 to 20 mg of HA in 24 h at room temperature. The complex protein binding was about 55–58 %. The high bone uptake ratios at all time intervals was in accordance with the HA test. Bone:kidney and bone:liver uptake ratios were significantly high for ZLD at 7 days post injection but not superior to 177Lu–EDTMP. 177Lu–ZLD can be an interesting new candidate for clinical trials for bone pain palliation therapy.
References
Serafini AN (2001) Therapy of metastatic bone pain. J Nucl Med 42:895–906
Bayouth JE, Macey DJ, Kasi LP, Fossella FV (1994) Dosimetry and toxicity of Samarium-153–EDTMP administered for bone pain to skeletal metastases. J Nucl Med 35:63–69
Campa JA, Rayne R (1992) The management of intractable bone pain: a clinician’s perspective. Semin Nucl Med 22:3–10
Eary JF, Collin C, Stabin M, Vernon C, Petersdorf S, Baker M, Hartnett S, Ferency S, Addison SJ, Appelbaum F (1993) Samarium-153–EDTMP biodistribution and dosimetry estimation. J Nucl Med 34:1031–1036
Luegmayr C-TE, Freedman LP, Guideon A, Rodan L (2006) Relative binding affinities of bisphosphonates for human bone and relationship to antiresorptive efficacy. Bone 38(5):628–636
http://www.theodora.com/drugs/zometa_for_intravenous_infusion_novartis.html
Lin J, Qiu L, Cheng W, Luo S, Xue L, Zhang S (2012) Development of superior bone scintigraphic agent from a series of 99mTc-labeled zoledronic acid derivatives. Appl Radiat Isot 70(5):848–855
Majkowska A, Neves M, Ines Antunes I, Aleksander Bilewicz A (2009) Complexes of low energy beta emitters 47Sc and 177Lu with zoledronic acid for bone pain therapy. Appl Radiat Isot 67:11–13
McKenzie K, Eng M, Bobyn JD, Roberts J, Karabasz D, Tanzer M (2011) Bisphosphonate remains highly localized after elution from porous implants. Clin Orthop Relat Res 469:514–522
Chakraborty S, Das T, Banerjee S, Balogh L, Chaudhari PR, Sarma HD, Polyák A, Máthé D, Venkatesh M, Janoki G, Pillai MR (2008) 177Lu–EDTMP: a viable bone pain palliative in skeletal metastasis. Cancer Biother Radiopharm 23:202–213
Manual for reactor produced radioisotopes, International Atomic Energy Agency (IAEA), Vienna, 2003, IAEA-TECDOC-1340, ISBN 92–0–101103–2, ISSN 1011–4289, pp 121–123, Printed by the IAEA in Austria, January 2003
cGRPP-guidelines (2007) EANM Radiopharmacy Committee, guidelines on current good radiopharmacy, practice (CGRPP) in the preparation of radiopharmaceuticals, version 2 March 2007
Dar UK, Khan IU, Javed M, Ahmad F, Ali M, Hyder SW (2012) Preparation and biodistribution in mice of a new radiopharmaceutical technetium-99m labeled methotrexate, as a tumor diagnostic agent. Hell J Nucl Med 15(2):120–124
Neves M, Gano L, Pereira N, Costa MC, Costa MR, Chandia M, Rosado M, Fausto R (2002) Synthesis, characterization and biodistribution of bisphosphonates Sm-153 complexes: correlation with molecular modeling interaction studies. Nucl Med Biol 29:329–338
Du XL, Zhang TL, Yuan L, Zhao YY, Li RC, Wang K, Yan SC, Zhang L, Sun H, Qian ZM (2002) Complexation of ytterbium to human transferrin and its uptake by K562 cells. Eur J Biochem 269:6082–6090
Yousefnia H, Anvari A, Bahrami-Samani A, Jalilian AR, Shirvani-Arani S, Aghamiri MR, Ghannadi Maragheh M (2012) Production, quality control and pharmacokinetic studies of 177Lu–EDTMP for human bone pain palliation therapy trials. Iran J Pharmaceut Res 11(1):137–144
Yuan J, Liu C, Liu X, Wang Y, Kuai D, Zhang G, Zaknun JJ (2013) Efficacy and safety of 177Lu–EDTMP in bone metastatic pain palliation in breast cancer and hormone refractory prostate cancer: a phase II study. Clin Nucl Med 38(2):88–92
Acknowledgments
We acknowledge the financial support of Iran National Science Foundation (INSF) for conducting this research project. The authors wish to thank Mr. M. Mazidi for performing animal tests as well as Dr. M. Erfani for HPLC experiments.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nikzad, M., Jalilian, A.R., Shirvani-Arani, S. et al. Production, quality control and pharmacokinetic studies of 177Lu–zoledronate for bone pain palliation therapy. J Radioanal Nucl Chem 298, 1273–1281 (2013). https://doi.org/10.1007/s10967-013-2490-2
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-013-2490-2