
Abstract
Proficiency testing is one of methods for regularly assessing the accuracy of the analytical data produced by laboratories for particular measurements. In 2008 and 2010, we participated in the IAEA 2008 and 2010 worldwide open proficiency tests on the determination of natural radionuclides in water spiked with 226Ra, 234U and 238U for activity analysis and with 90Sr and 230Th for gross alpha/beta analysis. Feedback statistics from the IAEA final report showed that the radioactivities of all of the samples fell within an acceptable range according to the IAEA. For 226Ra analysis, our result showed that 229Th–225Ra is suitable as a chemical tracer, although there are doubts that different co-precipitation efficient between parent 229Th and its daughter nuclide 225Ra in published literature. The impact factors of the analysis results, such as the lower limit of detection, standard substances, the background and efficiency for daily determination, are discussed in detail.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
A proficiency test (PT) is one of the series of activities organised every year by the IAEA terrestrial environment laboratory, and testing is one of the important ways for evaluating the accuracy of daily analytical procedures and final results from research laboratories. The accuracy of analytical data plays an important role not only in our research activities but also in our daily lives in fields, such as environmental protection, international trade, law enforcement and consumer health. In addition, measurements performed by laboratories located around the world should yield traceable and comparable results [1–3]. Therefore, frequent participation in international proficiency tests can identify the analytical problems of laboratories.
Based on IAEA experience with open worldwide laboratory intercomparison studies, a modified u-score evaluation and z-scores are applied in determining a laboratory’s performance, in which the trueness and precision of participants’ results are evaluated separately. In this approach, both accuracy and precision have to be ‘acceptable’ for the final results to also be acceptable. Otherwise, the reported results are ‘not acceptable’. Such an estimation approach for each set of reported results can identify not only methodological problems affecting accuracy but also shortcomings in uncertainty estimation [1, 4].
According to IAEA’s evaluation report [1], the initial step in evaluating a score for the reported data is estimating the bias of each laboratory. The relative bias (RB%) between the reported value (each laboratory, A lab) and the given value by IAEA (A ref) can be calculated by
The ‘trueness’ of the reported results will be determined to be ‘acceptable’ if the values of activities in final report can be as
A 1 as
and A 2 as
where A ref and A lab are the activities of each nuclide given by the IAEA (traceable from NIST) and lab analysis, respectively. Uncref and Unclab are concerned with uncertain error.
Participants’ results are scored as ‘acceptable’ for precision when (P < LAP) or (P = LAP). For evaluation of precision, estimator P is calculated for each participant with the following formula:
P is directly obtained from the reported measurement uncertainty by each laboratory. Based on the concentration or activity level of each nuclide and the complexity of the analytical problem, the acceptance limit for precision (LAP) for each nuclide is differently defined as showed in Table 1 [1, 5].
The estimation results of gross alpha/beta activities in the spiked water were applied using R (= Lab value/Lab uncertainty) and RB. More details are available for reference in the report by Shakhashiro et al. [1].
Experimental
The sample was prepared using tap water outsourced in one batch from Seibersdorf laboratories and then was spiked with an appropriate amount of radionuclide solution [1].
For Ra analysis, 10 dpm 229Th–225Ra tracer (code 6229, Eckert & Ziegler) was inserted into a 20 g spiked water sample after acidifying with HNO3. Here, we assume that the recoveries of radium and thorium from the sample were equal, as much of the original 225Ra decayed before further steps were completed. Pb(NO3)2 solution was added to the acidic sample to form a Pb(Ra)SO4 co-precipitation by adding dilute H2SO4 and solid K2SO4. The co-precipitate was centrifuged and redissolved in ethylene diamine tetraacetic acid (EDTA). This solution was passed through an anion exchange column (DOWEX 1X8-200, 100–200 mesh, chloride form, 50 mm height, 7 mm i.d.) and a cation exchange column (DOWEX 50WX8-200, 200–400 mesh, 80 mm height, 7 mm i.d.). Finally, Ra isotopes were electrodeposited onto a stainless steel disc and determined by alpha-spectrometry (Model: Canberra 7200-08). The activity of 226Ra was obtained immediately after the counting [6].
Similarly, a 232U tracer (code 7432, Eckert & Ziegler) was added into 30 g spiked water after acidifying the sample for 234,238U analysis. The water was converted to a 9 mol/L HCl solution and then loaded onto a resin column (DOWEX 1X8-200, 100–200 mesh, chloride form, 80 mm height, 7 mm i.d.). Uranium was then eluted using 0.1 mol/L HCl. Finally, uranium was electrodeposited onto a stainless steel disc. After electrodeposition onto stainless steel discs, the activity of uranium was determined by alpha-spectrometry.
For gross alpha/beta activity, analyses are referenced as reported by Zhang et al. [7]. Briefly, the radioactivity of gross alpha and gross beta were determined with a MPC 9604 ultra-low-level counter, while U3O8 (which was fired from UO2(NO3)2 and KCl) was selected as sub-standard and standard reference substances.
Results and discussions
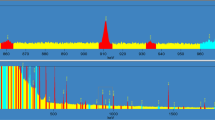
First, we used 500 mL water sample to determine whether activities in the spiked water can be quantified by He-gamma spectrometry. The one typical gamma-ray spectrum is showed in Fig. 1, which shows that there are almost no gamma-ray peaks after counting for 12 h. Generally, 226Ra and 234,238U or their daughter nuclides below 1 Bq/kg in water are difficult to quantify by gamma-ray spectroscopy because of a lower limit of detection (LLD) that is mostly caused by low detection efficiency due to the background contribution. Furthermore, 238U lacks direct gamma emissions and can be measured only assuming a radioactive equilibrium with the daughter nuclide 234Th (useful gamma emissions at 63.3 and 92.4–92.8 keV); 226Ra has a direct gamma emission at 186.2 keV strongly interfering with both 235U gamma emission and the background contribution [8–10].

The gamma-spectroscopy of water sample-1 of IAEA-CU-2010-03
As Table 1 shows, the laboratory has a good intercalibration result using α-spectroscopy. For 234U and 238U, most A 1 values were much lower than A 2 values, and P values were also much lower than LAP. For 226Ra, A 1 values were just a little lower than A 2 values. The absolute value of relative bias ranged from 1.79 to 39.76%, and most of the relative bias values were lower than 15% (Fig. 2). All of the lab values were acceptable, except one 226Ra value (because of the calculating mistake).

A comparison of the laboratory results with the IAEA value
As IAEA reported [1], there are 32 and 36 laboratories that had only one or two ‘not acceptable’ results, respectively. The unacceptable performance was mainly due to 226Ra.
Because the analytical system is a complex system, such cases can even include human factors, cultural effects and the specific conditions of each laboratory that can impact intercalibration results. Therefore, it is quite normal to make some mistakes in an analytical laboratory. However, their impact on the analysis data should be noted.
Tracer choice
Because of the complex radiochemical procedures for the analyses of 226Ra, 234U and 238U, yield tracers are necessary. The choice of yield tracer is important in an analysis to distinguish chemical recovery between the tracer and the nuclides. For the 226Ra measurement, the normal tracers are 225Ra, 224Ra, 223Ra and 133Ba [11–15]. Benedik et al. [14] showed that the activity concentrations of 226Ra are within measurement uncertainties for 225Ra, 223Ra, and 133Ba. A lower recovery was always found when 133Ba was used. 223Ra and 225Ra are generally used, but due to their short half-lives, their decays should be taken into account as well as the growth of their decay products.
For 234U and 238U measurement, 232U and 236U are widely used [16–22]. It is necessary to distinguish between the tracer and the nuclides. 232U and 236U can both be used as tracers for the measurement of uranium (Fig. 3). Figure 3 shows that the distance between 232U and 234U or 238U is larger than the distance between 236U and 234U or 238U. Therefore, 232U is better for tracer yield, especially when the chemical operation has some problems and the background and the tail of the peak is high.

The spectrum of uranium measurement using 232U and 236U as tracers
U3O8 and KCl were selected as sub-standard and standard reference substances for gross alpha analysis and gross beta analysis, respectively. The results indicate that when one selects UO2(NO3)2 as a sub-standard reference substance for gross α analysis, the total alpha activity of UO2(NO3)2 is much lower than that of the standard U3O8 that is recommended by GB 5750-85, only because the 238U and 234U do not reach the secular equilibrium for our laboratory sub-standard UO2(NO3)2 reagent. However, no effect of such UO2(NO3)2 on the analysis of gross alpha activity was observed after the activity of 234U and 238U was qualitatively measured by alpha-spectrometry.
Detector background
The background of alpha-spectrometry is very low in comparison with the background of gamma-spectrometry, as the system is a vacuum system. However, the background is important for the analysis of gross alpha and gross beta, especially when the environmental sample’s activity is small. The capacity of the detector is an important factor for the background, such as the lead block, the air and the tray. The filter is usually used in environmental sample analysis, and the material of the filter can greatly affect the background. The difference between various materials is too high as showed in Table 2. It can be seen that the background of the QMA filter (Whatman) is the highest, and the backgrounds of the acetate (Xinya), mixed cellulose ester (Xinya) and nuclear track membranes (Whatman) are almost the same. These results are comparable with the results reported by Ma et al. [23]. In addition, the results in Table 2 show that the background with and without air circulation in the instrument room did not change much. This result indicates that the air does not affect the background of the detector.
The background is related to the LLD and can be estimated as follows [24, 25]:
where B is the background counting number, η is the fractional counting efficiency, m is the sample mass, and t is the count time. For the analysis of environmental samples, the LLD should reduce the background, promote detectable efficiency and extend the detection time.
For the alpha-spectrometry vacuum, the background of alpha-spectrometry can be neglected. However, for gross alpha and gross beta procedures, the background is one of the most important factors. The background of trays without sample was detected for calculating the lower limit of detection. The change in the lower limit of detection (5 g sample) with the detected time was shown in Fig. 4. Figure 4 indicates that LLD decreased with the detected time. The LLD decreased sharply within 240 min, but the LLD did not change greatly after 240 min. For the environment sample, which has low activity, the LLD should be detected within at least 4–5 h.

The LLD of gross alpha/beta vs. detected time (the background count number was the background of trays without sample and the mass was assumed to be 5.00 g)
Shape and tail of the peak in alpha-spectroscopy
As many environmental samples have relatively low count rates for alpha-spectroscopy, the extent of the tailing of alpha peaks of lower energy is a critical factor for accurate spectral analysis.
The chamber gas pressure and source deposit thickness are the main factors affecting the tailing and peak resolution. Martin and Hancock [26] reported that full width at half maximum (FWHM) increased linearly with mass per unit area. Although the measurement has a procedure to separate the impurities, the sample may still have some impurities during the environmental sample analysis, and the deposit source may be thick. The unproficient operation can also have a thicker deposit source.
Estimated error of the results
In the final evaluation, both sources for trueness and precision were combined. The measurement uncertainty was used for calculating the trueness and precision, which are important for improving the measurement uncertainty. The laboratories’ results show that much of their uncertainty is higher than the IAEA’s uncertainty, which may influence the results.
The uncertainty in the measurement activity of gross alpha can be calculated using the equation [7]:
The uncertainty of the measurement activity of 226Ra, 234U, 238U and gross beta can be calculated as the following equation:
where m s and m are the mass of the standard solution and the mass of the sample; n x, n 0 and n s are the count rate of the sample, background and standard solution; \(S_{{m_{\text{S}}}}\) and S m are the uncertainty of the mass of the standard solution and the mass of the sample; and \(S_{n_{{x}}}\), \(S_{n_{{0}}}\) and \(S_{n_{{s}}}\) are the uncertainties in the sample count rate, background and standard solution.
Figure 5 shows the percentage of each part of the uncertainty in measured 238U activity with detected time. The results indicate that the uncertainty of the mass of the standard solution and of the sample accounted for only a small percentage of the uncertainty (less than 0.001%). The uncertainty of the mass of the standard solution and the sample is specific to a laboratory, and it can be reduced with as much sample as possible. The uncertainty in the measurement activity is most dependent on the uncertainty in the count rate. The sum of the uncertainties in the count rate of a sample and the standard solution can reach almost 100%. The percent of the uncertainty in the count rate of a sample increased with the detection time, and the percent of the uncertainty in the count rate of the standard solution decreased within 25 h. After 25 h, the percent of the uncertainty in the count rate of the sample and the standard solution were comparable. However, the uncertainty in the count rate decreased exponentially over time. To improve the precision, suitable materials and a suitable environment should be selected to reduce the background. A sample should be detected for as long as possible to obtain a reasonable measure of uncertainty in the count rate.

The percent of each part in calculating the measured uncertainty vs. the detected time (30.00 g sample-3 of IAEA-CU-2010-03 with 2.000 dpm 232U tracer)
Conclusions
Although the analytical system is a very complex system, good results can be obtained when care is taken with impact factors such as background and the environmental conditions of each laboratory.
Generally, proper equipment, human resources and material resources are important factors in obtaining reliable and high-quality results. Further improvement in the radiochemical analytical procedures for the determination of natural and transuranic radionuclides at a low level of radioactivity is necessary by capacity building with such analytical procedures.
To reduce the background and the lower limit of detection, the choice of a suitable material should be taken into account, especially when the activity is low. More samples and an extended analysis time can lower the uncertainty in measure activities.
Certain recommendations from the IAEA report can definitely enhance the analytical performance of the participating laboratories. In the present work, some comments and recommendations are given below.
The determination of chemical yield is also a limiting factor for the determination of radium and uranium isotopes by alpha-spectrometry. In the case of Ra, there are assumptions about its equilibrium, both with parent (229Th) and daughter (225Ac) nuclides. Although uranium measurement by alpha-spectrometry is a well-established procedure, it is not the easiest method. Moreover, special care should be taken to avoid cross-contamination from the repeated use of beakers and deposition cells. The internal yield tracers must be assured at all times once opened (and diluted), and internal procedures should be applied for their management (dilutions included), possibly including periodical monitoring of their actual concentrations.
The gross alpha/beta measurements are considered to be primarily screening determinations. Radionuclides were successfully used in calibrations, including 40K and 238U/234U.
References
Shakhashiro A, Sansone U, Trinkl A, Kim CK, Kis-Benedeck G, Forte M, Rusconi R, Tarjan S (2009) World wide open proficiency test: determination of naturally occurring radionuclides in phosphogypsum and water IAEA-CU-2008-03. International Atomic Energy Agency, Seibersdorf
Bleise AR, Smodis B, Glavic-Cindro D, Parr RM (2001) The updated IAEA database of natural matrix reference materials. J Radioanal Nucl Chem 248(1):205–209
Iyer RM, Oblozinsky P, Muir DW, Schwerer O (1999) IAEA nuclear data services for radiochemistry. J Radioanal Nucl Chem 239(1):139–141
Shakhashiro A, Fajgelj A, Sansone U (2007) Comparison of different approaches to evaluate proficiency test data. In: Combining and reporting analytical results. The Royal Society of Chemistry, pp 220–228
Shakhashiro A, Mabit L (2009) Results of an IAEA inter-comparison exercise to assess 137Cs and total 210Pb analytical performance in soil. Appl Radiat Isot 67(1):139–146
Su N, Du J, Moore WS, Liu S, Zhang J (2011) An examination of groundwater discharge and the associated nutrient fluxes into the estuaries of eastern Hainan Island, China using 226Ra. Sci Total Environ 409(19):3909–3918
Zhang Y, Zhao F, Wu M, Du J, Zhang J (2011) Gross α/β analysis of IAEA 2008 world-wide open proficiency test on determination of natural radionuclides in spiked water. J Nucl Radiochem 33(1):42–47
Dulaiova H, Burnett WC (2004) An efficient method for gamma-spectrometric determination of radium-226, 228 via manganese fibers. Limnol Oceanogr Meth 2:256–261
Justo J, Evangelista H, Paschoa AS (2006) Direct determination of Ra-226 in NORM/TENORM matrices by gamma-spectrometry. J Radioanal Nucl Chem 269(3):733–737
Ishikawa Y, Murakami H, Sekine T, Saito T, Yoshihara K (1994) Non-destructive determination of low-level 210Pb and 226Ra with an ordinary high-purity Ge-detector. J Radioanal Nucl Chem 178(2):301–310
Crespo MT (2000) On the determination of 226Ra in environmental and geological samples by α-spectrometry using 225Ra as yield tracer. Appl Radiat Isot 53(1–2):109–114
Hancock GJ, Webster IT, Ford PW, Moore WS (2000) Using Ra isotopes to examine transport processes controlling benthic fluxes into a shallow estuarine lagoon. Geochim Cosmochim Acta 64(21):3685–3699
Saueia C, Mazzilli B, Taddei M (2009) Sequential radioanalytical method for the determination of U and Th isotopes, 226Ra and 210Po using alpha spectrometry in samples of the Brazilian phosphate industry. J Radioanal Nucl Chem 281(2):201–204
Benedik L, Repinc U, Strok M (2010) Evaluation of procedures for determination of Ra-226 in water by α-particle spectrometry with emphasis on the recovery. Appl Radiat Isot 68(7–8):1221–1225
Zhang L (2007) Radium isotopes in Changjiang Estuary/East China Sea and their application in analysis on mixing among multiple water masses. Ph.D dissertation. East China Normal University, Shanghai
Kharkar DP, Tiiomson J, Turekian KK, Forster WO (1976) Uranium and thorium decay series nuclides in plankton from the Caribbean. Limnol Oceanogr Meth 21:294–299
Boulyga SF, Testa C, Desideri D, Becker JS (2001) Optimisation and application of ICP-MS and alpha-spectrometry for determination of isotopic ratios of depleted uranium and plutonium in samples collected in Kosovo. J Anal At Spectrom 16(11):1283–1289
El Mamoney MH, Khater AEM (2004) Environmental characterization and radio-ecological impacts of non-nuclear industries on the Red Sea coast. J Environ Radioact 73(2):151–168
Moore WS, Shaw TJ (2008) Fluxes and behavior of radium isotopes, barium, and uranium in seven southeastern US rivers and estuaries. Mar Chem 108(3–4):236–254
Saidou, Bochud F, Laedermann J, Njock MGK, Froidevaux P (2008) A comparison of alpha and gamma spectrometry for environmental natural radioactivity surveys. Appl Radiat Isot 66(2):215–222
Jia G, Torri G, Magro L (2009) Concentrations of 238U, 234U, 235U, 232Th, 230Th, 228Th, 226Ra, 228Ra, 224Ra, 210Po, 210Pb and 212Pb in drinking water in Italy: reconciling safety standards based on measurements of gross α and β. J Environ Radioact 100(11):941–949
Lee SH, La Rosa J, Gastaud J, Povinec PP (2005) The development of sequential separation methods for the analysis of actinides in sediments and biological materials using anion-exchange resins and extraction chromatography. J Radioanal Nucl Chem 263(2):419–425
Ma Q, Chen M, Qiu Y, Huang Y (2005) MnO2 co-precipitation and direct beta counting technique for determining 234Th in small-volume seawater. Acta Oceanol Sin 27(4):68–75
Biggin CD, Cook GT, MacKenzie AB, Pates JM (2002) Time-efficient method for the determination of 210Pb, 210Bi and 210Po activities in seawater using liquid scintillation spectrometry. Anal Chem 74(3):671–677
Hong Y, Kim G (2005) Measurement of cosmogenic 35S activity in rainwater and lake water. Anal Chem 77(10):3390–3393
Martin P, Hancock GJ (2004) Peak resolution and tailing in alpha-particle spectrometry for environmental samples. Appl Radiat Isot 61:161–165
Acknowledgments
This research was supported by the Natural Science foundation of China (41021064, 40976054), the State Key Laboratory of Estuarine and Coastal Research of China (2011KYYW04) and D.K. Huang was supported by ECNU Reward for Excellent Doctors in Academics (XRZZ2011020). We thanked the colleagues from the radioisotope group of SKLEC for their help in laboratory experiments.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Huang, D., Du, J. & Zhang, J. Intercalibrated radionuclide activities in spiked water samples of the IAEA worldwide open proficiency test. J Radioanal Nucl Chem 292, 1241–1248 (2012). https://doi.org/10.1007/s10967-011-1590-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-011-1590-0